
Abstract
Background
Artemisinin-based combination therapy (ACT) has been a major contributor to the substantial reductions in global malaria morbidity and mortality over the last decade. In Tanzania, artemether-lumefantrine (AL) was introduced as the first-line treatment for uncomplicated Plasmodium falciparum malaria in 2006. The World Health Organization (WHO) recommends regular assessment and monitoring of the efficacy of the first-line treatment, specifically considering that artemisinin resistance has been confirmed in the Greater Mekong sub-region. This study's main aim was to assess the efficacy and safety of AL for treating uncomplicated P. falciparum malaria in Tanzania.
Methods
This was a single-arm prospective antimalarial drug efficacy trial conducted in four of the eight National Malaria Control Programme (NMCP) sentinel sites in 2019. The trial was carried out in outpatient health facilities in Karume-Mwanza region, Ipinda-Mbeya region, Simbo-Tabora region, and Nagaga-Mtwara region. Children aged six months to 10 years with microscopy confirmed uncomplicated P. falciparum malaria who met the inclusion criteria were recruited based on the WHO protocol. The children received AL (a 6-dose regimen of AL twice daily for three days). Clinical and parasitological parameters were monitored during follow-up over 28 days to evaluate drug efficacy.
Results
A total of 628 children were screened for uncomplicated malaria, and 349 (55.6%) were enrolled between May and September 2019. Of the enrolled children, 343 (98.3%) completed the 28-day follow-up or attained the treatment outcomes. There were no early treatment failures; recurrent infections during follow-up were common at two sites (Karume 29.5%; Simbo 18.2%). PCR-corrected adequate clinical and parasitological response (ACPR) by survival analysis to AL on day 28 of follow-up varied from 97.7% at Karume to 100% at Ipinda and Nagaga sites. The commonly reported adverse events were cough, skin pallor, and abdominal pain. The drug was well tolerated, and no serious adverse event was reported.
Conclusion
This study showed that AL had adequate efficacy and safety for the treatment of uncomplicated falciparum malaria in Tanzania in 2019. The high recurrent infections were mainly due to new infections, highlighting the potential role of introducing alternative artemisinin-based combinations that offer improved post-treatment prophylaxis, such as artesunate-amodiaquine (ASAQ).
Similar content being viewed by others
Background
Artemisinin-based combination therapy (ACT) has been a major contributor to the substantial reductions in global malaria morbidity and mortality over the last decade [1]. The most commonly used artemisinin-based combinations in Africa are artemether-lumefantrine (AL) and artesunate-amodiaquine (ASAQ) [2]. AL was introduced as the first-line treatment for uncomplicated Plasmodium falciparum malaria in 2006 in Tanzania. Continuous use of a single ACT may result in unidirectional selection of resistant parasites [3, 4].
Despite the high ACT cure rates observed in Africa, studies conducted in Tanzania and other parts of Africa provide evidence for in vivo selection of lumefantrine tolerant/resistant parasites [5, 6]. Tolerance to AL has been linked to the selection of single nucleotide polymorphism (SNPs) associated with parasite drug tolerance/resistance in the P. falciparum multidrug resistance gene 1 (pfmdr1) at N86Y, Y184F and D1246Y; P. falciparum chloroquine resistance transporter gene (pfcrt) at K76T; and P. falciparum multidrug resistance-associated protein gene (pfmrp1) [7, 8]. Importantly, no clear evidence of increased pfmdr1 gene copy, previously linked to lumefantrine resistance in South-East Asia, exists to date in East Africa. After treatment with AL, lumefantrine selects for pfmdr1 N86, 184F, D1246, and pfcrt K76 [7, 8]. This hypothesis is supported by data from Bagamoyo District, indicating that reinfecting P. falciparum parasites harboring the pfmdr1 N86/184F/D1246 haplotype were able to withstand 15-fold higher lumefantrine blood concentrations than those with the alternative haplotype (86Y/Y184/1246Y) [6, 9].
The development of tolerance/resistance against the long-acting partner drugs in ACT, such as lumefantrine and amodiaquine, has been suggested to start through post-treatment selection among recurrent infections of less sensitive P. falciparum parasites as reinfecting parasites need to be able to survive the exposure of sub-therapeutic blood levels of the long-acting drug and its active metabolites. This may, in turn, lead to a gradually shortened post-treatment prophylactic period, long before clinical treatment failures are apparent, which is why temporal surveillance of efficacy and genetic anti-malarial drug resistance markers of P. falciparum has been proposed as an early warning system of evolution of ACT tolerance/resistance [10, 11].
Furthermore, gains in elimination efforts are threatened by the recent emergence of artemisinin and partner drug resistance in Southeast Asia [12]. This region has been the epicentre for the evolution and spread of resistance to every region. Evidence of reduced susceptibility to artemisinin in Western Cambodia was first reported in January 2007 and confirmed in subsequent detailed studies [3, 12, 13]. A major concern is that artemisinin and partner drug resistance may spread across a wider geographic area, as chloroquine resistance did in the 1960s and 1970s, moving from Southeast Asia to the Indian subcontinent and subsequently to Africa, which bears the vast majority of the global malaria burden. Partial artemisinin resistance has now been detected in several sub-Saharan African countries, including Uganda, Eritrea, and Rwanda [3, 4, 14, 15].
The development of partial resistance to artemisinin in Uganda, Eritrea, and Rwanda did not originate from the spread of genetic mutations from the Greater Mekong Subregion (GMS) but arose de novo [16]. Recently, partial artemisinin resistance was detected in the Kagera region of Tanzania [17]. The study revealed that 17% of patients in Kagera exhibited day three parasitaemia. Additionally, 10% of patients showed both day three parasitaemia and a WHO-validated k13 mutation (R561H), suggesting the presence of partial artemisinin resistance.
The National Malaria Control Programme (NMCP) in collaboration with its implementing partners conducts therapeutic and safety studies at selected sentinel sites at least every two years as recommended by the World Health Organization (WHO) [18]. Recent AL studies have shown a high cure rate of > 95% [5, 18,19,20]. This study was conducted to provide updated information on the AL’s efficacy and safety for treating uncomplicated P. falciparum malaria in Tanzania.
Methods
Study design
This was a single-arm prospective study for assessing the therapeutic efficacy and safety of AL for the treatment of uncomplicated falciparum malaria in children aged between six months and 10 years.
Study sites
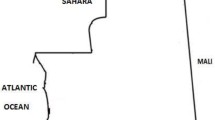
The study was conducted at four of the eight NMCP sentinel sites (Karume-Mwanza region, Ipinda-Mbeya region, Simbo-Tabora region, and Nagaga-Mtwara region) between May and September 2019. The study sites (Fig. 1) have been NMCP sentinel sites for monitoring of antimalarial efficacy since 1997 [5, 20, 21]. The eight primary or secondary health care facilities included in Fig. 1 represent basic features of geographic zones for mainland Tanzania. This geographical diversity has been considered as representative of the malaria epidemiology in Tanzania based on the past and current shift of malaria transmission intensity [22]. Malaria transmission in the majority of areas in Mainland Tanzania tends to be characterized as low to moderate [22]. However, seasonal peaks of malaria transmission occur subsequent to the primary long rainfall season, typically between March and June, although the specific timing may vary depending on the patterns of rainfall. Throughout Tanzania, P. falciparum is the predominant malaria species and Anopheles gambiae sensu stricto (s.s.), Anopheles arabiensis, and Anopheles funestus are now the primary vectors of human malaria in East Africa [23, 24].

The four study sites for antimalarial therapeutic efficacy in Tanzania in 2019
Study population
Children aged between six months and 10 years presenting with fever (axillary temperature ≥ 37.5 °C and/or reported history of fever in the past 24 h) were screened for possible enrollment into the study following inclusion and exclusion criteria as follows: mono-infection of P. falciparum detected by microscopy, parasitaemia between 250 and 200,000 asexual parasites/μl of blood, ability to swallow oral medications, ability and willingness to attend scheduled follow-up visits, informed consent provided by parent or guardian, and stable residence within the catchment area throughout the study period. Exclusion criteria included patients with negative malaria rapid diagnostic test (RDT) results and general danger signs or signs of severe malaria. Danger signs of malaria in children consisted of the following clinical presentation: child unable to drink or breastfeed, vomiting, recent history of convulsions, lethargic or unconscious state, unable to sit or stand, and difficulty in breathing [18]. Patients with mixed or mono-infections with another Plasmodium species, severe anaemia (Hb < 5 g/dL), or presence of severe malnutrition (defined as a child who had symmetrical edema involving at least the feet or mid-upper arm circumference < 110 mm) were excluded from the study. Other exclusion criteria included febrile conditions due to diseases other than malaria (e.g., measles, acute lower respiratory tract infection, severe diarrhoea with dehydration) or other known underlying chronic or severe diseases (including cardiac, renal, and hepatic diseases, and HIV/AIDS), regular medications that may interfere with anti-malarial pharmacokinetics, and history of hypersensitivity reactions or contraindications to AL. Excluded patients received appropriate treatment according to the national guidelines [25].
Sample size estimation
The sample size was determined based on the WHO 2009 standard protocol with the assumption that 5% of the patients treated with AL were likely to have treatment failure [18]. At a confidence level of 95% and an estimated precision of 5%, the minimum sample size was 73 patients at each site. With a 20% increase to allow for loss to follow-up and withdrawals during the 28-day follow-up period, 88 patients were targeted per site.
Sample collection
Blood samples were collected through a finger prick for malaria RDT (Carestart™ ACCESSBIO, USA) and thick and thin blood smears for detection of malaria parasites by microscopy. From each patient, dried blood spots (DBS) on Whatman III filter papers were collected for laboratory analysis of malaria parasites, including P. falciparum diversity, molecular markers of antimalarial resistance, and distinguishing recrudescent from new infections by PCR genotyping. Thick blood smears were stained with 3% Giemsa for 30–45 min and examined by microscopy to detect presence of malaria parasites and the level of parasitemia. Parasitaemia was measured by counting the number of asexual parasites against 200 white blood cells (WBCs) in thick blood films; thin blood films were examined for detection of the different parasite species as previously described [20]. A thick blood smear was declared negative when examination of 100 high power fields did not reveal the presence of any malaria parasite. For quality control, each slide was re-examined by a second microscopist and those with discrepancy were re-examined by a third microscopist. Further disagreement was resolved by a team of three microscopists who examined the same slide at the same time. Final parasitaemia was calculated as the average between the two closest readings.
Patient treatment and follow-up
During screening, patients were clinically examined before going to the laboratory for sample collection. Patients enrolled in the study were treated with AL (Coartem®, Beijing Novartis Pharma Ltd, Beijing China for Novartis Pharma AG, Basle, Switzerland, obtained from the WHO). This was a fixed dose combination of 20 mg of artemether and 120 mg lumefantrine in a tablet. The drugs were administered without food according to the recommended doses based on the weight of patient [25]. While treatment did not include the provision of food, caregivers were advised to supply fatty meals at home to enhance the drug's absorption. Patients were monitored for 30 min to confirm the absence of any vomiting related to the study drugs. One tablet was given to children weighing 5–14 kg; two tablets to children weighing 15–24 kg, and three tablets to children weighing 25–35 kg. A full course of AL consisted of 6-doses given twice daily (8 hourly apart on day 0 and 12 h apart on days 1 and 2). The study drugs were given under direct supervision of the study nurses at the health facility. In case of failure, the participants were treated following the National Malaria Treatment Guidelines [25].
All enrolled patients were followed for 28 days with scheduled visits on days 1, 2, 3, 7, 14, 21, and 28. During follow-up visits, clinical and safety assessments were performed, axillary temperature was measured, and a blood slide for parasite count was taken. On day 7, 14, and 28, DBS were collected on filter papers for genotyping. The patients and their parents or guardians were also informed to return on any day if the symptoms returned or any other danger signs were present. Patients who could not come for their scheduled visit by mid-day were visited at home by a member of the study team and asked to come to the health facility. In case a patient travelled outside the study area and could not be traced for scheduled follow-up within 2 days, he/she was withdrawn from the study. Patients who did not attend the scheduled visits or could not be found despite all reasonable efforts were classified as lost to follow-up.
Sample processing and analysis
Parasite DNA was extracted from DBS using QIAamp DNA blood Midi Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s instructions. All paired samples collected on day 0 and day of recurrent infection were genotyped by utilizing merozoite surface proteins 1 and 2 (msp1 and msp2) and glutamate rich protein (glurp) using gel electrophoresis. Bands were considered a match if the day 0 and day of failure fragment lengths were within 10 base pairs for msp1 and msp2 and within 50 base pairs for glurp.
Reinfection and recrudescence were differentiated using both the 3/3 and 2/3 methods as recommended by the WHO [26]. The 3/3 algorithm required at least one matching band in any sub-allele for all three makers. If Day 0 and recurrent samples shared alleles for at least 2/3 markers it was classified as recrudescence. If insufficient matches were identified, the recurrence was classified as reinfection. If there were no amplification products resulting in sharp, defined bands in both the day 0 and day of failure samples for a gene, that gene was not used to distinguish between recrudescence and reinfection. Determinations using the 3/3 method were used for analysis; 2/3 results were added in the supplemental materials [26, 27]. Raw genotyping data have been included as supplemental material (Additional file 1: Table S1).
The following molecular markers were genotyped to assess for anti-malarial drug resistance: multidrug resistance 1 (mdr1) and polymorphisms in kelch propeller domain (k13) [13] by capillary sequencing, and mdr1 copy number variants according to published protocols27. SNPs calling in mdr1 and k13 was performed using Geneious® analysis software (Biomatters, New Zealand; www.geneious.com) by mapping the sequence data on the 3D7 reference sequences. Raw sequence reads were cleaned using default settings and reads with high-quality scores (the percentage of high-quality bases) below 70% were discarded from further analysis. The pfk13 (codon positions 440–600) and pfmdr1 (codon positions: 86, 184 and 1246) were analysed for SNPs. SNPs were called only if they fit the following criteria: (1) they were found in both the forward and reverse reads, (2) they had a p-value of < 0.0001 (p-value represents the probability of a sequencing error resulting in observing bases with at least the given sum of qualities), and (3) they had a minimum strand bias p-value of < 0.0005 when exceeding 65% strand bias, as some errors from sequencing machines are more likely to happen on nearby upstream bases.
Study outcomes
The primary end point was parasitological cure on day 28 as per WHO protocol of 2009 [18], while secondary end points included occurrence and severity of adverse events and molecular markers of drug resistance. Treatment outcomes were classified as: (1) Early treatment failure (ETF) if the patient had presence of parasitaemia and danger signs on day 1, 2 or 3 or persistence of parasitaemia until day 3; (2) Late clinical failure (LCF) was defined as presence of danger signs or severe malaria, or axillary temperature of ≥ 37.5 °C with parasitaemia between days 4 and 28 in a patient who did not qualify as early treatment failure; (3) Late parasitological failure (LPF) if a patient had parasitaemia between days 7 and 28 in the absence of fever or other clinical symptoms, and was not classified as early treatment failure; (4) Adequate clinical and parasitological response (ACPR) in the absence of parasitaemia in a patient who was not classified as early, late clinical, or late parasitological failure; (5) lost to follow-up when despite all reasonable efforts, an enrolled patient did not attend the scheduled visits and could not be found, and thus the patient was withdrawn from the study; and (6) withdrawal when the patient consented to withdrawal, failed to complete treatment, or there was a protocol violation.
Data management and analysis
Single data entry was performed at the study sites; this was followed by second entry after the end of data collection. The data was entered into a Microsoft Access database, and later validated, cleaned, and analysed using STATA for Windows, version 11 (STATA Corporation, TX-USA). Descriptive statistics such as percentages, mean, median, standard deviation, and range were reported as appropriate. Treatment outcomes were analysed based on per protocol analysis[18]. In the per protocol analysis, patients with new infections, loss to follow up, withdrawal or protocol violations as well as those with indeterminate PCR results, were excluded. In Kaplan–Meier analysis patients were censored with new infections, lost to follow up, withdrawal, or protocol violations. Patients with indeterminate PCR results were excluded from the analysis of PCR-corrected treatment outcome[18]. Baseline characteristics and primary and secondary outcomes were presented descriptively for the four sites. Continuous variables such as parasite density and age among the four sites were compared using t-test (for normally distributed data) or Mann–Whitney U test (a non-parametric test for non-normally distributed data).
Ethical consideration
Ethical clearance was obtained from the Medical Research Coordinating Committee (MRCC) of the National Institute for Medical Research (NIMR) with reference number NIMR/HQ/R.8c/Vol.I/1149. Permission to conduct the study at the health facilities was sought in writing from the relevant regional and district medical authorities. Oral and written informed consent was obtained from parents or guardians of all eligible patients before they were screened for possible inclusion into the study.
Results
Basic characteristics
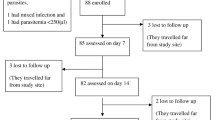
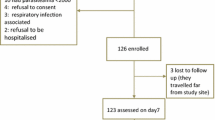
During the study period, 628 children were screened for uncomplicated malaria and 349 (55.6%) were enrolled between May and September 2019. Of the screened children, 279 (44.4%) were excluded because of presence of fever due to other causes (positive RDT but negative blood slide), low parasitaemia outside the defined interval, severe malaria, or living outside the study area (Fig. 2). Only three patients were lost lo follow up (Fig. 2). Table 1 summarizes the demographic and laboratory baseline data of the participants.

Trial profile showing the flow of patients at screening, enrollment, and follow-up at the four study sites Karume, Ipinda, Simbo, and Nagaga. ACPR Adequate clinical and parasitological response, LCF Late clinical failure, LPF Late parasitological failure
The Karume site recruited more females and children with lower age and body weight compared to other sites. The average axillary temperature was similar at all sites (Table 1). The geometric mean parasite density was comparable across the study sites (Fig. 3).

Distribution of geometric mean parasite density between microscopy readers by study site on day 0. Error bars are 95% Confidence intervals
Efficacy outcomes
The treatment outcomes are summarized in Table 2. As per the Kaplan–Meier analysis, the PCR-uncorrected ACPR with 95% CI was: 73.9% (63.3–81.8) in Karume, 92% (83.9–96.1) in Ipinda, 80.8% (70.6–87.8) in Simbo, and 90.6% (82.1–95.2) in Nagaga. No early treatment failure was recorded. The PCR results for two patients (one in Ipinda and one in Nagaga) with undetermined results were excluded from PCR corrected analysis. A total of three patients were withdrawn from the study, two from Simbo and one from Ipinda due to protocol violation. The Kaplan–Meier PCR-corrected results showed the ACPR was: 97.7% (91.0–99.4) in Karume, 98.9% (92.2–99.8) in Ipinda, and 100% in Simbo and Nagaga sites. The per protocol PCR-corrected cure rate was 97.0% (88.6–99.3) in Karume, 98.8% (91.5–99) in Ipinda, and 100% in Simbo and Nagaga sites. The recently recommended WHO guidelines on genotyping to detect recrudescent infections after antimalarial treatment (2/3 algorithm)[27] was more sensitive in detecting recrudescent infections resulting in lower cures rates (Additional file 2: Table S2).
Parasite clearance
Karume and Simbo sites had many patients with parasitaemia on day 2 compared to other sites (Fig. 4). One patient from Karume site had parasitaemia on day 3 (272 asexual parasites/μL); however, the parasitaemia was not greater than that of day 0 (11,320 asexual parasites/μL).

Microscopy positivity among enrolled patients at the four study sites. N (%) of patents enrolled
Occurrence of adverse events
A total of 46 adverse events were reported; no serious adverse events (SAE) were recorded. The most common adverse events were cough 24 (52.2%) and skin pallor 11 (23.9%). Distribution of adverse events is shown in Table 3.
Prevalence of molecular markers of drug resistance before and after treatment with AL
Out of 349 samples collected on day 0, 311 (89.1%) were successfully sequenced for pfk13 and 328 (94%) were successfully sequenced for pfmdr1. Out of 54 samples collected on the day of recurrent infection, 42 (77.8%) were successfully sequenced for pfk13 and 43 (79.6%) were successfully sequenced for pfmdr1. Among the 311 sequenced samples collected on day 0, 10 (3.2%) had mutations in the pfk13 gene; there was no mutation detected on the days of recurrent infection (Table 4). Of the 10 pfk13 mutations detected on day 0, nine samples had synonymous mutations and one sample had non-synonymous mutation (Table 5). However, these mutations were neither candidate nor validated mutations according to the WHO list published in November 2020 (https://www.who.int/publications/i/item/9789240012813). All successfully sequenced samples on day 0 and on days of recurrent infection were carrying the wild type pfmdr1 N86. The frequency of parasites carrying mutant type pfmdr1 184F increased from 32.9% on day 0 to 55.8% on the days of recurrent infection (Table 4). For pfmdr1 D1246, 372/403 (92.3%) of samples were successfully sequenced and all had wild type D1246 polymorphisms (Table 4).
Discussion
The study findings show that AL, the recommended first-line ACT for treatment of uncomplicated falciparum malaria in Tanzania, is still highly efficacious, with a PCR-corrected cumulative cure rate ranging from 97% at the Karume site to 100% at the Ipinda and Nagaga sites (per protocol analysis). These results are consistent with those obtained using the Kaplan–Meier method and are similar to our previous findings [5, 19, 23, 28, 29] in the same surveillance areas. Following the 2/3 match method for genotyping msp1, msp2, and glurp genotyping, lower cure rates, as more recrudescent infections were detected; this was similar to previous reports in high transmission settings that observed the main marker leading to discordance between the two analyses was glurp [27, 30].
However, the risk of recurrent parasitemia was high in Karume and Simbo, similar to the previous study [20]. The majority of patients with a recurrent infection presented at or after day 21. Potential contributing factors for recurrent infections could be high transmission in the study areas [31] and reduced prophylactic effect after treatment with AL, which is known to have a short elimination half-life.
Artemisinin resistance, defined as partial resistance by the WHO, is phenotypically characterized by prolonged P. falciparum clearance time after treatment with an artesunate monotherapy or ACT [32]. Resistance has been linked to specific mutations in the P. falciparum Kelch 13 propeller gene (pfk13) [3, 13, 33]. In this study, 10 pfk13 mutations were identified, but none were validated or candidate mutations according to the WHO artemisinin resistance protocol [32]. However, mutations have recently been documented in Rwanda, the first report of locally arising pfk13 mutations in Africa, without affecting the efficacy of AL [15]. From the mutations reported in Rwanda, one was among validated markers (561H) and three were candidate markers (469F, 441L and 449A) [15]. Evidence of partial artemisinin resistance, characterized by delayed parasite clearance with > 10% of patients remaining parasitaemic on day 3 and possessing the kelch13 R561 mutation, has been found in recent research conducted in the Kagera region of Tanzania, which shares a border with Rwanda [17]. Another report from a recent survey in Kibindu-Bagamoyo district show low uncorrected (73.8%) and low PCR-corrected (89.9%) AL efficacy (NMCP unpublished data). Furthermore, a study in Southeast Tanzania found mutations in k13-propeller gene, including one sample with R561H, a mutation that has been associated with delayed parasite clearance in Southeast Asia [34].
In this study parasite clearance determined by microscopy remained high, with only one patient having persistent parasitemia of 272 asexual parasites/μL on day 3, similar to other studies in Tanzania [9, 35]. However, there are concerns about the future long-term efficacy of AL in Tanzania, where this artemisinin-based combination has been used as first-line treatment for uncomplicated P. falciparum malaria since 2006. Several observations from Bagamoyo district contribute to this concern. High PCR-determined positivity on day 3 after supervised AL treatment in the magnitude of 28–84% has been reported [9, 35].
Widescale use of AL treatment has been associated with selection of wild type alleles (pfmdr1 N86 and pfcrt K76) [10, 36]. In this study only pfmdr1 polymorphism was analysed. Pfmdr1 N86 was detected in all successfully sequenced samples collected at enrollment and on the days of recurrent infection. Results from previous study showed higher prevalence of 184F in recurrent infection than in baseline (enrollment) samples [19]. Nonetheless, the presence of mutations does not always correlate with the measured cure rate[37]. However, molecular analysis for pfmdr1 and pfcrt mutation together with k13 mutations might contribute to understanding the factors underlying causes of tolerance/reduced efficacy [33, 37].
This study also showed that AL was well tolerated with mild adverse events, the most common being cough, skin pallor, and abdominal pain, which were similar to other studies [5, 19, 38]. No serious adverse events were reported in this study. Thorough clinical and laboratory assessment prevented inclusion of patients with suspected severe malaria (severe anemia or parasitemia > 100,000 asexual parasites/µl) or other disease conditions.
This study's limitations included the absence of a fatty meal in conjunction with AL treatment, as advised, potentially impacting lumefantrine absorption. Additionally, differential success in sub-allelic family amplification for msp1, msp2, and glurp might introduce variability in the PCR-corrected outcomes.
Conclusion
While high cure rates over 97% were observed in these sentinel sites, the notable high rates of reinfection could also indicate that AL is not the best drug to use from the standpoint of preventing spread of resistant parasites. Recent reports of partial artemisinin resistance in Kagera region are of great concern. Pfmdr1 N86 was detected in all samples and increased selection of pfmdr1 184F was observed. These markers have been associated with increased tolerance to AL. Continued surveillance of these markers, along with the markers associated with partial artemisinin resistance, is warranted.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect the views of the President’s Malaria Initiative, the U.S. Agency for International Development, the U.S. Centres for Disease Control and Prevention, or other employing organizations or sources of funding. Marian Warsame and Ritha Njau were staff member of the World Health Organization, and they alone are responsible for the views expressed in this publication and do not necessarily represent the decisions, policy or views of the World Health Organization.
Data availability
No datasets were generated or analysed during the current study.
References
WHO. World malaria report 2019. Geneva, World Health Organization, 2019. https://iris.who.int/handle/10665/330011.
WHO. Guidelines for malaria. Geneva, World Health Organization, 2021. https://iris.who.int/handle/10665/339609. WHO/UCN/GMP/2021.012021.
Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2014;371:411–23.
Boni MF, White NJ, Baird JK. The community as the patient in malaria-endemic areas: preempting drug resistance with multiple first-line therapies. PLoS Med. 2016;13: e1001984.
Kakolwa MA, Mahende MK, Ishengoma DS, Mandara CI, Ngasala B, Kamugisha E, et al. Efficacy and safety of artemisinin-based combination therapy, and molecular markers for artemisinin and piperaquine resistance in Mainland Tanzania. Malar J. 2018;17:369.
Mwaiswelo R, Ngasala BE, Jovel I, Gosling R, Premji Z, Poirot E, et al. Safety of a single low-dose of primaquine in addition to standard artemether-lumefantrine regimen for treatment of acute uncomplicated Plasmodium falciparum malaria in Tanzania. Malar J. 2016;15:316.
Mårtensson A, Strömberg J, Sisowath C, Msellem MI, Gil JP, Montgomery SM, et al. Efficacy of artesunate plus amodiaquine versus that of artemether-lumefantrine for the treatment of uncomplicated childhood Plasmodium falciparum malaria in Zanzibar. Tanzania Clin Infect Dis. 2005;41:1079–86.
Sisowath C, Strömberg J, Mårtensson A, Msellem M, Obondo C, Björkman A, et al. In vivo selection of Plasmodium falciparum pfmdr1 86N coding alleles by artemether-lumefantrine (Coartem). J Infect Dis. 2005;191:1014–7.
Mwaiswelo R, Ngasala B, Jovel I, Xu W, Larsson E, Malmberg M, et al. Prevalence of and risk factors associated with polymerase chain reaction-determined Plasmodium falciparum positivity on Day 3 after initiation of artemether–lumefantrine treatment for uncomplicated malaria in Bagamoyo District, Tanzania. Am J Trop Med Hyg. 2019;100:1179–86.
Bretscher MT, Dahal P, Griffin J, Stepniewska K, Bassat Q, Baudin E, et al. The duration of chemoprophylaxis against malaria after treatment with artesunate-amodiaquine and artemether-lumefantrine and the effects of pfmdr1 86Y and pfcrt 76T: a meta-analysis of individual patient data. BMC Med. 2020;18:47.
Malmberg M, Ngasala B, Ferreira PE, Larsson E, Jovel I, Hjalmarsson A, et al. Temporal trends of molecular markers associated with artemether-lumefantrine tolerance/resistance in Bagamoyo district. Tanzania Malar J. 2013;12:103.
Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–67.
Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois A-C, Khim N, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–5.
Balikagala B, Fukuda N, Ikeda M, Katuro OT, Tachibana S-I, Yamauchi M, et al. Evidence of artemisinin-resistant malaria in Africa. N Engl J Med. 2021;385:1163–71.
Uwimana A, Umulisa N, Venkatesan M, Svigel SS, Zhou Z, Munyaneza T, et al. Association of Plasmodium falciparum kelch13 R561H genotypes with delayed parasite clearance in Rwanda: an open-label, single-arm, multicentre, therapeutic efficacy study. Lancet Infect Dis. 2021;21:1120–8.
WHO. Strategy to respond to antimalarial drug resistance in Africa. Geneva, World Health Organization, 2022. https://iris.who.int/handle/10665/364531.
Ishengoma DS, Mandara CI, Bakari C, Fola AA, Madebe RA, Seth MD, et al. Evidence of artemisinin partial resistance in North-western Tanzania: clinical and drug resistance markers study. medRxiv 2024.
WHO. Methods for surveillance of antimalarial drug efficacy. Geneva, World Health Organization, 2009. https://iris.who.int/handle/10665/44048.
Ishengoma DS, Mandara CI, Francis F, Talundzic E, Lucchi NW, Ngasala B, et al. Efficacy and safety of artemether-lumefantrine for the treatment of uncomplicated malaria and prevalence of Pfk13 and Pfmdr1 polymorphisms after a decade of using artemisinin-based combination therapy in mainland Tanzania. Malar J. 2019;18:88.
Mandara CI, Kavishe RA, Gesase S, Mghamba J, Ngadaya E, Mmbuji P, et al. High efficacy of artemether–lumefantrine and dihydroartemisinin–piperaquine for the treatment of uncomplicated falciparum malaria in Muheza and Kigoma Districts, Tanzania. Malar J. 2018;17:261.
East African Network for Monitoring Antimalarial Treatment. The efficacy of antimalarial monotherapies, sulphadoxine–pyrimethamine and amodiaquine in East Africa: implications for sub-regional policy. Trop Med Int Health. 2003;8:860–7.
Tanzania Ministry of Health CD, Gender, Elderly and Children. National malaria strategic plan 2021–2025: transitioning to malaria elimination in phases. Tanzania, Dodoma, 2020.
Mwalimu CD, Kiware S, Nshama R, Derua Y, Machafuko P, Gitanya P, et al. Dynamics of malaria vector composition and Plasmodium falciparum infection in mainland Tanzania: 2017–2021 data from the national malaria vector entomological surveillance. Malar J. 2024;23:29.
Sinka ME, Bangs MJ, Manguin S, Coetzee M, Mbogo CM, Hemingway J, et al. The dominant Anopheles vectors of human malaria in Africa, Europe and the Middle East: occurrence data, distribution maps and bionomic précis. Parasit Vectors. 2010;3:117.
Tanzania Ministry of Health and Social Welfare. National Guidelines for Diagnosis and Treatment of Malaria. Tanzania, Dar es Salaam, 2006.
Medicines for Malaria Venture & WHO. Methods and techniques for clinical trials on antimalarial drug efficacy : genotyping to identify parasite populations : informal consultation organized by the Medicines for Malaria Venture and cosponsored by the World Health Organization, 29–31 May 2007, Amsterdam, The Netherlands. Geneva, World Health Organization, 2008. https://iris.who.int/handle/10665/43824.
WHO Informal consultation on methodology to distinguish reinfection from recrudescence in high malaria transmission areas: report of a virtual meeting, 17–18 May 2021. Geneva, World Health Organization, 2021. https://iris.who.int/handle/10665/348385.
Kabanywanyi AM, Mwita A, Sumari D, Mandike R, Mugittu K, Abdulla S. Efficacy and safety of artemisinin-based antimalarial in the treatment of uncomplicated malaria in children in southern Tanzania. Malar J. 2007;6:146.
Mandara CI, Francis F, Chiduo MG, Ngasala B, Mandike R, Mkude S, et al. High cure rates and tolerability of artesunate–amodiaquine and dihydroartemisinin–piperaquine for the treatment of uncomplicated falciparum malaria in Kibaha and Kigoma. Tanzania Malar J. 2019;18:99.
Hastings IM, Felger I. WHO antimalarial trial guidelines: good science, bad news? Trends Parasitol. 2022;38:933–41.
Shayo A, Buza J, Ishengoma DS. Monitoring of efficacy and safety of artemisinin-based anti-malarials for treatment of uncomplicated malaria: a review of evidence of implementation of anti-malarial therapeutic efficacy trials in Tanzania. Malar J. 2015;14:135.
WHO. Artemisinin resistance and artemisinin-based combination therapy efficacy: status report. Geneva, World Health Organization, 2018. https://iris.who.int/handle/10665/274362.
WWARN. Association of mutations in the Plasmodium falciparum Kelch13 gene (Pf3D7_1343700) with parasite clearance rates after artemisinin-based treatments—a WWARN individual patient data meta-analysis. BMC Med. 2019;17:1.
Bwire GM, Ngasala B, Mikomangwa WP, Kilonzi M, Kamuhabwa AA. Detection of mutations associated with artemisinin resistance at k13-propeller gene and a near complete return of chloroquine susceptible falciparum malaria in Southeast of Tanzania. Sci Rep. 2020;10:3500.
Mhamilawa LE, Aydin-Schmidt B, Mmbando BP, Ngasala B, Morris U. Detection of Plasmodium falciparum by light microscopy, loop-mediated isothermal amplification, and polymerase chain reaction on day 3 after initiation of artemether-lumefantrine treatment for uncomplicated malaria in Bagamoyo District, Tanzania: a comparative trial. Am J Trop Med Hyg. 2019;101:1144–7.
Ehrlich HY, Bei AK, Weinberger DM, Warren JL, Parikh S. Mapping partner drug resistance to guide antimalarial combination therapy policies in sub-Saharan Africa. Proc Natl Acad Sci USA. 2021;118: e2100685118.
Venkatesan M, Gadalla NB, Stepniewska K, Dahal P, Nsanzabana C, Moriera C, et al. Polymorphisms in Plasmodium falciparum chloroquine resistance transporter and multidrug resistance 1 genes: parasite risk factors that affect treatment outcomes for P. falciparum malaria after artemether-lumefantrine and artesunate-amodiaquine. Am J Trop Med Hyg. 2014;91:833–43.
Salvador C, Rafael B, Matsinhe F, Candrinho B, Muthemba R, De Carvalho E, et al. Efficacy and safety of artemether–lumefantrine for the treatment of uncomplicated falciparum malaria at sentinel sites in Mozambique, 2015. Acta Trop. 2017;171:146–50.
Acknowledgements
Acknowledgements are extended to parents/guardians of children for taking part in the study. Participating health facility staff, stakeholders, and colleagues from implementing partners and local health authorities are thanked for their support. The support provided by MUHAS, Catholic University of Health and Allied Sciences (CUHAS), Ifakara Health Institute (IHI), Kilimanjaro Christian Medical Centre (KCMC), National Institute for Medical Research (NIMR), NMCP Management and the Ministry Health during planning, implementation of the study and reporting/sharing of the results is highly appreciated. Many thanks to the WHO for providing the test drugs and filter papers through its Global Malaria Programme. The support provided by RTI International through Okoa Maisha Dhibiti Malaria (OMDM) activity (Cooperative agreement number: 72062118CA-00002) in collaboration with the President's Malaria Initiative, the U.S. Agency for International Development and the U.S. Centres for Disease Control and Prevention is much appreciated.
Funding
This study was funded by USAID under the US President’s Malaria Initiative through RTI subaward Okoa Maisha Dhibiti Malaria (OMDM) activity (Cooperative agreement number: 72062118CA-00002).
Author information
Authors and Affiliations
Contributions
BEN, MGC, BPM, CIM DSM EK, MA MKM, RAK and FM designed the study and took part in field data collection interpreted the findings, and wrote the paper. BEN, SB, MGC and T.M coordinated clinical study and sample collection, B.P.M and F. F performed data analysis. D. S. I performed molecular analysis All authors participated in study supervision, interpretation of findings, read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Locus-by-locus calls and final classification of recrudescence versus new infection
Additional file 2: Table S2.
PCR corrected cure rates based on 2/3 >= algorithm as recommended by WHO 2021
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ngasala, B.E., Chiduo, M.G., Mmbando, B.P. et al. Efficacy and safety of artemether-lumefantrine for the treatment of uncomplicated falciparum malaria in mainland Tanzania, 2019. Malar J 23, 101 (2024). https://doi.org/10.1186/s12936-024-04931-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-024-04931-0