
Abstract
The World Health Organization (WHO) recommends surveillance of molecular markers of resistance to anti-malarial drugs. This is particularly important in the case of mass drug administration (MDA), which is endorsed by the WHO in some settings to combat malaria. Dihydroartemisinin-piperaquine (DHA-PPQ) is an artemisinin-based combination therapy which has been used in MDA. This review analyses the impact of MDA with DHA-PPQ on the evolution of molecular markers of drug resistance. The review is split into two parts. Section I reviews the current evidence for different molecular markers of resistance to DHA-PPQ. This includes an overview of the prevalence of these molecular markers in Plasmodium falciparum Whole Genome Sequence data from the MalariaGEN Pf3k project. Section II is a systematic literature review of the impact that MDA with DHA-PPQ has had on the evolution of molecular markers of resistance. This systematic review followed PRISMA guidelines. This review found that despite being a recognised surveillance tool by the WHO, the surveillance of molecular markers of resistance following MDA with DHA-PPQ was not commonly performed. Of the total 96 papers screened for eligibility in this review, only 20 analysed molecular markers of drug resistance. The molecular markers published were also not standardized. Overall, this warrants greater reporting of molecular marker prevalence following MDA implementation. This should include putative pfcrt mutations which have been found to convey resistance to DHA-PPQ in vitro.
Similar content being viewed by others
Background
The evolution of anti-malarial drug resistance presents an alarming threat to eliminating malaria; a disease which causes over 500,000 deaths every year [1]. Malaria is caused by the protozoan parasite Plasmodium, with most fatal cases caused by Plasmodium falciparum [1]. The role of Mass Drug Administration (MDA) in the evolution of anti-malarial resistance is not well understood. MDA is defined by the WHO as the mass treatment of all, or a section of, the population, whether or not symptoms are present [2]. Historical MDA practices, such as the addition of the anti-malarial chloroquine to table salt, have been correlated with a subsequent rise in chloroquine resistance [3]. However, there is a lack of evidence linking more recent use of MDA with the evolution of anti-malarial resistance in P. falciparum. This review is focused on dihydroartemisinin-piperaquine (DHA-PPQ); an increasingly used artemisinin-combination therapy (ACT) in MDA for malaria. The first section of this review details current evidence on the molecular mechanisms behind resistance of P. falciparum to DHA-PPQ. This section also includes a comprehensive overview of the prevalence of molecular markers associated with DHA-PPQ resistance globally, using Whole Genome Sequence data from the MalariaGEN Pf3k project. The second section of this review systematically evaluates the impact of MDA with DHA-PPQ on the evolution of anti-malarial resistance. The authors systematically reviewed the evidence from the available literature reporting molecular markers of anti-malarial resistance following MDA with DHA-PPQ.
Section I: the evolution of drug resistance
Anti-malarial drugs can be grouped into broad classes (Table 1). Widespread resistance to monotherapies led the World Health Organization (WHO) to recommend artemisinin-based combination therapy (ACT) as first-line treatment in all malaria endemic countries for uncomplicated malaria, with artesunate recommended to treat severe cases [4]. ACT includes a combination therapy of an artemisinin derivative (artesunate, artemether or dihydroartemisinin) with a partner drug (either lumefantrine, amodiaquine, piperaquine, mefloquine or sulfadoxine-pyrimethamine) [4]. The most common artemisinin-based combinations used in Africa are artemether-lumefantrine (AL), artesunate-amodiaquine (AS-AQ) and dihydroartemisinin-piperaquine DHA-PPQ [4]. All currently have a high clinical efficacy in Africa, achieving 98%, 98.4% and 99.4%, respectively [4].
Resistance to many anti-malarial drugs has now evolved, but the speed at which resistance has emerged has differed depending on the drug (Fig. 1). Understanding the molecular mechanisms behind the evolution of resistance in these different drugs is crucial in understanding why resistance has evolved at different rates. Furthermore, the methods used for detecting and classifying anti-malarial resistance have changed over time [5]. In addition to in vivo, in vitro and ex vivo methods, molecular methods for detecting resistance have now been developed. This includes identifying genomic polymorphisms in the malaria parasite genome which are associated with resistance to anti-malarial drugs [5]. Historically, anti-malarial drug resistance has often spread from Southeast Asia to Africa, so monitoring molecular markers of resistance in different continents may enable the scientific community to pre-empt the spread of drug resistant malaria in Africa [6], depending on the mechanisms of resistance.

Dihydroartemisinin-piperaquine: how does it work?
Dihydroartemisinin-piperaquine (DHA-PPQ) is an artemisinin-based combination composed of fast acting dihydroartemisinin, and slow acting piperaquine. Dihydroartemisinin (DHA) is a synthetic derivative of artemisinin, which is a sesquiterpene lactone first extracted from the plant Artemisia annua in 1972 [11, 12]. DHA is activated by iron, which is likely supplied by haem [13] which is taken into the parasite digestive vacuole (DV) through endocytosis of cytosol [14]. The most popular hypothesis for DHA’s mechanism of action is that, once activated by haem, DHA produces radical oxygen species which cause oxidative damage in the parasite cell, killing the parasite [13, 15]. DHA is also hypothesized to act through the formation of covalent bonds with multiple targets in compartments external to the DV [16]. These mechanisms of action are illustrated in Fig. 2. DHA acts quickly and has a short half-life of approximately 0.85–2 h in adults [17,18,19]. To clear any residual parasites following the rapid action of DHA, it is paired in combination therapy with the long-acting partner drug, piperaquine [20].

A diagram illustrating how DHA is predicted to attack the parasite cell. Created with BioRender.com
Piperaquine (PPQ) was first introduced as a monotherapy in the 1960s, and later as the partner drug in DHA-PPQ combination therapy [20]. Piperaquine is thought to act by accumulating in high concentrations in the parasite’s digestive vacuole. Here, it inhibits the conversion of toxic haem to non-toxic haemozoin crystals during parasite haemoglobin digestion, which is an essential metabolic process for the parasite. Inhibiting the conversion of haem to haemozoin results in high concentrations of toxic haem accumulating in the digestive vacuole, leading to parasite death [21, 22]. Furthermore, in vitro studies have demonstrated that P. falciparum exposed to PPQ accumulate more undigested haemoglobin, suggesting that PPQ decreases the rate of haemoglobin digestion, possibly killing the parasite through ‘starvation’ [23]. There is also evidence suggesting that PPQ binds directly to the P. falciparum chloroquine resistance transporter, PfCRT [24], where it may inhibit PfCRT’s usual function as a transporter protein. These mechanisms of action are illustrated in Fig. 3.

A diagram illustrating how PPQ is predicted to attack the parasite cell. Created in Biorender.com
Dihydroartemisinin-piperaquine: what are the resistance mechanisms?
Resistance to DHA-PPQ has emerged in Southeast Asia [25, 26] but the molecular mechanism of resistance is not fully understood. Molecular markers for partial resistance to DHA include single nucleotide polymorphisms (SNPs) on the pfk13 Plasmodium gene, including the mutations F446I, N458Y, M476I, Y493H, R539T, Y543T, P553L, R561H, P574L and C580Y, which have been validated by the WHO [27]. Resistance to the partner drug PPQ is less well understood. Resistance to PPQ is associated with gene duplication of the plasmepsins pfpm2 and pfpm3 [28,29,30,31,32], and inactivation of either of these genes increases sensitivity to PPQ [33]. Plasmepsins II and III are proteases which work in a complex with other proteases in the digestive vacuole (DV) to digest haemoglobin and produce essential amino acids for the parasite [34]. Plasmepsin duplications may facilitate resistance to PPQ by increasing the rate of haemoglobin digestion. This may counteract the inhibitory effects of PPQ on haemoglobin digestion. Of note, resistance to PPQ has also been shown without duplication of pfpm2 [35,36,37] and there is some evidence that increased expression of pfpm2 and pfpm3 does not alter PPQ susceptibility [38]. Therefore, although PPQ resistance is correlated with pfpm2 and pfpm3 duplication, this is unlikely to be the sole mechanism of PPQ resistance. Considering this, plasmepsin copy number should not be used as the only indicator for surveying PPQ resistance. As with other anti-malarial drugs, such as mefloquine [39], all cases of resistance cannot usually be explained completely by one specific genetic polymorphism.
Resistance to DHA-PPQ has been associated with other genetic polymorphisms, including on the pfexo and pfcrt genes. Plasmodium falciparum Exonuclease (pfexo) is a putative exonuclease encoding gene. The E415G polymorphism on pfexo is strongly linked to increased copy number of pfpm2 and pfpm3 and has been correlated with treatment failure of DHA-PPQ in Cambodia [29, 40]. However, the functional role of this protein is uncertain.
Multiple polymorphisms in pfcrt are correlated with PPQ resistant phenotypes. The mutations T93S, H97Y, F145I and I218P have been associated with DHA-PPQ resistance in Cambodian isolates. Furthermore, in vitro studies have found that H97Y, F145I, M343L, G353V [21] and C101F [22] cause the Dd2 (“Indo-China”) laboratory strain to be PPQ resistant and CQ sensitive. In vitro data has shown that F145I and C350R can mediate efflux of PPQ from the DV in the 7G8 (“Brazil”) laboratory strain at the same time as reducing CQ transport, suggesting that PPQ resistance may arise through efflux of PPQ from the digestive vacuole via PfCRT, in a similar mechanism to CQ resistance [24]. The mutations T93S and I218P have been shown to confer resistance to PPQ without the presence of pfpm2 duplications [35]. Furthermore, T93S, H97Y, F145I and I128F each conferred resistance to PPQ on a background of pfexo E145G, but again without pfpm2 duplication [36]. These data further show that plasmepsin duplications are not required for PPQ resistance. Many of these pfcrt mutations also resulted in a swollen digestive vacuole [21, 22, 36, 41]. This vacuole swelling indicates that the PfCRT protein has a role in maintaining vacuole morphology, and that mutations in pfcrt disrupt this function [41]. The structure of PfCRT has recently been elucidated [24], showing that pfcrt mutations associated with PPQ resistance are at moderately conserved sites in selected helices of the protein, including T93S, H97Y and C101F. This work has highlighted a number of other amino acid sites with similar properties, which may be under similar selection pressures from PPQ and may be useful for future PPQ resistance surveillance [24].
One hypothesis for the mechanism of PPQ resistance is that pfcrt mutations enable PfCRT to transport PPQ out of the digestive vacuole, away from its putative site of action, similarly to CQ resistance. However, some of these pfcrt mutations conveyed PPQ resistance without changing the rate of PPQ transport out of the digestive vacuole [21]. A competing hypothesis is that PPQ binds to PfCRT as part of its mode of action, disrupting its role as a transporter and DV morphology regulator, causing parasite death. These pfcrt mutations may inhibit PPQ from binding, causing PPQ resistance, whilst simultaneously removing the transporter’s ability to transport CQ, leading to CQ susceptibility [22].
Polymorphisms in pfmdr1 have been associated with PPQ sensitivity. Conrad et al. found that DHA-PPQ treatment selected for the pfmdr1 haplotype 86Y/Y184/Y1246. Interestingly, treatment with Artemether-Lumefantrine (AL) selected for opposite alleles; N86/184F/D1246 [42]. These opposing selection pressures suggest that DHA-PPQ may be a good choice of partner drug in areas where AL was previously used. Furthermore, increased pfmdr1 copy number has been associated with enhanced sensitivity to piperaquine [29, 43]. Veiga et al. hypothesized that increased pfmdr1 copy number is associated with enhanced accumulation of PPQ in the DV, leading to increased sensitisation to PPQ [43].
In summary, PPQ is likely to kill parasites by disrupting haemoglobin digestion and may also act by binding to PfCRT, disrupting its role as a transporter. Pfpm2 and pfpm3 duplications correlate with PPQ resistance, but duplication is not essential for resistance. Therefore, plasmepsin copy number should not be used as a sole indicator of PPQ resistance. Additionally, some polymorphisms in pfcrt can confer resistance to PPQ in Dd2 parasites and the E415G pfexo mutation has been correlated with DHA-PPQ resistance in Cambodian isolates. Finally, increased pfmdr1 copy number has been associated with enhanced sensitivity to PPQ.
How prevalent are these putative PPQ-resistance conferring mutations?
As part of this review, whole genome sequence data was used to analyse the prevalence of the above-mentioned SNPs in P. falciparum samples from studies worldwide [44,45,46] (n = 4001) (Fig. 4). For frequency calculations, the authors considered isolates with monoclonal infections based on the Fws metric. The pfmdr1 N86Y mutation was found at a prevalence of between 21.1% and 23.7% in samples from Central, West and East Africa, and a lower prevalence of 11.4% in the Horn of Africa and 8.2% in Southern Africa. A higher prevalence was found in Southern Central Africa, at 43%. The prevalence of N86Y was also higher in Oceania, at 78.4%. Whereas, the prevalence was lower in samples from South America, at 2.3% and Southeast Asia, at 0.9%. The pfmdr1 Y184F mutation was found at a prevalence between 37.3% and 51.4% in samples from South Central Africa, East Africa, Southeast Asia and Southern Africa. The prevalence was 65.4% in West Africa and 68.3% in Central Africa. This mutation was found at a much higher prevalence of 95.5% in South America and in the Horn of Africa, where it was found to be 100%. In comparison, the prevalence of Y184F was very low in Oceania, at a prevalence of 3.1%. The D1246Y mutation was not found in samples from Oceania or the Horn of Africa, and ranged from a very low prevalence of 0.2% in Southeast Asia, to 29.5% in South America. Pfcrt mutations of interest, including T93S, H97Y, F145I, I218F, M343I and G353V, were only present in samples from Southeast Asia, with a mutation prevalence of 0% in the other global regions. The prevalence of these muations was low, with T93S, F145I, M343L in under 1% of the samples analysed. I218F had a prevalence of 1% and G353V had a prevalence of 1.2%, while H97Y had a prevalence of 2.6%. The pfexo mutation E415G was only found in samples from Southeast Asia, at a prevalence of 16.2%.

A diagram showing the global frequencies of mutations in the pfmdr1, pfcrt and pfexo genes, which are potential markers of DHA-PPQ resistance. These frequencies were calculated using whole genome sequence data from recent studies [44,45,46]. n is the number of samples containing a mutant allele, and N is the total number of successfully sequenced samples. Total sample size = 4001
Section II: how has mass drug administration with DHA-PPQ impacted molecular markers of resistance?
Monitoring the prevalence of molecular markers associated with DHA-PPQ resistance enables widespread surveillance of P. falciparum markers in populations undergoing mass drug administration (MDA). This molecular surveillance can then be used to inform treatment policy specific to different populations. This review has investigated the impact of MDA with DHA-PPQ on the evolution of molecular markers associated with anti-malarial resistance.
Methodology
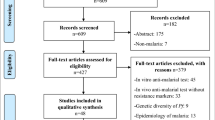
This review included relevant studies from clinicaltrails.gov, EMBASE, MEDLINE and the Infectious Diseases Data Observatory (IDDO). Searches were dated back to 2011, when DHA-PPQ was first approved as the ACT Eurartesim®, by the European Medicines Agency. A detailed search strategy and methodology can be found in Appendix 1, which follows PRISMA guidelines. In brief, MDA studies were included from www.clinicaltrials.gov which were completed, with results, which used DHA-PPQ or PPQ as an intervention. Associated publications with reference to molecular markers of resistance were included in this review. Two further publications were included from IDDO, following search terms ‘malaria’ and ‘piperaquine’. Search terms for malaria, mass drug administration, anti-malarial resistance and PPQ or DHA-PPQ were used to extract publications from EMBASE and MEDLINE. These were then filtered for publications which included analysis of molecular markers of resistance. The methodology flowchart can be seen in Fig. 5.

A flowchart demonstrating how studies were idendified for this review following PRISMA guidelines
Results
A total of 20 studies passed the screening criteria and were included for analysis in this systematic review. Each study was analysed to understand the reported impact of treatment with DHA-PPQ or PPQ on molecular markers associated with resistance to DHA-PPQ. Molecular marker data extracted from these studies included pfpm2 copy number, pfexo E415G, pfcrt mutations, pfmdr mutations and copy number variations, and artemisinin-resistance associated mutations. Of the 20 studies reviewed, 7 included analysis of pfpm2 copy number, 2 included analysis of pfexo E415G, 9 included analysis of mutations in pfcrt, 13 included analysis of mutations and/or copy number variation in pfmdr1 and 11 included analysis of mutations associated with reduced artemisinin-sensitivity. 16 of the studies were associated with clinical trials which included the use of DHA-PPQ. 2 used piperaquine phosphate and 1 used arteminisinin-piperaquine. 14 studies were associated with MDA trials, 7 of which were associated with IPTp, which is a form of targeted MDA. 5 studies were associated with clinical trials for treatment of confirmed malaria, but were retained in this review to provide the breadth of genomic data available related to DHA-PPQ use. A summary of the relevant molecular markers reported in each study can be seen in Table 2, sorted by molecular marker of interest. One report analysed molecular markers in the pfdhfr and pfdhps genes associated with SP resistance, but did not analyse polymorphisms associated with resistance to DHA-PPQ in pfpm2, pfmdr1, pfcrt or pfkelch13 [49]. Therefore, this study is not included in Table 2.
Discussion
Advances in sequencing technology have resulted in an explosion in the generation, availability, and analysis of sequencing data. This includes genomic data from the deadliest malaria parasite, P. falciparum. Genomic surveillance has consequently gained an increasingly important role in monitoring anti-malarial drug resistance, through the surveillance of molecular markers in the P. falciparum genome. The surveillance of molecular markers associated with drug resistance is recognized as a surveillance tool by the WHO [47]. Genomic surveillance is particularly important in the case of mass drug administration programmes, where drug treatment is given to members of a population whether or not they are symptomatic for malaria. MDA is endorsed by the WHO in certain settings, such as endemic island communities, where there is limited risk of importation of infection, good access to treatment and implementation of vector control and surveillance [2]. Furthermore, randomized controlled trials with MDA using DHA-PPQ have been shown to be safe and to significantly lower the burden of malaria in pre-elimination settings [48]. Therefore, with continued use of MDA, surveillance of molecular markers of resistance is crucial.
What impact did these studies have on molecular markers of drug resistance?
Pfpm2 copy number
There was no evidence for selection for increased pfpm2 copy number following MDA with DHA-PPQ in Kayin state, Myanmar [51] or after MDA for 2 months over 2 consecutive years in Mozambique [52] or after MDA taken for 3 days for 3 months in Myanmar [55]. Conrad et al. found modest increases in pfpm2 copy number in 1 of 18 samples from patients receiving DHA-PPQ IPTp, where participants received DHA-PPQ every 8 weeks or every 4 weeks during pregnancy [53]. Taken together, this suggests that short term MDA treatments are unlikely to select for amplification in pfpm2 copy number. However, Imwong et al. have found pfpm2 amplification in their longitudinal observational studies in the eastern Greater Mekong subregion [51, 57]. This is an area where DHA-PPQ has been used extensively for many years, and may suggest that longer periods of DHA-PPQ use can select for increased pfpm2 copy number.
Pfexo E415G
Two studies included in this review measured the frequency of the pfexo E415G mutation. Conrad et al. sequenced this locus and did not detect the pfexo E415G mutation in the samples that they analysed [53]. Son et al. identified the pfexo E415G mutation in their study population prior to MDA, but found no statistically significant increase in the prevalence of this mutation following MDA with forest rangers in Vietnam [58]. This evidence does not demonstrate a correlation between MDA with DHA-PPQ and increased prevalence of the pfexo E415G mutation. However, only 2 of the studies analysed this marker. Of note, some of the 20 studies analysed in this review were published before the association between the pfexo mutation and PPQ resistance was reported. This includes Ochong et al., Conrad et al., Somé et al., Zongo et al., Tumwebaze et al., Taylor et al. and Madanitsa et al. [42, 49, 61,62,63,64,65].
Pfcrt mutations
Nayebare et al. found that pfcrt K76T prevalence was higher in samples collected from women in Uganda during IPTp with DHA-PPQ than in parasites collected prior to the start of IPTp, or while women received IPTp with SP [59]. Similarly, Conrad et al. found that the prevalence of the K76T mutation was higher in samples collected from the DHA-PPQ arm of IPTp in Uganda, than in the SP arm or in samples collected prior to the start of IPTp [53]. This increase in K76T prevalence was also correlated with increased PPQ exposure.
In contrast, Somé et al. found no significant selection for pfcrt K76T following Seasonal Malaria Chemoprevention (SMC) with DHA-PPQ for 3 months in Burkina Faso [62]. Imwong et al. analysed pfcrt mutations F145I, I218F, N326S, M343I/L and G353V and found no evidence of selection of pfcrt mutations associated with PPQ resistance following MDA in Kayin State, Myanmar [51]. In support of this, Gupta et al. found no statistically significant difference in the prevalence of pfcrt polymorphisms after MDA with DHA-PPQ for 2 months, for 2 years, in Mozambique [52]. Tumwebaze et al. found that monthly MDA with DHA-PPQ in Uganda was not associated with changes in the prevalence of pfcrt polymorphisms [63]. Finally, Zongo et al. found no significant difference in the prevalence of the K76T polymorphism following SMC with DHA-PPQ in Burkina Faso [61].
Overall, evidence for selection of pfcrt K76T following MDA with DHA-PPQ is mixed. Few studies analysed pfcrt markers other than K76T. Other key polymorphisms have been associated with PPQ resistance in vitro. Pfcrt H97Y, F145I, M343L, G353V [21] and C101F [22] mutations have been shown to confer PPQ resistance and CQ sensitivity in vitro, and F145I and C350R have been shown to efflux PPQ from the DV at the same time as reducing CQ transport [24]. Furthermore, T93S and I218P have been shown to confer PPQ resistance [35] along with T93S, H97Y, F145I and I128F. Each of these mutations conferred resistance to PPQ on a background of pfexo E145G [36]. This evidence suggests that that there may be other, more relevant pfcrt markers than K76T, and that future studies would benefit from monitoring this range of identified putative pfcrt polymorphisms which have conferred DHA-PPQ resistance in vitro.
Pfmdr1 mutations and copy number
Following IPTp with DHA-PPQ in Uganda, both Nayebare et al. and Conrad et al. found increased prevalence of pfmdr1 N86Y and Y184F mutations in samples collected during treatment than in samples collected before treatment [53, 59]. Increased exposure to PPQ also correlated with increased prevalence of N86Y [53, 63] and D1246Y [63] in Uganda. Mixed results were found regarding the D1246Y polymorphism. Nayebare et al. found that the prevalence of pfmdr1 D1246Y was similar in samples collected before and after IPTp [59]. Whereas, Conrad et al. found that D1246Y prevalence decreased in samples collected during IPTp with DHA-PPQ, compared with samples collected before treatment or during treatment with SP [53]. Somé et al. found borderline selection for wild-type D1246Y following treatment with DHA-PPQ in Burkina Faso [62]. Taylor et al. and Conrad et al. monitored changes in polymorphisms and haplotypes in pfmdr1 in Uganda between 2007 and 2012 after treatment with AL or DHA-PPQ [42, 64]. Conrad et al. found that treatment with DHA-PPQ was associated with increased prevalence of N86Y and D1246Y, and a lower prevalence of Y184F [42]. Taylor et al. used a haplotype frequency estimation model and found that treatment with DHA-PPQ only selected for N86Y when this allele was found with D1246Y and Y184F, in the haplotype YYY, and that it selected against haplotypes NFD and NYY [64].
In contrast, Gupta et al. found no change in pfmdr1 polymorphisms following MDA with DHA-PPQ in Mozambique [52]. Furthermore, Zongo et al. found no significant difference in the prevalence of pmdfr1 mutations following SMC with DHA-PPQ in Burkina Faso [61]. Gupta et al., Son et al., and Conrad et al., found no association between MDA with DHA-PPQ and increased pfmdr1 copy number [42, 52, 53, 58]. This small number of studies suggests that MDA with DHA-PPQ may select for mutations in pfmdr1, particularly N86Y.
Artemisinin partial resistance associated mutations
No evidence for increased selection of pfkelch13 mutations was found after MDA with DHA-PPQ in Kayin State, Myanmar [51] in Mozambique [52], Uganda [53], or in Eastern Myanmar [55], or following treatment with artemisinin-piperaquine in the Comoros [67]. This suggests that although the prevalence of some pfkelch13 polymorphisms have become widespread in the GMS [51, 57], MDA with DHA-PPQ has not increased the prevalence of pfkelch13 mutations associated with reduced artemisinin sensitivity, as least in the studies which have monitored molecular markers of resistance following MDA.
Conclusion
Despite molecular markers of drug resistance being a recognized surveillance tool by the WHO [27], the level of reporting of molecular markers associated with DHA-PPQ resistance found in this study was low. Of the total 96 papers screened for eligibility in this review, only 20 analysed molecular markers of drug resistance. This highlights considerations for future studies with DHA-PPQ, where further analysis and reporting of molecular markers related to DHA-PPQ resistance would greatly assist understanding of how MDA impacts polymorphisms associated with resistance. Importantly, molecular markers associated with DHA-PPQ have different prevalences in different geographic regions. Molecular surveillance data from future studies in different geographies will further increase understanding of how treatment with DHA-PPQ is impacting the evolution of resistance in these different geographies.
The choice of markers analysed is not currently standardized, which may be partly because pfexo E415G and pfpm2 copy number have only recently emerged as molecular markers associated with PPQ resistance. Future studies with DHA-PPQ should monitor a broader range of molecular markers which have been associated with resistance to DHA-PPQ. This includes pfexo E415G, pfpm2 copy number and pfmdr1 copy number and N86Y, Y184F and D1246Y mutations. In addition to these markers, putative polymorphisms in pfcrt should also be monitored, including mutations T93S, H97Y, F145I, I218P, M343L, C350R, G353V. This would contribute to a more comprehensive analysis of resistance polymorphisms following MDA implementation.
To really understand the impact that DHA-PPQ MDA has had on the evolution of drug resistance, there needs to be much greater focus and investment on genomic surveillance in trial and programmatic settings. This would enable the research community to build on the already growing field of genomic surveillance to better understand the impact of using anti-malarial drugs on a large scale. Phenomenal steps have already been made, including through the Pan-African Malaria Genetic Epidemiology Network (PAMGEN). However, the lack of published molecular surveillance data from trials highlights the need for increasing focus on genomic surveillance if MDA is used as a population-based strategy for malaria control.
Availability of data and materials
The processed datasets are available from the corresponding author. All data is available from the individual studies cited, including ENA accession numbers for raw sequences.
Abbreviations
- MDA:
-
Mass drug administration
- DHA-PPQ:
-
Dihydroartemisinin-piperaquine
- WGS:
-
Whole genome sequencing
- SNPs:
-
Single nucleotide polymorphisms
- WHO:
-
World Health Organization
- ACT:
-
Artemisinin-combination therapy
- AL:
-
Artemether-lumefantrine
- AS-AQ:
-
Artesunate-amodiaquine
- ITN:
-
Insecticide-treated nets
- IPTp:
-
Intermittent preventive treatment in pregnancy
- LLIN:
-
Long-lasting insecticidal nets
- SP:
-
Sulfadoxine-pyrimethamine
- AS:
-
Artesunate
- QN:
-
Quinine
- IVM:
-
Ivermectin
- DHA:
-
Dihydroartemisinin
- DV:
-
Digestive vacuole
- PPQ:
-
Piperaquine
- CQ:
-
Chloroquine
- CNV:
-
Copy number variant
- PCR:
-
Polymerase chain reaction
- DNA:
-
Deoxyribose nucleic acid
References
WHO. World malaria report 2021. Geneva: World Health Organization; 2021.
WHO. Mass drug administration, mass screening and treatment and focal screening and treatment for malaria. Malaria policy advisory committee meeting. Geneva: World Health Organization; 2015.
Payne D. Did medicated salt hasten the spread of chloroquine resistance in Plasmodium falciparum? Parasitol Today. 1988;4:112–5.
WHO. World malaria report 2020. Geneva: World Health Organization; 2020.
Slater L, Betson M, Ashraf S, Sargison N, Chaudhry U. Current methods for the detection of antimalarial drug resistance in Plasmodium parasites infecting humans. Acta Trop. 2021;216: 105828.
Takala-Harrison S, Laufer MK. Antimalarial drug resistance in Africa: key lessons for the future. Ann N Y Acad Sci. 2015;1342:62–7.
Looareesuwan S, Viravan C, Webster HK, Kyle DE, Hutchinson DB, Canfield CJ. Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am J Trop Med Hyg. 1996;54:62–6.
Edgcomb JH, Arnold J, Yount Eh Jr, Alving AS, Eichelberger L, Jeffery GM, et al. Primaquine, SN 13272, a new curative agent in vivax malaria; a preliminary report. J Natl Malar Soc. 1950;9:285–92.
Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM, Artemisinin Resistance in Cambodia 1 (ARC1) Study Consortium. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med. 2008;359:2619–20.
Institute of Medicine (US) Committee on the Economics of Antimalarial Drugs. Saving lives, buying time: economics of malaria drugs in an age of resistance. Washington (DC): National Academies Press (US); 2004. p. 1125–7.
van Agtmael MA, Eggelte TA, van Boxtel CJ. Artemisinin drugs in the treatment of malaria: from medicinal herb to registered medication. Trends Pharmacol Sci. 1999;20:199–205.
You-you T, Mu-yun N, Yurong Z, Lan-na L, Shu-lian G, Mu-qun Z, et al. Studies on the constituents of Artemisia annua. J Med Plant Res. 1982;44:143–5.
Wang J, Zhang CJ, Chia WN, Loh CC, Li Z, Lee YM, He Y, Yuan LX, Lim TK, et al. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat Commun. 2015;6:10111.
Spielmann T, Gras S, Sabitzki R, Meissner M. Endocytosis in Plasmodium and toxoplasma parasites. Trends Parasitol. 2020;36:520–32.
Heller LE, Roepe PD. Artemisinin-based antimalarial drug therapy: molecular pharmacology and evolving resistance. Trop Med Infect Dis. 2019;4:89.
Eckstein-Ludwig U, Webb RJ, Van Goethem ID, East JM, Lee AG, Kimura M, et al. Artemisinins target the SERCA of Plasmodium falciparum. Nature. 2003;424:957–61.
Le Thi DT, Le NH, Nguyen CH, Phan Thi D, Na-Bangchang K. Pharmacokinetics of a five-day oral dihydroartemisinin monotherapy regimen in patients with uncomplicated falciparum malaria. Drug Metab Pharmacokinet. 2008;23:158–64.
Nguyen DV, Nguyen QP, Nguyen ND, Le TT, Nguyen TD, Dinh DN, Nguyen TX, et al. Pharmacokinetics and ex vivo pharmacodynamic antimalarial activity of dihydroartemisinin-piperaquine in patients with uncomplicated falciparum malaria in Vietnam. Antimicrob Agents Chemother. 2009;53:3534–7.
Newton PN, van Vugt M, Teja-Isavadharm P, Siriyanonda D, Rasameesoroj M, et al. Comparison of oral artesunate and dihydroartemisinin antimalarial bioavailabilities in acute falciparum malaria. Antimicrob Agents Chemother. 2002;46:1125–7.
Wirth DF, Alonso PL. Malaria biology in the era of eradication. Cold Spring Harbor Perspect Med. 2017. https://doi.org/10.1101/cshperspect.a025510.
Ross LS, Dhingra SK, Mok S, Yeo T, Wicht KJ, Kümpornsin K, et al. Emerging Southeast Asian PfCRT mutations confer Plasmodium falciparum resistance to the first-line antimalarial piperaquine. Nat Commun. 2018;9:25–8.
Dhingra SK, Redhi D, Combrinck JM, Yeo T, Okombo J, Henrich PP, et al. A variant PfCRT isoform can contribute to Plasmodium falciparum resistance to the first-line partner drug piperaquine. mBio. 2017;8:3.
Sachanonta N, Chotivanich K, Chaisri U, Turner GD, Ferguson DJ, Day NP, et al. Ultrastructural and real-time microscopic changes in P. falciparum-infected red blood cells following treatment with antimalarial drugs. Ultrastruct Pathol. 2011;35:214–25.
Kim J, Tan YZ, Wicht KJ, Erramilli SK, Dhingra SK, Okombo J, Vendome J, et al. Structure and drug resistance of the Plasmodium falciparum transporter PfCRT. Nature. 2019;576:315–20.
Saunders DL, Vanachayangkul P, Lon C, U.S. Army Military Malaria Research Program, National Center for Parasitology, Entomology, and Malaria Control (CNM), Royal Cambodian Armed Forces. Dihydroartemisinin-piperaquine failure in Cambodia. N Engl J Med. 2014;371:484–5.
van der Pluijm RW, Imwong M, Chau NH, Hoa NT, Thuy-Nhien NT, Thanh NV, et al. Determinants of dihydroartemisinin-piperaquine treatment failure in Plasmodium falciparum malaria in Cambodia, Thailand, and Vietnam: a prospective clinical, pharmacological, and genetic study. Lancet Infect Dis. 2019;19:952–61.
WHO. Report on antimalarial drug efficacy, resistance and response: 10 years of surveillance (2010–2019). Geneva: World Health Organization; 2020.
Witkowski B, Duru V, Khim N, Ross LS, Saintpierre B, Beghain J, et al. A surrogate marker of piperaquine-resistant Plasmodium falciparum malaria: a phenotype–genotype association study. Lancet Infect Dis. 2017;17:174–83.
Amato R, Lim P, Miotto O, Amaratunga C, Dek D, Pearson RD, et al. Genetic markers associated with dihydroartemisinin–piperaquine failure in Plasmodium falciparum malaria in Cambodia: a genotype–phenotype association study. Lancet Infect Dis. 2017;17:164–73.
Bopp S, Magistrado P, Wong W, Schaffner SF, Mukherjee A, Lim P, Dhorda M, et al. Plasmepsin II–III copy number accounts for bimodal piperaquine resistance among Cambodian Plasmodium falciparum. Nat Commun. 2018;9:1769.
Agrawal S, Moser KA, Morton L, Cummings MP, Parihar A, Dwivedi A, et al. Association of a novel mutation in the Plasmodium falciparum chloroquine resistance transporter with decreased piperaquine sensitivity. J Infect Dis. 2017;216:468–76.
Ansbro MR, Jacob CG, Amato R, Kekre M, Amaratunga C, Sreng S, Suon S, et al. Development of copy number assays for detection and surveillance of piperaquine resistance associated plasmepsin 2/3 copy number variation in Plasmodium falciparum. Malar J. 2020;19:181.
Mukherjee A, Gagnon D, Wirth DF, Richard D. Inactivation of plasmepsins 2 and 3 sensitizes Plasmodium falciparum to the antimalarial drug piperaquine. Antimicrob Agents Chemother. 2018;62:e02309-e2317.
Nasamu AS, Polino AJ, Istvan ES, Goldberg DE. Malaria parasite plasmepsins: more than just plain old degradative pepsins. J Biol Chem. 2020;295:8425–41.
Dhingra SK, Small-Saunders JL, Ménard D, Fidock DA. Plasmodium falciparum resistance to piperaquine driven by PfCRT. Lancet Infect Dis. 2019;19:1168–9.
Boonyalai N, Vesely BA, Thamnurak C, Praditpol C, Fagnark W, Kirativanich K, et al. Piperaquine resistant Cambodian Plasmodium falciparum clinical isolates: in vitro genotypic and phenotypic characterization. Malar J. 2020;19:269.
Chebore W, Zhou Z, Westercamp N, Otieno K, Shi YP, Sergent SB, et al. Assessment of molecular markers of anti-malarial drug resistance among children participating in a therapeutic efficacy study in western Kenya. Malar J. 2020;19:291.
Loesbanluechai D, Kotanan N, de Cozar C, Kochakarn T, Ansbro MR, et al. Overexpression of plasmepsin II and plasmepsin III does not directly cause reduction in Plasmodium falciparum sensitivity to artesunate, chloroquine and piperaquine. Int J Parasitol Drugs Drug Resist. 2019;9:16–22.
Price RN, Uhlemann AC, Brockman A, McGready R, Ashley E, Phaipun L, et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet. 2004;364:438–47.
Parobek CM, Parr JB, Brazeau NF, Lon C, Chaorattanakawee S, Gosi P, et al. Partner-drug resistance and population substructuring of artemisinin-resistant Plasmodium falciparum in Cambodia. Genome Biol Evol. 2017;9:1673–86.
Pulcini S, Staines HM, Lee AH, Shafik SH, Bouyer G, Moore CM, Daley DA, et al. Mutations in the Plasmodium falciparum chloroquine resistance transporter, PfCRT, enlarge the parasite’s food vacuole and alter drug sensitivities. Sci Rep. 2015;5:14552.
Conrad MD, LeClair N, Arinaitwe E, Wanzira H, Kakuru A, Bigira V, Muhindo M, et al. Comparative impacts over 5 years of artemisinin-based combination therapies on Plasmodium falciparum polymorphisms that modulate drug sensitivity in Ugandan children. J Infect Dis. 2014;210:344–53.
Veiga MI, Ferreira PE, Malmberg M, Jörnhagen L, Björkman A, Nosten F, et al. pfmdr1 amplification is related to increased Plasmodium falciparum in vitro sensitivity to the bisquinoline piperaquine. Antimicrob Agents Chemother. 2012;56:3615–9.
Osborne A, Manko E, Takeda M, Kaneko A, Kagaya W, Chan C, Ngara M, et al. Characterizing the genomic variation and population dynamics of Plasmodium falciparum malaria parasites in and around Lake Victoria. Kenya Sci Rep. 2021;11:19809.
Turkiewicz A, Manko E, Sutherland CJ, Diez Benavente E, Campino S, Clark TG. Genetic diversity of the Plasmodium falciparum GTP-cyclohydrolase 1, dihydrofolate reductase and dihydropteroate synthetase genes reveals new insights into sulfadoxine-pyrimethamine antimalarial drug resistance. PLoS Genet. 2020;16: e1009268.
MalariaGEN, Ahouidi A, Ali M, Almagro-Garcia J, Amambua-Ngwa A, et al. An open dataset of Plasmodium falciparum genome variation in 7,000 worldwide samples. Wellcome Open Res. 2021;6:42.
WHO and Technical Expert Group. Technical expert group (TEG) on drug efficacy and response. Geneva: World Health Organization; 2015.
Eisele TP. Mass drug administration can be a valuable addition to the malaria elimination toolbox. Malar J. 2019;18:281.
Madanitsa M, Kalilani L, Mwapasa V, van Eijk AM, Khairallah C, Ali D, et al. Scheduled intermittent screening with rapid diagnostic tests and treatment with dihydroartemisinin-piperaquine versus intermittent preventive therapy with sulfadoxine-pyrimethamine for malaria in pregnancy in Malawi: an open-label randomized controlled trial. PLoS Med. 2016;13: e1002124.
Huang F, Takala-Harrison S, Jacob CG, Liu H, Sun X, Yang H, Nyunt MM, et al. A Single mutation in K13 predominates in southern China and is associated with delayed clearance of Plasmodium falciparum following artemisinin treatment. J Infect Dis. 2015;212:1629–35.
Imwong M, Dhorda M, Myo Tun K, Thu AM, Phyo AP, Proux S, Suwannasin K, et al. Molecular epidemiology of resistance to antimalarial drugs in the Greater Mekong subregion: an observational study. Lancet Infect Dis. 2020;20:1470–80.
Gupta H, Galatas B, Chidimatembue A, Huijben S, Cisteró P, Matambisso G, et al. Effect of mass dihydroartemisinin-piperaquine administration in southern Mozambique on the carriage of molecular markers of antimalarial resistance. PLoS ONE. 2020;15: e0240174.
Conrad MD, Mota D, Foster M, Tukwasibwe S, Legac J, Tumwebaze P, et al. Impact of intermittent preventive treatment during pregnancy on Plasmodium falciparum drug resistance-mediating polymorphisms in Uganda. J Infect Dis. 2017;216:1008–17.
von Seidlein L, Peto TJ, Landier J, Nguyen TN, Tripura R, Phommasone K, et al. The impact of targeted malaria elimination with mass drug administrations on falciparum malaria in Southeast Asia: a cluster randomised trial. PLoS Med. 2019;16: e1002745.
Landier J, Kajeechiwa L, Thwin MM, Parker DM, Chaumeau V, Wiladphaingern J, et al. Safety and effectiveness of mass drug administration to accelerate elimination of artemisinin-resistant falciparum malaria: a pilot trial in four villages of eastern Myanmar. Wellcome Open Res. 2017;2:81.
Leroy D, Macintyre F, Adoke Y, Ouoba S, Barry A, Mombo-Ngoma G, et al. African isolates show a high proportion of multiple copies of the Plasmodium falciparum plasmepsin-2 gene, a piperaquine resistance marker. Malar J. 2019;18:126.
Imwong M, Suwannasin K, Kunasol C, Sutawong K, Mayxay M, Rekol H, et al. The spread of artemisinin-resistant Plasmodium falciparum in the Greater Mekong subregion: a molecular epidemiology observational study. Lancet Infect Dis. 2017;17:491–7.
Son DH, Thuy-Nhien N, von Seidlein L, Le Phuc-Nhi T, Phu NT, Tuyen NTK, et al. The prevalence, incidence and prevention of Plasmodium falciparum infections in forest rangers in Bu Gia Map National Park, Binh Phuoc province, Vietnam: a pilot study. Malar J. 2017;16:444.
Nayebare P, Asua V, Conrad MD, Kajubi R, Kakuru A, Nankabirwa JI, et al. Associations between malaria-preventive regimens and Plasmodium falciparum drug resistance-mediating polymorphisms in Ugandan pregnant women. Antimicrob Agents Chemother. 2020;64:e01047-e1120.
Wallender E, Zhang N, Conrad M, Kakuru A, Muhindo M, Tumwebaze P, et al. Modeling prevention of malaria and selection of drug resistance with different dosing schedules of dihydroartemisinin-piperaquine preventive therapy during pregnancy in Uganda. Antimicrob Agents Chemother. 2019;63:e01393-e1418.
Zongo I, Milligan P, Compaore YD, Some AF, Greenwood B, Tarning J, et al. Randomized noninferiority trial of dihydroartemisinin-piperaquine compared with sulfadoxine-pyrimethamine plus amodiaquine for seasonal malaria chemoprevention in Burkina Faso. Antimicrob Agents Chemother. 2015;59:4387–96.
Somé AF, Zongo I, Compaoré YD, Sakandé S, Nosten F, Ouédraogo JB, Rosenthal PJ. Selection of drug resistance-mediating Plasmodium falciparum genetic polymorphisms by seasonal malaria chemoprevention in Burkina Faso. Antimicrob Agents Chemother. 2014;58:3660–5.
Tumwebaze P, Jagannathan P, Legac J, Mota D, Tukwasibwe S, Whalen M, et al. Impact of antimalarial treatment and chemoprevention on the drug sensitivity of malaria parasites isolated from Ugandan Children. Antimicrob Agents Chemother. 2015;59:3018–30.
Taylor AR, Flegg JA, Holmes CC, Guérin PJ, Sibley CH, Conrad MD, et al. Artemether-lumefantrine and dihydroartemisinin-piperaquine exert inverse selective pressure on Plasmodium falciparum drug sensitivity-associated haplotypes in Uganda. Open Forum Infect Dis. 2016;4:ofw229.
Ochong E, Tumwebaze PK, Byaruhanga O, Greenhouse B, Rosenthal PJ. Fitness consequences of Plasmodium falciparum pfmdr1 polymorphisms inferred from ex vivo culture of ugandan parasites. Antimicrob Agents Chemother. 2013;57:4245–51.
Tripura R, Peto TJ, Chea N, Chan D, Mukaka M, Sirithiranont P, et al. A controlled trial of mass drug administration to interrupt transmission of multidrug-resistant falciparum malaria in Cambodian villages. Clin Infect Dis. 2018;67:817–26.
Deng C, Huang B, Wang Q, Wu W, Zheng S, Zhang H, et al. Large-scale artemisinin-piperaquine mass drug administration with or without primaquine dramatically reduces malaria in a highly endemic region of Africa. Clin Infect Dis. 2018;67:1670–6.
Macintyre F, Adoke Y, Tiono AB, Duong TT, Mombo-Ngoma G, Bouyou-Akotet M, et al. A randomised, double-blind clinical phase II trial of the efficacy, safety, tolerability and pharmacokinetics of a single dose combination treatment with artefenomel and piperaquine in adults and children with uncomplicated Plasmodium falciparum malaria. BMC Med. 2017;15:181.
Acknowledgements
Thank you to colleagues at LSHTM and SGUL for our many interesting and insightful discussions.
Funding
SM is funded by Medical Research Council UK (Grant No. MR/N013638/1). TGC is funded by Medical Research Council UK (Grant no. MR/M01360X/1, MR/N010469/1, MR/R025576/1, and MR/R020973/1) Grants. SC is funded by BloomsburySET and Medical Research Council UK Grants (MR/M01360X/1, MR/R025576/1, and MR/R020973/1). AL is funded by Joint Global Health Trials Scheme (MRC, Wellcome Trust, UKRI, NIHR, Grant no. MR/S005013/1).
Author information
Authors and Affiliations
Contributions
SM conceived, designed and conducted the study. EM, SC and TGC provided data and assisted with analysis. SM wrote the first draft of the manuscript. All authors commented on and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Appendices
Appendix 1
Detailed search methodology
Detailed search terms used in MEDLINE
Ovid MEDLINE(R) ALL < 1946 to May 24, 2021 >
-
1.
Malaria/or malaria, cerebral/or malaria, falciparum/or blackwater fever/or malaria, vivax/67560
-
2.
(Malaria* or falciparum or plasmodium infection* or remittent fever or blackwater fever).mp. 107016
-
3.
1 or 2 107016
-
4.
Chemoprevention/or mass drug administration/6658
-
5.
Mass screening/or anonymous testing/or multiphasic screening/108880
-
6.
[Mass adj3 (screening* or administration* or test* or treatment*)].mp.121669
-
7.
(MSAT or MTAT or FSAT or FEMSE or MPPT or TME or DOT or RACD or IPTp or SMC).mp.52228
-
8.
Direct observed therap*.mp.27
-
9.
Mass piperaquine prophylactic treat*.mp.0
-
10.
Seasonal malaria chemo*.mp.115
-
11.
(Focal screening and treat*).mp.6
-
12.
Intermittent preventive treatment in pregnancy.mp.148
-
13.
Reactive case detection.mp.71
-
14.
(Focal screening and treatment).mp.6
-
15.
4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 181089
-
16.
3 and 15 2086
-
17.
Exp Antimalarials/85757
-
18.
(Anti-malarial* or antimalarial*).mp.35587
-
19.
Drug resistance/or drug resistance, multiple/or drug tolerance/83191
-
20.
[(Drug or multidrug) adj3 (resist* or toleran* or tolerate* or sensit*)].mp.368770
-
21.
[(Drug or multidrug) adj3 (resist* or toleran* or tolerate* or sensit*)].mp.368770
-
22.
17 or 18 or 19 or 20 or 21 448688
-
23.
16 and 22 1195
-
24.
(Piperaquine or dihydroartemisinin or dihydroartemisinin-piperaquine).mp. [mp = title, abstract, original title, name of substance word, subject heading word, floating sub-heading word, keyword heading word, organism supplementary concept word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier, synonyms] 2129
-
25.
23 and 24 117
These studies were filtered to included publication dates of 2011 onwards, resulting in 99 papers in total. Detailed search terms used in EMBASE. Embase <1974 to 2021 May 24>
-
1.
Malaria falciparum/or Plasmodium vivax malaria/ or malaria/ or Plasmodium knowlesi malaria/ or Plasmodium ovale malaria/88321
-
2.
(Malaria* or falciparum or plasmodium infection* or remittent fever or blackwater fever).mp.130248
-
3.
1 or 2 130248
-
4.
Mass drug administration/or chemoprophylaxis/26856
-
5.
Mass screening/56612
-
6.
[Mass adj3 (screening* or administration* or test* or treatment*)].mp.76289
-
7.
Direct observed therap*.mp.65
-
8.
Mass piperaquine prophylactic treat*.mp.0
-
9.
Seasonal malaria chemo*.mp.255
-
10.
(Focal screening and treat*).mp.16
-
11.
Intermittent preventive treatment in pregnancy.mp.226
-
12.
Reactive case detection.mp.157
-
13.
(Focal screening and treatment).mp.15
-
14.
4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 102462
-
15.
3 and 14 3684
-
16.
Antimalarial agent/24465
-
17.
(Anti-malarial* or antimalarial*).mp.41035
-
18.
Antimalarial drug resistance/ or antimalarial drug susceptibility/ or drug resistance/ or multidrug resistance/143743
-
19.
[(Drug or multidrug) adj3 (resist* or toleran* or tolerate* or sensit*)].mp.470284
-
20.
16 or 17 or 18 or 19 502467
-
21.
15 and 20 1461
-
22.
(Dihydroartemisinin or dihydroartemisinin plus piperaquine or piperaquine).mp. [mp = title, abstract, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword, floating subheading word, candidate term word] 4363
-
23.
21 and 22 192
These studies were filtered to included publication dates of 2011 onwards, resulting in 129 papers in total. Removing duplicates resulted in 184 papers. These 134 papers were excluded because they either did not include reference to PPQ or DHA-PPQ, or they did not include reference to drug resistance/efficacy/tolerability or sensitivity in the abstract or title. This resulted in 50 publications which were assessed for eligibility. 35 studies were excluded because they did not include measurement of molecular markers of resistance, leaving 15 studies from MEDLINE or EMBASE which were included in this review.
IDDO search
Search date: 7/7/2021.
In the IDDO database (https://www.iddo.org), the terms ‘Malaria’ and ‘Piperaquine’ were used to identify publications. This resulted in 9 publications, 2 of which included analysis of molecular markers of resistance and were included in this review.
Clinicaltrials.gov search
Search date: 7/7/2021.
The following search (Disease: malaria, Terms: Piperaquine Status: Has results) resulted in 13 clinical studies. Four studies were excluded as these were pharmacokinetic studies of interventions in healthy volunteers. Three studies were excluded as these were studies involving induced blood stage malaria in healthy participants, as opposed to vector transmitted malaria. This resulted in 1 Phase 2 clinical trial and 5 Phase 3 clinical trial remaining. There were 7 publications associated with these clinical trials which included analysis of molecular markers of resistance. Four of these were unique and included in this review.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Moss, S., Mańko, E., Krishna, S. et al. How has mass drug administration with dihydroartemisinin-piperaquine impacted molecular markers of drug resistance? A systematic review. Malar J 21, 186 (2022). https://doi.org/10.1186/s12936-022-04181-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-022-04181-y