
Abstract
Chemotherapy resistance remains a major cause of therapeutic failure in gastric cancer. The combination of genetic material such as interference RNAs (iRNAs) to silence cancer-associated genes with chemotherapeutics has become a novel approach for cancer treatment. However, finding the right target genes and developing non-toxic, highly selective nanocarrier systems remains a challenge. Here we developed a novel sialyl-Tn-targeted polylactic acid—didodecyldimethylammonium bromide nanoparticle (PLA-DDAB) nanoparticles (NPs) loaded with dsRNA targeting ST6GalNac-I and/or galectin-3 genes. Using single photon emission computed tomography (SPECT), we have demonstrated that 99mtechnetium radiolabeled sialyl-Tn-targeted nanoparticles can reach the tumor site and downregulate ST6GalNAc-I and galectin-3 RNA expression levels when injected intravenously. Furthermore, using an in vivo gastric tumor model, these nanoparticles increased the effectiveness of 5-FU in reducing tumor growth. Our findings indicate that cancer-associated glycan-targeted NPs loaded with dsRNA targeting ST6GalNAc-I and/or galectin-3 in combination with standard chemotherapy, have the potential to become a novel therapeutic tool for gastric cancer.
Similar content being viewed by others


Introduction
Gastric cancer is the fifth most common cancer and the fourth leading cause of cancer death worldwide in 2020 (Sung et al. 2020). Because gastric cancer is often diagnosed at an advanced stage, systemic chemotherapy is the mainstay of treatment for these patients (Hatta et al. 2020; Kim et al. 2019; Katai et al. 2020). 5-fluorouracil (5-FU) currently remains the most widely accepted first-line chemotherapeutic option for gastric cancer (Smyth et al. 2016; Ajani et al. 2016), but most patients develop intrinsic or acquired resistance to 5-FU, which leads to therapeutic failure. In the last few years, many efforts have been made to identify more specific therapeutic targets with the aim of reducing the side-effects associated with conventional chemotherapy and also overcoming cancer treatment resistance frequently developed by tumors.
Aberrant cell surface glycosylation, a known cancer hallmark, is currently known to actively drive tumor transformation, metastasis, angiogenesis, immune escape and resistance to therapy (Pinho and Reis 2015; Santos et al. 2016; Schultz et al. 2012). Cancer-associated glycans are often correlated with a worse prognosis and have been considered important clinical tumor markers useful for diagnostic and therapeutic purposes (Kim and Varki 1997; Reis et al. 2010). In human cells, these glycans can be found covalently attached to asparagines residues in a Asn-X-Ser/Thr motif in N-glycans, or to hydroxyl groups of a serine, threonine or hydroxylysine in O-glycans (Moremen et al. 2012). ST6GalNAc-I is the major sialyltransferase responsible for the synthesis of sialyl-Tn (sTn) antigens, which are shortened or truncated O-glycans associated to tumor progression (Marcos et al. 2011). It has been demonstrated that blocking the posterior elongation of O-glycans by ST6GalNAc-I can disrupt the binding of glycan-binding proteins (or lectins) such as galectins, which are signaling molecules involved in cell–cell communication, adhesion, migration, cancer metastasis and drug resistance (Song et al. 2014; Takenaka et al. 2004; Ochieng et al. 1998; Dumic et al. 2006).
Galectin-3 (gal-3), a glycan-binding protein, binds to lactosamine sequences (Galβ1,4GlcNAc) on both N- and O-glycans via the carbohydrate recognition domain (CRD) (Barondes et al. 1994). Galectin-3 can be found in the nucleus, cytoplasm, and extracellularly bound to cells or extracellular matrix. Intracellularly, gal-3 has been shown to protect breast cancer cells against cisplatin, anthracycline, adriamycin and 5-FU-induced apoptosis (Takenaka et al. 2004). Both Galectin-3 and ST6GalNAc-I have been found to be upregulated in several tumors types such as breast, colon, pancreatic, ovary, thyroid and gastric (Cheng et al. 2015; Mayoral et al. 2008; Zaia Povegliano et al. 2011; Barut et al. 2010; Xie et al. 2012; Pinho et al. 2007; Xu et al. 2015; Akita et al. 2012; Julien et al. 2006; Kim et al. 2002) when compared to healthy tissue. In addition, Galectin-3 and ST6GalNAc-I overexpression have been widely associated with tumorigenesis and the acquisition of a metastatic phenotype. In a previous study, our group demonstrated that overexpression of sTn in gastric cancer cells, induced by ST6GalNAc-I overexpression, decreased galectin-3 (gal-3) cell surface binding sites, leading to gal-3 intracellular accumulation, and protecting tumor cells to chemotherapeutics-induced cytotoxicity (Santos et al. 2016). The in vitro inhibition of gal-3 or ST6GalNAc-I expression in sTn-expressing tumor cells, was able to increase sensitivity of cells to chemotherapeutic drugs such as cisplatin and 5-FU (Santos et al. 2016). All of the above-mentioned data highlight the importance of cellular glycosylation machinery in drug resistance, which often leads to treatment failures and disease progression.
Since resistance to chemotherapy is a major obstacle to clinical treatment of cancer, our hypothesis is that targeting gal-3 and/or ST6GalNAc-I enzyme may be a potential therapeutic strategy to overcome chemotherapy resistance in gal-3 and/or sTn-expressing-gastric tumors. As such, the use of interference RNA (RNAi) has become an attractive agent for the development of novel specific therapeutics to overcome drug resistance. Currently, nanoparticles (NPs) have received considerable attention as nanocarriers for RNAi delivery (Dizaj et al. 2014). Indeed, NPs can protect RNAi molecules from degradation in vivo and can be efficiently decorated with molecules such as antibodies or peptides to target specific tissue. For example, by directing a NP loaded with Gli1 siRNA to the CD44 receptor present in gastric cancer stem cells, Yao et al. (2020) demonstrated significant anti-tumor recurrence efficacy in vivo. In another study, a NP directed to the CD320 receptor overexpressed in gastric cancer containing miR-532-3p was found to induce apoptosis and suppress proliferation of gastric cancer cells both in vitro and in vivo (Chen et al. 2021). Although several cancer biomarkers have been considered for directing NPs to the tumor site and silencing target gene expression, so far, no NPs have been directed to the cancer-associated glycan sTn, nor aimed at silencing gal-3 and ST6GalNAc-I target genes in vivo.
Here, we developed a dsRNA-loaded nanocarrier to knockdown ST6GalNAc-I or gal-3 in sTn-expressing tumors in order to reverse the molecular mechanisms underlying chemoresistance. This nanocarrier system is expected to become a novel, potential therapeutic strategy for cancers with already known mechanisms of chemoresistance, in combination with standard chemotherapy.
Results
Development of sTn-targeted nanoparticles loaded with dsRNA targeting ST6GalNAc-I or galectin-3
To further evaluate the interplay between ST6GalNAc-I and galectin-3 in increasing the sensitivity of sTn-expressing tumor cells to chemotherapy (Santos et al. 2016), we developed sTn-targeted nanoparticles for the in vivo delivery of dsRNA against ST6GalNAc-I and galectin-3. To this end, poly-lactic acid (PLA)—polyvinyl alcohol (PVA)—didodecyldimethylammonium bromide (DDAB) nanoparticles (NPs), containing 1 nmol of dsRNA against ST6GalNAc-I, 1 nmol of dsRNA against galectin-3 or 1 nmol of random dsRNA were obtained by the double emulsion solvent evaporation method in the presence or absence of 100 µg of anti-sialyl-Tn antibody (TKH2) (Fig. 1A).

Development of sTn-targeted nanoparticles loaded with dsRNA targeting ST6GalNAc-I or galectin-3. A Schematic representation of poly-lactic acid nanoparticles (PLA-NP) development. PLA-NP were prepared by the double emulsion solvent evaporation method with poly-lactic acid polymer (PLA), dsRNA (double strand RNA against ST6GalNAc-I, galectin-3 or random), didodecyldimethylammonium bromide (DDAB) and anti-sTn antibody. B Table shows the mean size and polydispersity index of sTn-targeted and non-targeted NPs loaded with dsRNA-galectin-3, dsRNA-ST6GalNAc-I or dsRNA-random. C Representative images of sTn-targeted and non-targeted NPs by TEM
The dsRNAs targeting ST6GalNAc-I (dsRNA-ST6GalNAc-I) or galectin-3 (dsRNA-galectin-3) were obtained as described previously (Santos et al. 2016) and for subsequent experiments dsRNA1 for gal-3 and dsRNA2 for ST6GalNAc-I were chosen because they induced significant downregulation of gene expression levels (Additional file 1: Figure S1A and S1B). DDAB was used in the formulation as a cationic agent to neutralize the negative dsRNA charges and stabilize the nanoparticles. The developed NPs were spherical, showed a size range of 207.6 to 230.7 nm, which is satisfactory for in vitro and in vivo assays (Blanco et al. 2015; Hoshyar et al. 2016), and displayed a low polydispersity index (PDI) ranging from 0.060 to 0.109, indicating homogeneous particle-size distributions (Fig. 1B, C and Additional file 1: Figure S2), which is desirable for pharmaceuticals products (da Silva de Barros et al. 2021; Corrêa et al. 2022).
The NPs were subsequently tested for their ability to inhibit gal-3 or ST6GalNAc-I using MKN45-Mock (gastric cancer cell line negative for sTn) and MKN45-ST6GalNAc-I (gastric cancer cell line, which overexpress ST6GalNAc-I, the enzyme responsible for the synthesis of sTn antigen). As shown in Fig. 2A, 10 nM of naked dsRNA-galectin-3 or encapsulated in NPs (with or without the anti-sialyl-Tn antibody) was able to significantly reduce the mRNA levels of gal-3 in MKN45-Mock and MKN45-ST6GalNAc-I cells when compared to dsRNA-random (naked or encapsulated). Similarly, dsRNA-ST6GalNAc-I, both naked and encapsulated, was able to significantly reduce ST6GalNAc-I mRNA expression in MKN45-ST6GalNAc-I cells in comparison to dsRNA-random (Fig. 2B). The specific binding of sTn-targeted NPs to sTn-expressing cells was also confirmed in vitro (Additional file 1: Figure S3). These data indicate that sTn-targeted NPs are able to efficiently and specifically deliver dsRNA to downregulate ST6GalNAc-I and gal-3 mRNA expression in gastric tumor cells.

STn-targeted nanoparticles inhibit galectin-3 and ST6GalNAc-I mRNA expression in vitro. mRNA levels of A galectin-3 or B ST6GalNAc-I in MKN45-Mock and MKN45-ST6GalNAc-I cells after treatment with naked dsRNA-galectin-3, dsRNA-ST6GalNAc-I, dsRNA-random or sTn-targeted NP loaded with dsRNA-galectin-3, dsRNA-ST6GalNAc-I or dsRNA-random for 48 h. Values were normalized to β-actin. Data are the mean ± SEM, n = 3, **p < 0.01 and ***p < 0.001
STn-targeted nanoparticles containing dsRNA-galectin-3 and/or dsRNA-ST6GalNAc-I increase gastric cancer cells susceptibility to 5-FU in vitro
The sTn-targeted NPs were then evaluated for toxicity by treating tumor cells (MKN45-Mock and MKN45-ST6GalNAc-I) and non-tumoral HUVEC and CHO cells with increasing concentrations of NPs loaded with dsRNA-ST6GalNAc-I or dsRNA-galectin-3 (ranging from 0.3 to 160 nM). After 48 h of treatment no reduction in cell viability was observed, suggesting that sTn-targeted NPs were not toxic for both tumor and non-tumor cells (Fig. 3A–D).

STn-targeted nanoparticles containing dsRNA against galectin-3 and/or ST6GalNAc-I increase gastric cancer cells susceptibility to 5-FU in vitro. SRB assay showing the viability of MKN45-Mock, MKN45-ST6GalNAc-I, HUVEC and CHO cells under the treatment with sTn-targeted NP loaded with dsRNA-galectin-3 (A, B) or sTn-targeted NP loaded with dsRNA-ST6GalNAc-I (C, D). E SRB assay showing the viability of MKN45-ST6GalNAc-I cells in the presence of 10 nM of sTn-targeted NP loaded with dsRNA-galectin-3, dsRNA-ST6GalNAc-I or dsRNA-random, or a (1:1) combination of dsRNA-ST6GalNAc-I and dsRNA-galectin-3, in the presence of 5-FU (ranging from 2 to 0.001 mM). Results are presented as a percentage of viable cells relative to control (cells with no NPs or 5-FU treatment). F The average of IC50 values. Data are the mean ± SEM, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001
The ability of sTn-targeted NPs to increase MKN45-ST6GalNAc-I cells susceptibility to 5-FU chemotherapy was also evaluated in vitro. To this end, MKN45-ST6GalNAc-I cells were treated with increasing concentrations of 5-fluorouracil (5-FU), ranging from 0.001 to 2 mM, in the presence of sTn-targeted NPs loaded with 10 nM of dsRNA-ST6GalNAc-I, dsRNA-galectin-3, dsRNA-random or a (1:1) combination of dsRNA-ST6GalNAc-I and dsRNA-galectin-3. As expected, MKN45-ST6GalNAc-I cells incubated with sTn-targeted NPs loaded with dsRNA-ST6GalNAc-I displayed a higher susceptibility to the cytotoxic effect of 5-FU when compared to sTn-targeted NPs loaded with dsRNA-random (Fig. 3E, F). The downregulation of gal-3 mRNA expression with sTn-targeted NPs loaded either with dsRNA-galectin-3 or the (1:1) combination of dsRNA-ST6GalNAc-I + dsRNA-galectin-3 further increased MKN45-ST6GalNAc-I cells susceptibility to 5-FU, in comparison with sTn-targeted NPs loaded with dsRNA-ST6GalNAc-I (Fig. 3E, F). Overall, the data indicate that sTn-targeted NPs loaded with dsRNA-galectin-3, dsRNA-ST6GalNAc-I or a combination of both can efficiently increase the susceptibility of gastric cancer cells to 5-FU treatment.
STn-targeted nanoparticles reach sTn-expressing tumors in vivo
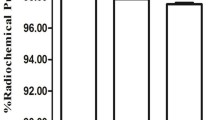
We next sought to evaluate whether the sTn-targeted NPs reach sTn-expressing tumor microenvironment by using SPECT/CT imaging to visualize the uptake of NPs by MKN45-ST6GalNAc-I-derived tumors. To accomplish this, sTn-targeted NPs were radiolabeled with 99mTc (99mTc-sTn-targeted NPs) using a direct labeling method, achieving a high radiochemical purity (> 98%) (Fig. 4A, B). 99mTc-radiolabeling was stable for at least 6 h in both serum and saline (Additional file 1: Figure S4). Radiolabeled sTn-targeted NPs (37 MBq) were then injected intravenously in MKN45-ST6GalNAc-I tumor-bearing mice (Fig. 4C) and SPECT/CT images were acquired 60 min post-injection. SPECT/CT images revealed that the uptake of 99mTc-sTn-targeted NPs by sTn-expressing tumors was significantly higher than that of non-targeted NPs (99mTc-NP), with an SUV of 0.051 and 0.013, respectively (Fig. 4D).

STn-targeted nanoparticles reach sTn-expressing tumors in vivo. Radio thin-layer chromatogram of A 99mTc-non-targeted or B 99mTc-sTn-targeted NPs with acetone as mobile phase. The radiochemical purity (RCP) > 98%. C Schematic representation of the radiolabeling process followed by tail vein injection of NPs in mice. D Representative static small animal SPECT/CT images 1-h after injection of 37 MBq of 99mTc-non-targeted or 99mTc-sTn-targeted NPs in MKN45-ST6GalNAc-I tumor-bearing Balb/c nude mice. Images are displayed as representative SPECT/CT images of tumor. Standard uptake values (SUV) for tumors are shown. Semiquantitative data are shown as mean ± SEM from 3 individual experiments
The uptake of 99mTc-sTn-targeted NPs was also quantified in relevant organs 1-h post-intravenous administration through an ex vivo biodistribution study. 99mTc-sTn-targeted NPs were mostly found in the liver, spleen and blood of MKN45-ST6GalNAc-I tumor-bearing mice, while 99mTc-non-targeted NPs were mainly found in the kidneys and liver of mice (Fig. 5A). The increased uptake of sTn-targeted NPs (which contain antibodies in their composition) by the liver and spleen in comparison with non-targeted NPs is a result of specific uptake by tissue-resident macrophages (Foroozandeh and Aziz 2018; Behzadi et al. 2017; Santos et al. 2017).

Biodistribution of 99mTc-non-targeted and 99mTc-sTn-targeted NPs in MKN45-ST6GalNAc-I tumor-bearing mice. A 99mTc-non-targeted or 99mTc-sTn-targeted NPs (3.7 MBq) were injected intravenously in MKN45-ST6GalNAc-I tumor-bearing Balb/c nude mice. One-hour post-injection, mice were euthanized, organs of interest were collected, weighted and radioactivity in each organ was measured. Data are presented as the mean (% ID/g of tissue) ± SD of n = 5. B Tumor-to-muscle ratio uptake of 99mTc-non-targeted and 99mTc-sTn-targeted NPs. Data were analyzed by one unpaired t-test (multiple t tests), *p < 0.05 and **p < 0.01
In agreement with the SPECT/CT imaging studies, sTn-expressing tumors showed a statistically significant increase in uptake of 99mTc-sTn-targeted NPs in comparison with 99mTc-non-targeted NPs (0.56 ± 0.12% ID/g and 0.206 ± 0.05% ID/g, respectively). The tumor-to-muscle ratio was significantly higher in 99mTc-sTn-targeted NPs in comparison with 99mTc-non-targeted NPs (Fig. 5B). Altogether, our data demonstrate that sTn-targeted NPs display a specific accumulation in sTn-expressing tumors.
STn-targeted NPs downregulate galectin-3 and/or ST6GalNAc-I in tumor cells in vivo
After demonstrating that sTn-targeted NPs reach the tumor site in vivo, we next sought to investigate whether these NPs could efficiently deliver dsRNA-galectin-3 or dsRNA-ST6GalNAc-I to the tumor microenvironment. To this end, MKN45-ST6GalNAc-I bearing mice were injected intratumorally or intravenously with sTn-targeted NPs loaded with dsRNA-ST6GalNAc-I, dsRNA-galectin-3, dsRNA-random or a (1:1) combination of dsRNA-ST6GalNAc-I and dsRNA-galectin-3. Seventy hours post-injection, tumors were collected to evaluate the mRNA levels of gal-3 and ST6GalNAc-I. As expected, intratumoral injection of sTn-targeted NPs loaded with either dsRNA-ST6GalNAc-I or dsRNA-galectin-3 led to a significant downregulation of ST6GalNAc-I (75%) (Fig. 6A) or gal-3 (77%) mRNA expression (Fig. 6B), respectively, in comparison with control NPs (sTn-targeted NPs loaded with dsRNA-random). When the NPs were injected intravenously, we could still observe a statistically significant downregulation of ST6GalNAc-I (52%) (Fig. 6C) and gal-3 (48%) (Fig. 6D) mRNA expression when compared to sTn-targeted NPs loaded with dsRNA-random. Overall, we have demonstrated that sTn-targeted NPs can reach sTn-expressing tumors and efficiently release their content in the tumor microenvironment.

STn-targeted NPs inhibit galectin-3 or ST6GalNAc-I in sTn-expressing tumor cells in vivo. ST6GalNAc-I (A, C) or galectin-3 (B, D) mRNA expression in MKN45-ST6GalNAc-I-derived tumors after intratumoral (A, B) or intravenous (C, D) injection of sTn-targeted NP (dsRNA-ST6GalNAc-I), sTn-targeted NP (dsRNA-galectin-3) or sTn-targeted NP (dsRNA-random). Values were normalized to β-actin. Data are the mean ± SEM, n = 3, *p < 0.05 and **p < 0.01
STn-targeted NPs containing dsRNA-galectin-3 and/or dsRNA-ST6GalNAc-I increase 5-FU therapeutic effect in vivo
We finally evaluated whether the downregulation of ST6GalNAc-I and/or gal-3 expression in the tumor microenvironment could increase the sensitivity of sTn-expressing tumor cells to 5-FU. Using a mouse model of gastric cancer, we treated MKN45-ST6GalNAc-I-derived tumors with sTn-targeted NPs loaded with dsRNA-ST6GalNAc-I, dsRNA-galectin-3, dsRNA-random or a (1:1) combination of dsRNA-ST6GalNAc-I and dsRNA-galectin-3. The NPs were injected intravenously (loaded with150 nM of dsRNA) in combination with 5-FU (25 mg/Kg; injected intraperitoneally), three times a week for a total period of 23 days (Fig. 7A). Our results demonstrated that the treatment with dual combination of sTn-targeted NPs (loaded with dsRNA-ST6GalNAc-I) and 5-FU led to a significant decrease (48% reduction at day 43) in the tumor growth when compared to control sTn-targeted NPs (loaded with dsRNA-random) combined with 5-FU treatment (Fig. 7B). Treatment of mice with sTn-targeted NPs loaded with dsRNA-galectin-3 further enhance the sensitivity of MKN45-ST6GalNAc-I-derived tumors to 5-FU in comparison to control (68% reduction at day 43) and; the dual treatment with sTn-targeted NPs (loaded with a (1:1) combination of dsRNA-ST6GalNAc-I and dsRNA-galectin-3) showed a synergistic effect with an additional reduction in tumor growth when compared to control (reduction of 77%) and to all other treatment groups (Fig. 7B).

STn-targeted NPs loaded with dsRNA-galectin-3 and/or dsRNA-ST6GalNAc-I increase 5-FU therapeutic effect in vivo. A Schematic representation of the in vivo experimental design and treatment schedule. On day-10, mice were brought from the vivarium to the laboratory animal research facility. On day zero, 1 × 106 MKN45-ST6GalNAc-I cells were injected subcutaneously in the right flank of mice. On day 20, mice were treated intravenously with sTn-targeted NPs and intraperitoneally with 25 mg/Kg of 5-FU for 3 weeks, 3 times a week and; on day 45 mice were euthanized. B Tumor growth of MKN45-ST6GalNAc-I-overexpressing cells in Balb/c nude mice treated or not with 5-FU (25 mg/Kg) plus sTn-targeted NPs loaded with dsRNA-galectin-3, dsRNA-ST6GalNAc-I, dsRNA-random or a (1:1) combination of dsRNA-ST6GalNAc-I and dsRNA-galectin-3. C Immunohistochemical staining of sTn and galectin-3 in MKN45-ST6GalNAc-I-derived tumors treated or not with 5-FU (25 mg/Kg) plus sTn-targeted NPs loaded with dsRNA-galectin-3, dsRNA-ST6GalNAc-I, dsRNA-random or a (1:1) combination of dsRNA-ST6GalNAc-I and dsRNA-galectin-3, bar = 20 μm. Data are the mean ± SEM (C) or representative (D) of n = 6, **p < 0.01 and ****p < 0.0001
At the end of the experiment, tumors were collected and sTn and galectin-3 expression were assessed by immunohistochemical staining (Fig. 7C). STn was found to be strongly expressed in MKN45-ST6GalNAc-I-derived tumors (control) treated or not with 5-FU and sTn-targeted NPs loaded with dsRNA-random and dsRNA-galectin-3. When treated with 5-FU + sTn-targeted NPs (dsRNA-ST6GalNAc-I) w/o sTn-targeted NPs loaded with dsRNA-galectin-3, we detected lower level of sTn expression in the tumor microenvironment, when compared to control group. Similarly, MKN45-ST6GalNAc-I-derived tumors from mice treated with sTn-targeted NPs loaded with dsRNA-galectin-3 or a (1:1) combination of dsRNA-ST6GalNAc-I and dsRNA-galectin-3 displayed reduced protein levels of galectin-3 when compared to the other groups of treatment. Altogether our data indicate that the downregulation of ST6GalNAc-I and/or galectin-3 expression in MKN45-ST6GalNAc-I-derived tumors lead to enhanced chemosensitivity to 5-FU treatment.
Material and methods
dsRNA synthesis
The plasmid pcDNA3.1 containing the full-length human ST6GalNAc-I was used as a template for the synthesis of two dsRNA sequences targeting ST6GalNAc-I mRNA using specific primers containing the T7 promoter sequences. For dsRNA1: forward primer (5’-3’): TAATACGACTCACTATAGGGAGAAGGCCGCCAACTTCAAATCT, reverse primer (5’-3’): TAATACGACTCACTATAGGGAGACTGCTGGGGCACTGGAG. For dsRNA2: forward primer (5’-3’): TAATACGACTCACTATAGGGAGAGCACTGCTTATGAATCAGACGG, reverse primer (5’-3’): TAATACGACTCACTATAGGGAGAATCCCTTCATCGT GTAGCCG.
For the synthesis of two dsRNA sequences targeting galectin-3 mRNA, we used the plasmid pET11a containing the full-length human galectin-3 coding sequence. The following specific primers containing the T7 promoter sequences were used. For dsRNA1: forward primer (5’-3’): TAATACGACTCACTATAGGGAGAACAATTCTGGGCACGGTGAA, reverse primer (5’-3’): TAATACGACTCACTATAGGGAGACCTTCCCCAGTTATTATCCAGC. For dsRNA2: forward primer (5’-3’): TAATACGACTCACTATAGGGAGAGAAGGGAAGAAAGACAGTCGGT, reverse primer (5’-3’): TAATACGACTCACTATAGGGAGAGCACTGGTGAGGTCTATGTCA.
The dsRNAs were synthetized by in vitro transcription using Megascript RNAi kit (Ambion) according to the manufacturer’s instructions. The dsRNA-random (non-targeting) sequence was generated by the “Random DNA Sequence Generator” (http://www.faculty.ucr.edu/~mmaduro/random.htm). Random sequence (5’-3’): GTAAAACGACGGCCAGTGATGAGTCTGGGTGGAGCGCGCCCCATTTATACCGTGAGTAGGGTCGACCAAGAACCGCAAGATGCGTCGGTGTACAAATAATTGTCAACAGACCGTCGTGTTTTGAAAATGGTACCAGCATCTTCGGGCGGTCTCAATCAAGCATGGATTACGGTTGAACTAATACGTATACTTTGCACGGGTTCACTGCGGTCCGTTCAGAGTCGACCAAGGACACAATCGAGCTCCCATCTGTATGCTCGA.
Poly-lactic acid nanoparticles (PLA-NP) development
Poly-lactic acid nanoparticles were prepared by the double emulsion solvent evaporation method with the poly-lactic acid polymer via super-sonic agitation. Briefly, 2 mL of poly vinyl alcohol (PVA) 1% aqueous solution (Sigma-Aldrich) was dripped in an organic solution containing 1 nmol of dsRNA, 490,86 µg of didodecyldimethylammonium bromide (DDAB) (Sigma-Aldrich), 100 µg anti-sTn antibody (TKH2, Creative Biolabs) and 50 mg of PLA (Sigma) previously solubilized in 3 mL of dichloromethane under ultra-sonic agitation (UP100H, Hielscher) for 10 min at 55 W. This first emulsion was further emulsified with 20 mL of PVA 0.1% solution under ultra-sonic agitation (UP100H, Hielscher) for 5 min at 55 W to produce a W/O/W emulsion. The organic phase was removed by evaporation under reduced pressure for 1.5 h at 25 ºC. The obtained nanoparticles were then washed three times with milli-Q water with centrifugation at 10.000g for 30 min and resuspended in 1 mL of saline (0.9% NaCl). dsRNA incorporation into the developed NPs was > 98% (Additional file 1: Figure S5) and remained consistent inside the developed NPs (Additional file 1: Figure S6).
The size distribution, mean size and polydispersity index (PDI) of the developed NPs were determined by dynamic light scattering (DLS) using the equipment Zetasizer Nano ZS (Malvern Instruments, UK). Measurements were performed in triplicate at 25 ºC and the laser incidence angle in relation to the sample was 173º using a 12-mm3 quartz cuvette. The morphology of the developed nanoparticles was analyzed with a transmission electron microscopy (TEM).
Cell culture
MKN45-Mock and MKN45-ST6GalNAc-I cells (Marcos et al. 2004) were cultured in RPMI (Gibco, Life technologies, MD, USA) while HUVEC (ATCC: CRL-2922) and CHO-K1 (ATCC: CCL-61) cells were cultured in DMEM (Gibco, Life technologies, MD, USA). All cells were supplemented with 10% of fetal bovine serum (Gibco, Life technologies, MD, USA) and 50 μg/mL of gentamicin (Gibco, Life technologies, MD, USA). Mycoplasma contamination in cultured cells was excluded by using Lonza Mycoplasma Detection Kit.
Cellular viability tests
Cells (2000) were seeded in a 96-well plate for 24 h. Next, cells were incubated with or without sTn-targeted NPs loaded with dsRNA (ranging from 0.3 to 160 nM) targeting galectin-3 (dsRNA-galectin-3), ST6GalNAc-I (dsRNA-ST6GalNAc-I) or random (dsRNA-random) for a total period of 72 h. Additionally, cells were incubated with 10 nM of sTn-targeted NPs loaded with dsRNA-galectin-3, dsRNA-ST6GalNAc-I, dsRNA-random or a (1:1) combination of dsRNA-ST6GalNAc-I and dsRNA-galectin-3 (5 nM each) for a period of 6 h. Then cells were treated with a serial dilution of 5-FU (Sigma) (ranging from 2 to 0.001 mM) for 72 h. Cell mass at the end of the experiment was determined based on the SRB cell protein stain (Voigt 2005). After this period, cells were fixed with 10% trichloroacetic acid at 4 °C for 1 h. The plates were then washed with distilled water and dried. SRB solution (150 μL) at 0.4% (w/v) in 1% acetic acid was added and incubated for 30 min at room temperature. The well plates were then washed with 1% acetic acid and dried. Tris base (100 μL of a 10 mM solution) was added to the wells to solubilize the bound SRB, and absorbance was then read at 515 nm on an automated microplate reader (VERSAmax, Molecular Devices, Sunnyvale, CA, USA). Data were analyzed with GraphPad Prism 6.0 software. The experiments were performed at least three times, with each condition plated in triplicate.
Ds-RNA-mediated gene silencing assay in vitro
MKN45-Mock or MKN45-ST6GalNAc-I cells were cultured in a 6-well plate and then treated with: (1) 10 nM of naked dsRNA against galectin-3, ST6GalNAc-I or random using the RNAiMAX reagent (Invitrogen) in Opti-MEM reduced serum medium (Invitrogen) or, (2) 10 nM of dsRNA against galectin-3, ST6GalNAc-I or random encapsulated in sTn-targeted NP). Six hours after transfection, medium was replaced with complete RPMI medium. Forty-eight hours after treatment, cells were collected for gene expression analysis.
Gene expression analysis
Total RNA from cell cultures or tumor tissue was isolated with Tri-Reagent (Sigma) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized using the High-capacity cDNA RT kit (Applied Biosystems), according to the manufacturer’s protocols. Quantitative PCR analysis was performed in triplicate using the SYBR Green (Applied Biosystem). Relative quantification was done using the ΔΔCt method normalizing to β-actin gene expression. The following primers were used. Forward primer human ST6GalNAc-I: TCCAAGGGAACACTTGAACCA; reverse primer human ST6GalNAc-I: GCCTCAGGACCTACAGCAAT; forward primer human galectin-3: TGTTTGCAATACAAAGCTGGA; reverse primer human galectin-3: GCAACCTTGAAGTGGTCAGG; forward primer human β-actin: GCCAGGTCATCACCATTGG; reverse primer human β-actin: GGTAGTTTCGTGGATGCCACA.
Radiolabeling of nanoparticle radiolabeling with 99mTc
Nanoparticles were radiolabeled with 99mTc by a direct method, as described previously (Santos et al. 2017). Briefly, 50 µL of sTn-targeted or non-targeted NP loaded with dsRNA-random were incubated with a stannous chloride (SnCl2, Sigma-Aldrich) solution (30 µg/mL) for 15 min at room temperature. Then 185 MBq of technetium-99 m (99mTc) (from IPEN-SP/CNEN, Brazil) was added to the solution for 10 min. The radiochemical purity (RCP) of the radiolabeled NPs was evaluated by radio-iTLC (instant thin-layer chromatography—iTLC) using glass microfiber chromatography paper impregnated with a silica gel (Agilent) and acetone (Sigma-Aldrich) as mobile phase. The radioactivity in the paper was analyzed in a TLC scanner (scan-RAM radio-TLC Scanner, LabLogic’s).
Tumor induction in Balb/c nude mice
Balb/c nude mice were bred at the animal facility of IPEN-SP/CNEN and all experiments were performed in compliance with the relevant laws and were approved by local animal ethics committees (protocol number: 181/17). For tumor induction, Balb/c nude mice were subcutaneously injected with 1 × 106 MKN45-ST6GalNAc-I cells. When tumors reached 150–300 mm3 mice were used for imaging, biodistribution, and treatment with sTn-targeted NPs for inhibition studies and evaluation of tumor growth.
Imaging and biodistribution studies
The in vivo binding specificity of radiolabeled 99mTc-NPs was evaluated in Balb/c nude mice bearing MKN45-ST6GalNAc-I tumors. Briefly, mice were injected intratumorally or intravenously with 37 MBq of 99mTc-sTn-targeted or 99mTc-non-targeted NPs (dsRNA-random). Imaging experiments were conducted 1-h post-injection on a Bruker Albira microPET/SPECT/CT imaging system (Bruker Biospin Corporation, Woodbridge, CT, USA). MicroSPECT/CT images were acquired under general anesthesia (1.8% isoflurane/O2) and heating at 37 °C. SPECT data for each mouse were recorded via static scans: SPECT scan—FOV 80 mm, 30 s/projection), followed by a 10 min CT scan (FOV 80 mm, 35 kV, 400 μA). The microSPECT/CT scans were reconstructed with Albira software (Bruker Biospin Corporation, Woodbridge, CT, USA) with Ordered Subsets Expectation Maximization (OSEM) and Filtered Back Projection (FBP) algorithms, for SPECT and CT, respectively, and images were processed with the PMOD software (PMOD Technologies, Zurick, CH). An elliptical volume-of-interest that enclosed the entire tumor was positioned manually on the SPECT images for the determination of the tumor volume. Then, 3-dimensional isocontours were drawn automatically. For each VOI, the standard uptake value (SUV) could be calculated directly using the output parameters from the μSPECT.
For biodistribution studies, 3.7 MBq/0.1 mL of 99mTc-sTn-targeted or 99mTc-non-targeted NPs (random) were injected in the tail vein of Balb/c nude mice bearing MKN45-ST6GalNAc-I tumors (5 animals/group). One hour after NPs administration mice were euthanized and organs of interest were immediately dissected out and weighed for quantitative estimation of gamma counts using a Perkin Elmer (Waltham, MA, USA) Wizard2 2480 automatic gamma counter to quantify the percent of injected dose per gram of tissue (% ID/g).
Intratumoral and intravenous injection of NPs loaded with dsRNA
STn-targeted or non-targeted NPs loaded with dsRNA-galectin-3, dsRNA-ST6GalNAc-I or dsRNA-random (150 nM) were intratumorally or intravenously injected in Balb/c nude mice bearing MKN45-ST6GalNAc-I tumors at day 0 and day 3. On day 6, tumors were collected for RNA extraction and quantification of ST6GalNAc-I or galectin-3 mRNA levels.
Evaluation of tumor growth upon treatment of MKN45-derived tumor with sTn-targeted NPs loaded with dsRNA plus 5-FU
For tumor growth evaluation, Balb/c nude mice bearing MKN45-ST6GalNAc-I tumors (6 mice per group) were treated 3 times a week (during 25 days) with a dual combination of sTn-targeted NPs loaded with dsRNA-galectin-3, dsRNA-ST6GalNAc-I, dsRNA-random (150 nM) or a combination 1:1 of dsRNA-galectin-3:dsRNA-ST6GalNAc-I (75 nM each) intravenously plus 5-FU (25 mg/kg) intraperitoneally. Tumor growth was assessed by caliper measurement.
Immunostaining
Tissue sections were deparaffinized in xylene and rehydrated in serial alcohol dilutions. Tissue sections were stained with anti-sialyl-Tn antibody (TKH2, creative biolabs) or anti-galectin-3 (M3/38, ATCC TIB166) followed by a secondary anti-mouse or anti-rat biotinylated antibodies (DAKO). Then, streptavidin-peroxidase (DAKO) was added, and color development was done with DAB (DAKO). Nuclei were counterstained with hematoxylin. Tissue samples were washed in PBS and mounted in Vectashield (Vector Laboratories, Inc). Representative areas were digitalized by digital camera (axioskop Plus, Zeiss, Germany).
Statistical analysis
Data are expressed as the mean ± SEM of at least three independent experiments. Statistical analysis including t-test, one-way ANOVA and two-way ANOVA were done using GraphPad Prism 8.0 software. A p-value < 0.05 was considered statistically significant.
Discussion
In the last two decades, substantial efforts have been directed at identifying specific therapeutic targets for cancer aimed at overcoming the chemoresistance mechanisms frequently developed by tumors. Indeed, there has been a shift from relatively non-specific cytotoxic medicines to selective, mechanism-based therapeutics. Additionally, the combination of anticancer therapies has become a standard practice in medical oncology. For example, the use of conventional chemotherapeutics with immunotherapies that boost the immune system to eradicate tumor cells can be an alternative approach for cancer treatment (Varayathu et al. 1844). In this direction, the use of nanosystems has shown to be an effective and viable alternative for combinatorial therapy. For example, nanopolymers can be used to encapsulate and delivery in vivo a variety of therapeutic agents such as nucleic acids, peptides, proteins and small hydrophilic or hydrophobic molecules, increasing the therapeutic bioavailability of drugs in the target area, and minimizing the toxic effects in healthy cells (Sun et al. 2017). In this study, we developed for the first time a nanosystem directed to sTn-expressing tumor cells aimed at delivering dsRNAs (targeting ST6GalNAc-I and galectin-3) as a combinatorial strategy to overcome current resistance to cancer chemotherapy.
Therapeutic agents using RNA, such as messenger RNAs (mRNAs), antisense oligonucleotides (ASOs), small interfering RNAs (siRNAs), microRNA (miRNAs) or aptamers have been widely studied over the last decades (Zogg et al. 2022). A few of them have already been approved by the U.S. Food and Drug Administration (FDA) such as patisiran, givosiran, lumasiran, inclisiran, casimersen and many are still under clinical trials (Zogg et al. 2022). Besides being extremely versatile, therapy using RNA is cost-effective and easier to develop than traditional small molecule or protein-based therapeutics (Winkle et al. 2021). Despite this, the current challenges for RNA therapeutics include the systemic delivery of the RNA molecule specifically to the target cell, without off-target effect, in a carrier stable enough to efficiently reach the target. The use of nanoparticles for this purpose shows great promise as an option for cancer treatment.
A wide variety of nanocarriers have been developed for the delivery of oligonucleotides to the tumor site as an attempt to overcome the in vivo obstacles. Recent literature has focused on lipid-based NPs and polymeric-based NPs because of their biocompatibility and low systemic side-effects. Indeed, lipid-based NPs containing RNA-targeted therapeutics have been widely found in clinical studies for cancer treatment (Barata et al. 2016; Kara et al. 2022) and pre-clinical research. At the time of writing this study, over 35 clinical studies were underway. For example, a neutral LNPs-liposomes loaded with siRNA against EphA2 is under investigation for the treatment of advanced solid tumors (NCT01591356). The drug NBF-006 (NP loaded with siRNA against GSTP) is under investigation for the treatment of Non-Small Cell Lung, Pancreatic, or Colorectal Cancer (NCT03819387) and the drug STP705 is under investigation for the treatment of cutaneous squamous cell carcinoma (in situ) skin cancer using siRNA against TGF-β1 and COX-2 (NCT 04,844,983). Regarding pre-clinical studies, for example, Lee and Ahn (2018) recently developed a PEGylated DC-Chol/DOPE-siRNA lipoplexes loaded with siRNA against kinesin spindle protein (KSP) gene and showed a significant suppression of tumor growth. In another example, the systemic administration of the cationic liposome/pVAX-miR-143 complex (CL-pVAX-miR-143) resulted in a remarkable inhibition of tumor metastasis in lung cancer metastasis mouse models (Jiang et al. 2019). Also, a cationic self-assembled DOTAP and methoxy poly(ethylene glycol)-poly(e-caprolactone) (MPEG–PCL) hybrid micelles (DMP) containing Bcl-xl-siRNA and Mcl1-siRNA was found to suppress the growth of subcutaneous xenograft of colon cancer model (Lu et al. 2019). More recently, Lu et al. (2020) designed a Th17 antibody-modified liposome polycation–DNA complex (LPD) encapsulated with TSPAN1 small interfering RNA (siRNA) (Th17-LPDT) and showed a decreased in the development of a subcutaneous xenograft of gastric cancer in mice.
Polymer-based nanoparticles, such as chitosan, proteins, PEI, poly(lactic-co-glycolic acid) (PLGA), poly-lactic acid (PLA), and dendrimers, are one of the most extensively investigated nanocarriers because of their chemical flexibility of functionalization. In this study, we used poly-lactic acid (PLA), a Food and Drug Administration approved polymer, as a drug delivery vehicle for dsRNA against ST6GalNAc-I and/or gal-3. PLA has been explored for several years in many therapeutic application because of its versatility, biocompatibility and biodegradability (Essa et al. 2013; Legaz et al. 2016; Casalini et al. 2019; Buhecha et al. 2019). Yang et al., for example, showed the fabrication assembly of a NP delivery system to encapsulate siRNA composed of a cationic lipid and amphiphilic block copolymer of poly(ethylene glycol)-b-poly(d,l-lactide) (mPEG–PLA) (Yang et al. 2011). Besides PLA, we also used the dichain cationic surfactant didodecyldimethylammonium bromide (DDAB) in our NP composition, not only to encapsulate our dsRNA, as previously reported (Gossmann et al. 2015) but also to facilitate the NP–biophysical interaction with membrane lipids of cancer cells, increasing in vivo tumor accumulation (Sharma et al. 2013; Peetla and Labhasetwar 2009).
It has long been known that aberrant and de novo sTn expression has been associated with tumor growth, invasion and metastasis (Julien et al. 2006; Ozaki et al. 2012) and lately with chemoresistance to cisplatin and 5-FU (Santos et al. 2016). In fact, we have already demonstrated that treatment of sTn-expressing cells in vitro with dsRNA-ST6GalNAc-I or dsRNA-galectin-3 could restore chemotherapeutic sensitivity (Santos et al. 2016). In this study, we have demonstrated that sTn-targeted NPs loaded with dsRNA-ST6GalNAc-I and/or dsRNA-galectin-3 is a promising strategy to overcome drug resistance in gastric cancer. Indeed, a large number of studies using polymeric NPs for the simultaneous delivery of iRNA and chemotherapeutics have been reported. For example, Qian et al., used a dendrimer analogs with an amphiphilic star-branched copolymers comprising PLA and polydimethylaminoethyl methacrylate (PDMAEMA) for the co-delivery of the chemotherapeutic doxorubicin and a microRNA for glioma therapy (Qian et al. 2014). In a study published by Su et al. (2012), PEI-coated polylactic-co-glycolic acid (PLGA) NPs were used for the simultaneous delivery of paclitaxel (chemotherapeutic) and siRNA to Stat3, demonstrating its effectivity in reducing tumor growth when compared to either treatment alone. In another study, using a pancreatic cancer, Zhao et al. tested a combination of gemcitabine (chemotherapeutic) and siRNA against HIF-1α encapsulated in polymeric PLGA NPs, containing PEG (polyethylene glycol) and PEI (Zhao et al. 2015). This approach also led to a reduction in the target gene expression (HIF-1α) and the combinatorial treatment showed a synergistic effect in reducing metastasis formation. In a recent study, human serum albumin (HSA) NPs modified with cetuximab was selected as the carrier for the multi drug resistance gene (MDR) siRNA and doxorubicin chemotherapeutic (DOX). The Cex-HSA/DOX/MDR1 siRNA NPs reached the tumor microenvironment and inhibited the tumor growth rate of MCF7-resistant tumor cells in vivo (Yang et al. 2021). Regarding gastric cancer, it has been found that the co-delivery of HIF-1α siRNA and 5-FU chitosan nanoparticles is an attractive strategy for overcoming multidrug resistance of gastric cancer (Chen et al. 2017), and in vitro, silencing CFL1 with magnetite iron oxide NPs containing siRNA in AGS gastric cancer cells suppressed migration and induced apoptosis (Daryabari et al. 2020). More recently, Zhou et al. (2022) have found the chitosan-gelatin-EGCG NPs containing an siRNA for the LncRNA TMEM44-AS1 was able to activate the P53 signaling pathway reversing 5-FU resistance in a gastric cancer mouse model. In a clinical setting, for example, a Phase 2 Study is being conducted with the delivery of siRNA against the mutated KRAS oncogene, in combination with systemic chemotherapy in patients with locally advanced pancreatic cancer (PROTACT) (NCT01676259). Similar to other studies, the nanocarrier developed here showed a great promise as an adjuvant therapy to systemic non-encapsulated chemotherapeutic treatment.
Although nanoparticles seem to be of great potential as nanocarriers to deliver inhibitory RNA targeting tumors, specific targeting is another important aspect to be considered when producing nanopolymers for the simultaneous administration of different classes of anti-tumor molecules. In order to direct nanoparticles directly to the tumor microenvironment, NPs have been modified with molecules that will specifically interact with receptors or ligands specifically expressed by tumor cells. Monoclonal antibodies are among the most used molecules for specific targeting (Bazak et al. 2015), because they specifically bind to tumor antigens overexpressed on the surface of many types of tumor cells. Yang et al. (2018), for example, constructed a SATB1 siRNA-encapsulated immunoliposomes conjugated with CD44 antibodies (CD44-SATB1-ILs) in order to target gastric cancer-initiating cells in in vitro models of gastric cancer. Hu et al. (2017), on the other hand, coated a poly(ε-caprolactone) (PCL)-poly (ethylene glycol) (PEG) NP, (PEG-PCL NPs) loaded with anti-miRNA-21 (AMO-21) and 5-FU, with trastuzumab to target gastric cancer with HER2 receptor overexpression and found a reduced growth of gastric cancer cell in vitro and in vivo. The NP developed here radiolabeled with 99mTc allowed us to visualize in vivo their accumulation in the tumor site, besides being able to increase tumor sensitivity to 5-FU in vitro and in vivo. In fact, radiolabeled NPs have been widely used to study NPs biodistribution in small animals through SPECT and PET imaging, reviewed recently (Das et al. 2021; Skotland et al. 2022), because of the advantage to obtain a quantitative whole-body biodistribution data on NPs. Certainly, the glycan-targeted NPs developed here still warrant further exploration and optimization and several questions still need to be answered before clinical translation: (1) will the tumor be completely eradicated after the combinatorial treatment of sTn-targeted NPs (for downregulating gal-3 and ST6GalNAc-I expression) and chemotherapeutic, or will it change its clonal dominance to select for clones that do not express gal-3 nor sTn, leading again to drug resistance? Further studies will have to address whether new cell surface biomarkers or new target genes will be required for a complete cure from cancer. (2) Can we improve tumor targeting by reducing the accumulation of sTn-targeted NPs in the liver and spleen? Indeed, small immune complexes containing antibodies are often eliminated by the classical reticuloendothelial system of liver and splenic macrophages because of their interaction with the Fc region of antibodies (Benacerraf et al. 1959; Arend and Sturge 1979; Kurlander et al. 1974; Skogh et al. 1985). Additional studies using NPs containing anti-sTn antibodies depleted of their Fc region (Fab’) might avoid sTn-targeted NPs uptake by the liver and spleen, making NPs more available to reach the tumor. Still, an exciting future of opportunities lies ahead and technologies that allows assessing and locating tissue-specific accumulation of NPs in vivo using molecular imaging techniques, such as PET and SPECT, are of main interest in the field. This would allow the development of more specific delivery systems, minimize the doses to be administered and evaluate the off-target effects.
Conclusions
Resistance to chemotherapy is a major obstacle to clinical treatment of cancer and, the combination of anticancer therapies is becoming a standard practice in the medical oncology. Interestingly, the combination of chemotherapeutics with genetic material such as dsRNA has proven to have great potential in enhancing cancer treatment efficacy by lowering the dose of drugs alleviating chemotherapeutic side-effects, and decreasing drug resistance (Kara et al. 2022). Sialyl-Tn is rarely expressed in normal tissues, and its de novo expression in cancer tissues makes it the perfect target for the delivery of dsRNA to cancer tissues. Moreover, the development of glycan-targeted dsRNA-loaded NPs for clinical applications such as SPECT/CT imaging and combinatorial therapy has the potential to revolutionize future therapeutic strategies.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reason- able request.
References
Ajani JA, D’Amico TA, Almhanna K, Bentrem DJ, Chao J, Das P et al (2016) Gastric Cancer, Version 3.2016, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 14:1286–1312
Akita K, Yoshida S, Ikehara Y, Shirakawa S, Toda M, Inoue M et al (2012) Different levels of sialyl-Tn antigen expressed on MUC16 in patients with endometriosis and ovarian cancer. Int J Gynecol Cancer 22:531–538
Arend WP, Sturge JC (1979) Composition and biologic properties of soluble IgG-Anti-IgG immune complexes: effects of variations in the specificity of rabbit antibodies to different structural components of human IgG. J Immunol 123:9
Barata P, Sood AK, Hong DS (2016) RNA-targeted therapeutics in cancer clinical trials: Current status and future directions. Cancer Treat Rev 50:35–47
Barondes SH, Castronovo V, Cooper DNW, Cummings RD, Drickamer K, Felzi T et al (1994) Galectins: A family of animal β-galactoside-binding lectins. Cell 8:597–598
Barut F, Kandemir NO, Bektas S, Bahadir B, Keser S, Ozdamar SO (2010) Universal markers of thyroid malignancies: Galectin-3, HBME-1, and Cytokeratin-19. Endocr Pathol 21:80–89
Bazak R, Houri M, El Achy S, Kamel S, Refaat T (2015) Cancer active targeting by nanoparticles: a comprehensive review of literature. J Cancer Res Clin Oncol 141:769–784
Behzadi S, Serpooshan V, Tao W, Hamaly MA, Alkawareek MY, Dreaden EC et al (2017) Cellular uptake of nanoparticles: journey inside the cell. Chem Soc Rev 46:4218–4244
Benacerraf B, Sebestyen M, Cooper NS (1959) The clearance of antigen antibody complexes from the blood by the reticulo-endothelial system. J Immunol 82:9
Blanco E, Shen H, Ferrari M (2015) Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol 33:941
Buhecha MD, Lansley AB, Somavarapu S, Pannala AS (2019) Development and characterization of PLA nanoparticles for pulmonary drug delivery: Co-encapsulation of theophylline and budesonide, a hydrophilic and lipophilic drug. J Drug Deliv Sci Technol 53:101128
Casalini T, Rossi F, Castrovinci A, Perale G. A perspective on polylactic acid-based polymers use for nanoparticles synthesis and applications. Front Bioeng Biotechnol; 2019;7:3168
Chen Y, Sun L, Guo D, Wu Z, Chen W (2017) Co-delivery of hypoxia inducible factor-1α small interfering RNA and 5-fluorouracil to overcome drug resistance in gastric cancer SGC-7901 cells. J Gene Med 19:9
Chen Z, Liang Y, Feng X, Liang Y, Shen G, Huang H et al (2021) Vitamin-B12-conjugated PLGA-PEG nanoparticles incorporating miR-532–3p induce mitochondrial damage by targeting apoptosis repressor with caspase recruitment domain (ARC) on CD320-overexpressed gastric cancer. Mater Sci Eng C Mater Biol Appl 120:111722
Cheng D, Liang B, Li Y (2015) Serum galectin-3 as a potential marker for gastric cancer. Med Sci Monit 21:755–760
Corrêa LB, Pinto SR, Alencar LMR, Missailidis S, Rosas EC, Henriques M et al (2022) Nanoparticle conjugated with aptamer anti-MUC1/Y for inflammatory arthritis. Colloids Surfaces B Biointerfaces. 211:112280
da Silva AO, Portilho FL, Dos SantosMatos AP, Ricci-Junior E, Alencar LMR, Dos Santos CC et al (2021) Preliminary studies on drug delivery of polymeric primaquine microparticles using the liver high uptake effect based on size of particles to improve malaria treatment. Mater Sci Eng 128:112275
Daryabari SS, Fathi M, Mahdavi M, Moaddab Y, Hosseinpour Feizi MA, Shokoohi B et al (2020) Overexpression of CFL1 in gastric cancer and the effects of its silencing by siRNA with a nanoparticle delivery system in the gastric cancer cell line. J Cell Physiol 235:6660–6672
Das S, Imlimthan S, Airaksinen AJ, Sarparanta M (2021) Radiolabeling of theranostic nanosystems. Adv Exp Med Biol 1295:49–76. https://doi.org/10.1007/978-3-030-58174-9_3
Dizaj SM, Jafari S, Khosroushahi AY (2014) A sight on the current nanoparticle-based gene delivery vectors. Springer, New York, pp 1–9
dos Santos SN, dos Reis SRR, Pinto SR, Cerqueira-Coutinho C, Nigro F, Barja-Fidalgo TC et al (2017) Anti-inflammatory/infection PLA nanoparticles labeled with technetium 99m for in vivo imaging. J Nanoparticle Res 19:1–8. https://doi.org/10.1007/s11051-017-4037-x
dos Santos SN, Rezende SR, Pires LP, Helal-Neto E, Sancenón F, Barja-Fidalgo TC et al (2017) Avoiding the mononuclear phagocyte system using human albumin for mesoporous silica nanoparticle system. Microporous Mesoporous Mater 251:181–189
Dumic J, Dabelic S, Flögel M. Galectin-3: An open-ended story. Biochim Biophys Acta; 2006. p. 616–35.
Essa S, Louhichi F, Raymond M, Hildgen P (2013) Improved antifungal activity of itraconazole-loaded PEG/PLA nanoparticles. Microencapsul 30:205–217
Foroozandeh P, Aziz AA (2018) Insight into cellular uptake and intracellular trafficking of nanoparticles. Nanoscale Res Lett 131:1–12. https://doi.org/10.1186/s11671-018-2728-6
Gossmann R, Langer K, Mulac D (2015) New perspective in the formulation and characterization of didodecyldimetylammonium bromide (DMAB) Stabilized Poly(Lactic-co-Glycolic Acid) (PLGA) Nanoparticles. PLoS ONE 10:26147338
Hatta W, Gotoda T, Koike T, Masamune A (2020) History and future perspectives in Japanese guidelines for endoscopic resection of early gastric cancer. Dig Endosc 32:180–190
Hoshyar N, Gray S, Han H, Bao G (2016) The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 11:673
Hu N, Yin JF, Ji Z, Hong Y, Wu P, Bian B et al (2017) Strengthening gastric cancer therapy by Trastuzumab-conjugated nanoparticles with simultaneous encapsulation of anti-MIR-21 and 5-fluorouridine. Cell Physiol Biochem 44:2158–2173
Jiang Q, Yuan Y, Gong Y, Luo X, Su X, Hu X et al (2019) Therapeutic delivery of microRNA-143 by cationic lipoplexes for non-small cell lung cancer treatment in vivo. J Cancer Res Clin Oncol 145:2951–2967
Julien S, Adriaenssens E, Ottenberg K, Furlan A, Courtand G, Vercoutter-Edouart AS et al (2006) ST6GalNAc I expression in MDA-MB-231 breast cancer cells greatly modifies their O-glycosylation pattern and enhances their tumourigenicity. Glycobiology 16:54–64
Kara G, Calin GA, Ozpolat B (2022) RNAi-based therapeutics and tumor targeted delivery in cancer. Adv Drug Deliv Rev 182:114113
Katai H, Mizusawa J, Katayama H, Morita S, Yamada T, Bando E et al (2020) Survival outcomes after laparoscopy-assisted distal gastrectomy versus open distal gastrectomy with nodal dissection for clinical stage IA or IB gastric cancer (JCOG0912): a multicentre, non-inferiority, phase 3 randomised controlled trial. Lancet Gastroenterol Hepatol 5:142–151
Kim YJ, Varki A (1997) Perspectives on the significance of altered glycosylation of glycoproteins in cancer. Glycoconj J 8:569–576
Kim GE, Bae HI, Park HU, Kuan SF, Crawley SC, Ho JJL et al (2002) Aberrant expression of MUC5AC and MUC6 gastric mucins and sialyl Tn antigen in intraepithelial neoplasms of the pancreas. Gastroenterology 123:1052–1060
Kim HH, Han SU, Kim MC, Kim W, Lee HJ, Ryu SW et al (2019) Effect of laparoscopic distal gastrectomy vs open distal gastrectomy on long-term survival among patients with stage i gastric cancer: the KLASS-01 Randomized Clinical Trial. JAMA Oncol 5:506–513
Kurlander RJ, Ellison DM, Hall J (1984) The blockade of Fc receptor-mediated clearance of immune complexes in vivo by a monoclonal antibody (2.4G2) directed against Fc receptors on murine leukocytes. J Immunol 133:9
Lee J, Ahn HJ (2018) PEGylated DC-Chol/DOPE cationic liposomes containing KSP siRNA as a systemic siRNA delivery Carrier for ovarian cancer therapy. Biochem Biophys Res Commun 503:1716–1722
Legaz S, Exposito JY, Lethias C, Viginier B, Terzian C, Verrier B (2016) Evaluation of polylactic acid nanoparticles safety using Drosophila model. Nanotoxicology 10:1136–1143
Lu Z, Pang T, Yin X, Cui H, Fang G, Xue X et al (2020) Delivery of TSPAN1 siRNA by Novel Th17 targeted cationic liposomes for gastric cancer intervention. J Pharm Sci 109:2854–2860
Lu Y, Zhong L, Jiang Z, Pan H, Zhang Y, Zhu G, et al. Cationic micelle-based siRNA delivery for efficient colon cancer gene therapy. Nanoscale Res Lett; 2019;14:5329
Marcos NT, Pinho S, Grandela C, Cruz A, Samyn-Petit B, Harduin-Lepers A et al (2004) Role of the human ST6GalNAc-I and ST6GalNAc-II in the synthesis of the cancer-associated Sialyl-Tn antigen. Cancer Res 64:7050–7057
Marcos NT, Bennett EP, Gomes J, Magalhaes A, Gomes C, David L et al (2011) ST6GalNAc-I controls expression of sialyl-Tn antigen in gastrointestinal tissues. Front Biosci 3:1443–1455
Mayoral MA, Mayoral C, Meneses A, Villalvazo L, Guzman A, Espinosa B et al (2008) Identification of Galectin-3 and mucin-type O-glycans in breast cancer and its metastasis to brain. Cancer Invest 26:615–623
Moremen KW, Tiemeyer M, Nairn AV (2012) Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol 9:448–462
Ochieng J, Leite-Browning ML, Warfield P (1998) Regulation of cellular adhesion to extracellular matrix proteins by galectin-3. Biochem Biophys Res Commun 246:788–791
Ozaki H, Matsuzaki H, Ando H, Kaji H, Nakanishi H, Ikehara Y et al (2012) Enhancement of metastatic ability by ectopic expression of ST6GalNAcI on a gastric cancer cell line in a mouse model. Clin Exp Metastasis 29:229–238
Peetla C, Labhasetwar V (2009) Effect of molecular structure of cationic surfactants on biophysical interactions of surfactant-modified nanoparticles with a model membrane and cellular uptake. Langmuir 25:2369–2377
Pinho SS, Reis CA (2015) Glycosylation in cancer: Mechanisms and clinical implications. Nature Publishing Group, New York, pp 540–555
Pinho S, Marcos NT, Ferreira B, Carvalho AS, Oliveira MJ, Santos-Silva F et al (2007) Biological significance of cancer-associated sialyl-Tn antigen: modulation of malignant phenotype in gastric carcinoma cells. Cancer Lett 249:157–170
Qian X, Long L, Shi Z, Liu C, Qiu M, Sheng J et al (2014) Star-branched amphiphilic PLA-b-PDMAEMA copolymers for co-delivery of miR-21 inhibitor and doxorubicin to treat glioma. Biomaterials 35:2322–2335
Reis CA, Osorio H, Silva L, Gomes C, David L (2010) Alterations in glycosylation as biomarkers for cancer detection. J Clin Pathol 16:322–329
Santos SN, Junqueira MS, Francisco G, Vilanova M, Magalhães A, Baruffi MD et al (2016) O-glycan sialylation alters galectin-3 subcellular localization and decreases chemotherapy sensitivity in gastric cancer. Oncotarget 7:83570–83587
Schultz MJ, Swindall AF, Bellis SL (2012) Regulation of the metastatic cell phenotype by sialylated glycans. Cancer Metastasis Rev 31:501–518
Sharma B, Peetla C, Adjei IM, Labhasetwar V (2013) Selective biophysical interactions of surface modified nanoparticles with cancer cell lipids improve tumor targeting and gene therapy. Cancer Lett 334:228–236
Skogh T, Blomhoff R, Eskildt W, Bergt T (1985) Hepatic uptake of circulating IgG immune complexes. Immunology 55:585
Skotland T, Iversen TG, Llorente A, Sandvig K (2022) Biodistribution, pharmacokinetics and excretion studies of intravenously injected nanoparticles and extracellular vesicles: Possibilities and challenges. Adv Drug Deliv Rev 186:114326
Smyth EC, Verheij M, Allum W, Cunningham D, Cervantes A, Arnold D et al (2016) Gastric cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 27:38–49
Song L, Tang J, Owusu L, Sun MZ, Wu J, Zhang J (2014) Galectin-3 in cancer. Clin Chim Acta 89:185–191
Su WP, Cheng FY, Shieh DB, Yeh CS, Su WC (2012) PLGA nanoparticles codeliver paclitaxel and Stat3 siRNA to overcome cellular resistance in lung cancer cells. Int J Nanomed 7:4269–4283
Sun H, Yarovoy I, Capeling M, Cheng C (2017) Polymers in the Co-delivery of siRNA and Anticancer Drugs for the Treatment of Drug-resistant Cancers. Top Curr Chem 89:7
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A et al (2021) Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 71:209–249
Takenaka Y, Fukumori T, Yoshii T, Oka N, Inohara H, Kim HRC et al (2004) Nuclear export of phosphorylated galectin-3 regulates its antiapoptotic activity in response to chemotherapeutic drugs. Mol Cell Biol 24:4395–4406
Varayathu H, Sarathy V, Thomas BE, Mufti SS, Naik R (2021) Combination strategies to augment immune check point inhibitors efficacy - implications for translational research. Front Oncol 11:1844
Voigt W (2005) Sulforhodamine B assay and chemosensitivity. Methods Mol Med 110:39–48
Winkle M, El-Daly SM, Fabbri M, Calin GA (2021) Noncoding RNA therapeutics - challenges and potential solutions. Nat Rev Drug Discov 20:629–651
Xie L, Ni WK, Chen XD, Xiao MB, Chen BY, He S et al (2012) The expressions and clinical significances of tissue and serum galectin-3 in pancreatic carcinoma. J Cancer Res Clin Oncol 138:1035–1043
Xu F, Fan C, Fan S, Liu F, Wen T, An G et al (2015) Expression profile of mucin-associated sialyl-Tn antigen in Chinese patients with different colorectal lesions (adenomas, carcinomas). Int J Clin Exp Pathol 8:11549–11554
Yang XZ, Dou S, Sun TM, Mao CQ, Wang HX, Wang J (2011) Systemic delivery of siRNA with cationic lipid assisted PEG-PLA nanoparticles for cancer therapy. J Control Release 156:203–211
Yang F, Zheng Z, Zheng L, Qin J, Li H, Xue X et al (2018) SATB1 siRNA-encapsulated immunoliposomes conjugated with CD44 antibodies target andeliminate gastric cancer-initiating cells. Onco Targets Ther 11:6811
Yang X, Wang Y, Chen S, Zhang S, Cui C (2021) Cetuximab-modified human serum albumin nanoparticles co-loaded with doxorubicin and MDR1 siRNA for the treatment of drug-resistant breast tumors. Int J Nanomed 16:7051–7069
Yao H, Sun L, Li J, Zhou X, Li R, Shao R et al (2020) A Novel Therapeutic siRNA Nanoparticle Designed for Dual-Targeting CD44 and Gli1 of Gastric Cancer Stem Cells. Int J Nanomedicine 15:7013–7034
Zaia Povegliano L, Oshima CTF, De Oliveira LF, Andrade Scherholz PL, Manoukian FN (2011) Immunoexpression of galectin-3 in colorectal cancer and its relationship with survival. J Gastrointest Cancer 42:217–221
Zhao X, Li F, Li Y, Wang H, Ren H, Chen J et al (2015) Co-delivery of HIF1α siRNA and gemcitabine via biocompatible lipid-polymer hybrid nanoparticles for effective treatment of pancreatic cancer. Biomaterials 46:13–25
Zhou M, Dong J, Huang J, Ye W, Zheng Z, Huang K et al (2022) Chitosan-Gelatin-EGCG Nanoparticle-Meditated LncRNA TMEM44-AS1 Silencing to Activate the P53 Signaling Pathway for the Synergistic Reversal of 5-FU Resistance in Gastric Cancer. Adv Sci 210:5077
Zogg H, Singh R, Ro S (2022) Current Advances in RNA Therapeutics for Human Diseases. Int J Mol Sci 23:9
Acknowledgements
We acknowledge the support of the Instituto de Pesquisas Energéticas e Nucleares (IPEN-CNEN/SP), São Paulo, Brazil and the Nuclear Engineering Institute, Rio de Janeiro, RJ, Brazil.
Funding
This research was supported by Grants from: the São Paulo Research Foundation—FAPESP, Brazil (Grant Number 2012/06875-6); The Brazilian National Council for Scientific and Technological Development—CNPQ (Visitor Researcher Grant Number 300376/2017-0) and; Carlos Chagas Filho Foundation for Research Support of Rio de Janeiro State (FAPERJ) (Cientista do Nosso Estado: E-26/200.815/2021; Rede NanoSaude: E-26/010.000981/2019).
Author information
Authors and Affiliations
Contributions
SNS, DSGJ, JPMP, AHS, TN and FROS performed the experiments and prepared the figures. dos SNS and ESB wrote the manuscript. ERJ, RSO and LAPD revised the manuscript critically for important intellectual content. ESB approved the version to be published. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary file: Aditional Figures and material and methods.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
dos Santos, S.N., Junior, D.S.G., Pereira, J.P.M. et al. Development of glycan-targeted nanoparticles as a novel therapeutic opportunity for gastric cancer treatment. Cancer Nano 14, 26 (2023). https://doi.org/10.1186/s12645-023-00161-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12645-023-00161-2