
Abstract
Background
Classification of disease severity is crucial for the management of COVID-19. Several studies have shown that individual proteins can be used to classify the severity of COVID-19. Here, we aimed to investigate whether integrating four types of protein context data, namely, protein complexes, stoichiometric ratios, pathways and network degrees will improve the severity classification of COVID-19.
Methods
We performed machine learning based on three previously published datasets. The first was a SWATH (sequential window acquisition of all theoretical fragment ion spectra) MS (mass spectrometry) based proteomic dataset. The second was a TMTpro 16plex labeled shotgun proteomics dataset. The third was a SWATH dataset of an independent patient cohort.
Results
Besides twelve proteins, machine learning also prioritized two complexes, one stoichiometric ratio, five pathways, and five network degrees, resulting a 25-feature panel. As a result, a model based on the 25 features led to effective classification of severe cases with an AUC of 0.965, outperforming the models with proteins only. Complement component C9, transthyretin (TTR) and TTR-RBP (transthyretin-retinol binding protein) complex, the stoichiometric ratio of SAA2 (serum amyloid A proteins 2)/YLPM1 (YLP Motif Containing 1), and the network degree of SIRT7 (Sirtuin 7) and A2M (alpha-2-macroglobulin) were highlighted as potential markers by this classifier. This classifier was further validated with a TMT-based proteomic data set from the same cohort (test dataset 1) and an independent SWATH-based proteomic data set from Germany (test dataset 2), reaching an AUC of 0.900 and 0.908, respectively. Machine learning models integrating protein context information achieved higher AUCs than models with only one feature type.
Conclusion
Our results show that the integration of protein context including protein complexes, stoichiometric ratios, pathways, network degrees, and proteins improves phenotype prediction.
Similar content being viewed by others

Background
COVID-19 caused by SARS-CoV-2 remains an ongoing pandemic [1]. Distinguishing severe and non-severe cases is crucial since only the severe cases require special treatment such as artificial ventilation [2]. Proteins from COVID-19 patients’ serum or plasma have been utilized to develop severity classifiers. In a TMT-based serum proteomics and metabolomics study, 118 sera samples from 65 COVID-19 patients and 53 controls were analyzed, resulting in a severity classifier based on characteristic proteins and metabolites [3]. Based on blood samples from early hospitalized cases, Messner et al. built a classifier including 27 proteins for the prediction of COVID-19 severity [4]. A longitudinal cohort from hospitalized COVID-19 patients identified a distinct proteomic trajectory associated with mortality in blood samples [5]. Another longitudinal cohort established an immune biomarker panel to gauge the severity of COVID-19 [6]. Demichev et al. presented a prognostic map of COVID-19 by linking clinical parameters to plasma proteomes [7] and established a proteomic survival predictor to distinguish severe cases [8]. A plasma-based proteomics study reported multiple modulated blood proteins of recovered COVID-19 patients 3 months after discharge [9]. Using the serum proteomics of COVID-19 patients with samples from different disease stages, Zhang et al. monitored disease progression and predicted viral nucleic acid positivity during COVID-19 [10].
However, all the above-mentioned protein classifiers for COVID-19 are based on individual biomolecules, mainly proteins, ignoring the fact that no protein functions in an isolated manner. As the ultimate effectors of diseases, protein complexes regulate many core biological processes. For example, mitochondrial complexes, such as the mitochondrial ribosomal small subunit, respiratory chain complex I and the mitochondrial pyruvate dehydrogenase complex, are involved in energy production and found to be highly conserved but dysregulated in diseases such as cancers [11]. Several tools for analyzing the protein complex features have been reported in recent years, including NetProt and Fuzzy-FishNET [12, 13]. PCprophet is a machine learning based software for identifying protein complex [14]. Protein stoichiometric ratios in a complex are also important since they are self-normalized and relatively conserved [15]. In addition, protein network degree is the number of edges connected to a protein in a network that includes all proteins in the matrix. Network degree reflects expression associations with other proteins [16]. In an in vitro cell culture experiment, 332 high-confidence protein–protein interactions between SARS-CoV-2 and host proteins were identified using affinity purification mass spectrometry, screening out two sets of pharmacological agents [17]. However, this study is limited to in vitro cell line culture, hence not directly transferrable to human plasma collected from patients with COVID-19.
As discussed above, although some studies have stratified COVID-19 severity based on protein levels, no protein functions in an isolated manner, and the information obtained from protein quantification alone may not be comprehensive. Besides the abundance, multidimensional information of proteins, including but not limited to protein complexes, protein topology, post translational modifications, etc., are essential to understand the disease biology. In this study, we tried to investigate the aspect of protein complex using several COVID-19 datasets. We performed machine learning based on three previously published datasets [3, 4, 10]. The first was the training dataset, which was the MS data matrix generated using 20 min SWATH [10], containing 331 proteins, and these samples were obtained prior to the onset of disease severity. The second was the test dataset 1, which was the MS data matrix using 35 min TMTpro 16plex DDA proteomics [3], containing 894 proteins. The third was the test dataset 2, a MS data matrix analyzed by 5 min fast flow SWATH [4], containing 229 proteins. Via this way, we evaluated the feasibility to utilize protein context information, including protein complexes, stoichiometric ratios in a complex, protein pathways and degree of protein networks, besides proteins, as key features to classify severe COVID-19 cases. The results showed that protein context could be exploited as integrative biomarkers for the stratification of COVID-19.
Methods
Patients and samples
The training set for this study consisted of 54 sera samples from 40 Chinese patients with COVID-19 (25 non-severe and 15 severe, according to the Chinese Government Diagnosis and Treatment Guideline 5th version), which were quantified using SWATH MS based proteomics [10]. Its performance was subsequently evaluated in two test datasets. One was a TMTpro 16plex dataset from our previous publication [3], containing 21 samples from 21 Chinese patients (6 non-severe and 15 severe). The other was the SWATH data set of 102 sera samples from 31 German patients with COVID-19 [4].
Serum sample collection, peptide preparation, and MS data acquisition
Regarding to the training set, the procedures for serum sample collection, peptides preparation and SWATH acquisition for the training set have been described in our previous study [10]. Briefly, these samples were collected from 40 patients with COVID-19 in stage 1, namely, the nucleic acid positive stage in the first 48 h after admission [10]. Most patients had only one blood test, while some of them had two blood tests as recorded in the medical history. In total, there were 54 sera samples collected from 40 patients.
Regarding to the test sets, the first test set used 35 min TMTpro 16plex proteomics. For each patient, the serum sample was obtained within 48 h after hospital admission [3]. The second test set used 5 min SWATH MS proteomics, and the samples were obtained from early hospitalized patients (nearly 1–2 days after hospital admission) [4]. The details of serum sample collection, peptide preparation and MS data acquisition in the two test sets have been described in the previously published studies [3, 4], respectively.
Three proteomic data sets
The training set consisted of 54 sera samples from 40 Chinese COVID-19 patients, and resulted in a MS data matrix generated using 20 min SWATH, containing 331 proteins and 3474 peptides [10]. The test set 1 included 21 sera samples from 21 Chinese COVID-19 patients [3], and 894 proteins and 7747 peptides were identified from the MS data matrix using 35 min TMTpro 16plex DDA proteomics. The test set 2 contained 102 sera samples from 31 early hospitalized German patients, analyzed by 5 min fast flow SWATH [4], and the resultant data matrix included 229 proteins and 3000 peptides.
The generation of protein complex, pathways, and stoichiometric ratios
The proteins in the training set of this study were from the dataset of previously published literature [10]. Subsequent features, including complexes, pathways, stoichiometric ratios and network degree, were all generated based on these proteins. Complexes were obtained from CORUM [18] and BioPlex Explorer 3.0 [19] using all proteins in the training cohort. Pathways were acquired by G:profiler (version e99_eg46_p14_f929183, database updated on 07/02/2020) from all proteins in the training cohort. The expression values of complexes and pathways were the sum of the Z-scores of proteins in complexes and pathways. The determination of Z-score was performed using the scale function of R package. In addition, stoichiometric ratios are the ratio of any two proteins in a complex. The values of stoichiometric ratios in a complex are the ratio of the two proteins treated with Z-score.
The generation of network degree
The network degrees, which shows the degree to which one protein is related to another, were also generated based on proteins from dataset published before [10]. The value of a protein’s network degree is the sum of all its edge values. The edges mean the protein–protein expression associations, and the calculation of protein–protein association is based on a reference that calculates gene–gene association [20]. Briefly, the protein–protein association was determined by statistical independence of two proteins. The threshold for significant level was set as 0.01. If the normalized statistic of an equation was greater than the significant level, null hypothesis that proteins x and y are independent to each other were rejected, and the edge for x and y was equal to 1, otherwise it was equal to 0.
The screening of differential features
The differential expression of proteins and other four features between severe cases and non-severe cases was determined using R package Limma (version 3.44) by fitting a linear model. Features with a p value < 0.05 were considered as differential features. It should be noted that although the adjustment of p values based on multiple hypothesis correction can avoid type-1 errors, the risk of introducing type-2 errors is also increased, eliminating some potentially differential proteins. Especially, when the sample size and number of proteins are not too large, there were few differential proteins left after p value adjustment, which was not enough for subsequent random forest machine learning model. Therefore, based on actual situation, multiple hypothesis correction was not performed in this study.
The input variables and process of random forest machine learning
We used five categories of variables as the input for the machine learning model, including proteins, complexes, pathways, stoichiometric ratios in a complex, and network degrees. The values of all differential features were normalized by Z-score for machine learning. The output predictor of the machine learning model was the non-severe or severe disease type, and we built a random forest machine learning model for the binary classification task. The random forest model was based on an R package random forest (version 4.6-14) with 5000 trees and 5 nodes as the minimum size of terminal nodes, while the type of prediction was chosen to be probabilistic. The best features were selected by 100 times random forest machine learning. Ten-fold cross validation was performed for each training process of the machine learning model. Receiver Operator Curve (ROC) was estimated by predicting results of the cross-validation using R package pROC (version 1.15.3).
Data visualization
PCAs were plotted using R package PCA. Heatmaps were plotted by R package heatmap (version 1.0.12). Density plots were performed by Kernel Density Estimation, a base function of R (version 4.0.0) (a Gaussian kernel with default bandwidth was used).
Results
Three sera proteomics data sets for modeling and testing
Three independently obtained proteomic data sets of sera from COVID-19 patients were utilized in this study (Fig. 1). The training data set was a matrix containing the relative expression of 331 proteins in 54 sera samples from 40 patients (25 non-severe and 15 severe) with 21.7% missing values. The mean age of the patients was 51.1 years and the mean body mass index (BMI) was 23.9. Severe patients exhibited a higher BMI (p < 0.01) and a higher incidence of hypertension and diabetes than the non-severe cases (Table 1 and Additional file 2: Table S1). In addition, two test datasets were included. The first is the TMTpro 16plex data set from our previous publication [3], containing a relative expression of 894 proteins in 21 sera samples from 21 patients (6 non-severe and 15 severe). The other is a 5-min gradient SWATH data containing the relative expression of 229 proteins in 102 sera samples from 31 German patients with 12.2% missing values [4]. The details of the protein matrix are summarized in Table 2.

Study overview. In general, 331 proteins were identified from 54 serum samples of COVID-19 patients. Subsequently, five kinds of 868 features were derived from these proteins. The top 25 differential features were selected for the machine learning model, which was further validated in two test datasets
Extraction of protein context features
We established a few protein context features including protein complexes, protein stoichiometric ratios in a protein complex, pathways, proteins and network degrees. A total of 868 features were obtained based on the quantification of 331 proteins (Additional file 3: Table S2). For protein complexes, two databases, namely BioPlex and CORUM, were utilized to retrieve the complex entities based on the 331 proteins. This led to identification of 27 potentially functional protein complexes from BioPlex, a database of human protein–protein interactions based on affinity purification mass spectrometry (AP-MS) [19]. In addition, 16 protein complexes were identified from the CORUM database, a manually curated and experimentally characterized protein complexes repository [18]. Therefore, BioPlex and CORUM together led to 43 protein complexes (Additional file 4: Table S3). For each protein complex, we computed the ratio of each protein pair, leading to 105 protein ratios (Additional file 4: Table S3). Subsequently, 58 pathway features were enriched by 71 differentially expressed proteins (Limma, adjust p < 0.05) between severe and non-severe patients by G:profiler [21]. Thus, we compiled a feature list containing 43 complexes, 71 differentially expressed proteins and 58 enriched pathways (Additional file 4: Table S3), which were utilized as input features for machine learning to stratify COVID-19 patients. Additionally, protein network degrees, which reveal the co-expression relationships with other proteins [16], were also applied as one type of feature. A total of 331 protein degrees were obtained as features (Additional file 3: Table S2). Finally, we focused on differential features between severe and non-severe patients using limma (p value < 0.05). A total of 192 differential features were obtained, including 7 complexes, 16 protein stoichiometry ratios, 27 pathways, 71 proteins, and 71 protein network degrees (Additional file 5: Table S4).
Classification of severe patients using machine learning
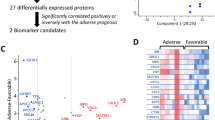
As shown in Fig. 2A, all the 192 identified features by Limma were ranked by log-scaled p values. To further identify biomarkers for the classification of severe cases, a random forest machine learning model based on the above-mentioned features was applied to the training cohort. The fit of the model was evaluated by the area under the curve (AUC) (Fig. 2A). The best classifier contained the top twenty-five features, including two complexes, one stoichiometric ratio, five pathways, twelve proteins, and five network degrees (Fig. 2B, Additional file 1: Fig. S1). The changes of the top 25 features in sera of severe cases (Chinese SWATH cohort) are visualized in a heatmap (Fig. 2C).

The selected features for classifying COVID-19. A All Identified features ranked by log p value; B The top 25 features identified; C The heatmap of top 25 features in the training set
As shown in Fig. 3A, severe and non-severe cases in training set and two independent test sets were not well separated when all features were used. However, by using the top 25 features, the severe and non-severe cases in the training set and two test sets can be well distinguished. We next tested this model in an independent TMT-based proteomic data set containing 6 non-severe and 15 severe cases (Test set 1). The model achieved an AUC of 0.900 in this dataset (Fig. 3B). It should be noted that not all the top 25 features were used in test sets due to some missing features. Actually, 18 features, including two complexes, five pathways, nine proteins, and two network degrees, were identified in the test data set 1. Next, this classifier was further evaluated using another independent SWATH data set from 12 non-severe and 19 severe German patients (Test set 2) [4]. In total, 16 features, including two complexes, five pathways, eight proteins, and one network degree were identified in the test data set 2, leading to an AUC of 0.908 (Fig. 3B). The AUC results reveal that, besides proteins, protein complexes, stoichiometric ratios, pathways and protein degrees could be potential biomarkers for stratification of COVID-19 patients.

The performance of the machine learning model. A The PCA map of the training set and test sets using all features; B The comparison of AUC between the model with five types of features and the model with only one type of feature in the training set, test set 1, and the test set 2
To validate whether the selected features were optimal for our classifier, we built 200 models with random features and validated them with the TMT data set and the SWATH-based German cohort dataset. The median AUC of these models is 0.756, which is significantly lower than the AUC of the model with top 25 features (0.900), indicating the superiority of our selected features.
Modeling with only one type of feature
To explore whether the machine learning models with five different types of features are superior to the models with only one type of feature, we trained models with only one type of feature and tested them with the TMT-based proteomics data set (test set 1) and the SWATH-based German cohort data set (test set 2). As shown in Fig. 3B, for the training set and test set 2, the AUC values for the model with all five types of features were 0.965 and 0.908, respectively, which were better than the AUC of the model only with proteins as the feature (0.949 and 0.883, respectively). For test set 1, the model with all types of features reached an AUC of 0.900, which was slightly lower than that of the model only with proteins. This may be because that test set 1 is based on the TMT-tagging data acquisition mode, which is different from the SWATH data of the training data set and test set 2. In addition, the difference in sample size may have also contributed.
The AUC for the model with only the protein network degree as features reached 0.932, 0.667, and 0.467 in the training set and the two test sets, respectively, indicating that it performs well in the training set, but not in the two test sets (Fig. 3B). Similar observation was found in the model with only the protein complex ratio as a feature (Fig. 3B). In addition, in the training set and two test sets, the AUC values of the model with five types of features were all better than those of the models with only complex or pathway. These findings together consolidate the benefit of integrating multiple model features for COVID-19 patient stratification.
Discussion
The highlight of this study is that we integrated five types of features including protein complexes, protein stoichiometric ratios, pathways, network degrees, and proteins, rather than using purely individual proteins, to build machine learning models for disease classification. Twenty-five predictive markers were identified to stratify COVID-19. Our work demonstrates that integrating protein expression levels with protein context improves COVID-19 patient stratification.
The 25 features highlighted by our analysis are all associated with the pathogenesis of COVID-19. As shown in Fig. 4, after the SARS-CoV-2 enters the alveolar, the macrophages subsequently phagocytose the virus and release cytokines, resulting in the release of acute phase proteins (APPs) from the liver [22]. These APPs stimulate the complement system response [23]. However, in severe cases, the complement system reacts abnormally, which can potentially trigger a cytokine storm [24, 25]. On one hand, cytokine storm leads to multi-organ damages, such as damages to the liver and testis [26]. On the other hand, more macrophages are recruited from the peripheral blood to the lungs, causing alveolar macrophage infiltration, lung damage, and respiratory failure [27].

The biological interpretation of the top 25 features. MAC, membrane attack complex. Red border, upregulation; green boarder, downregulation
Several studies have reported predictive blood markers for severe cases, such as ITIH4 [28, 29], M-CSF, CCL3 and CCL4 [30], as well as CMAs [31]. Studies utilizing MS-based proteomics also have found that proteins associated with complement system, acute phase protein response, inflammation system, macrophage dysregulation, antibody response, and coagulation system are altered in severe COVID-19 cases [3, 4, 7, 9, 32], which have also been confirmed by other proteomic approaches [33,34,35]. In this study, we found a complex, two pathways, seven proteins and one network degree are involved in the complement system, acute phase proteins and inflammation, including “SAA1, SAA2, YLPM1”, “complement activation”, “acute-phase response”, IGHG3, SAA1, SAA2, IGLV1-47, C9, ITIH4, C4BPA and IGHV3-73 (Additional file 1: Fig. S1). In addition, one pathway (phagocytosis, engulfment) associated with macrophage dysregulation was identified as a key feature. Our data uncovered previously hidden COVID-19-associated proteome context information.
Our study also identified other molecular features in severe patients. Several transport proteins were upregulated. Vitamin D-binding protein (GC) enhances the activity of C5a in the complement system [36], which may induce cytokine storms. MyRIP, another transport protein, participates in melanosomes and produces pigmented melanin to skins [37]. The upregulation of MyRIP may be related to skin hyperpigmentation in severe patients [38]. Transthyretin (TTR) is a marker for inflammation and a negative acute-phase reactant. Reduced TTR has been reported to be associated with acute-phase response induced by inflammation, and TTR is also a malnutrition marker, suggesting nutritional disorders in severe cases [39]. TTR-RBP complex consists of TTR and retinol-binding protein 4 (RBP4), and the upregulation of TTR-RBP complex suggests an improved inflammation state [40]. In this study, both TTR and TTR-RBP complexes decreased in the sera of severe cases, suggesting a more intense acute response and inflammatory state.
Notably, some proteins associated with the complement system were also altered in severe cases. Abnormal response of complement system can trigger cytokine storm, which can further develop into severe cases [24, 41]. Carvelli et al. found that C5 was the main effector of abnormal complement system, and blockade of C5 could prevent excessive lung inflammation [42]. Complement protein C3 was also associated with fatal outcome of COVID-19 [25]. Different from previous studies, we found that C9, another protein in the complement system, was elevated in severe cases, suggesting that it may also be a marker or potential therapeutic target. In addition, GC, which activates the activity of C5 [36], was also upregulated. C4BPA associated with C4 activity was abnormally expressed [43] (Fig. 4). In addition to changes in proteins associated with complement system, RPIA was downregulated in severe cases, which may indicate an impaired glucose metabolism and liver damage. The Tudor domain-containing protein 1 (TDRD1), which plays a central role in spermatogenesis [44], was also downregulated, which may contribute to impaired testis functions observed in severe cases [26].
In addition to proteins, other types of protein context feature further shed light on the mechanism of severe COVID-19 cases. Cytolysis pathway is induced after viral infection and serves as a clearance mechanism for infected cells [45]. The alteration of the phosphatidylcholine binding pathway may contribute to the inflammatory process [46]. The increased ratio of SAA2/YLPM1 in the "SAA2, SAA1, YLPM1" complex in severe cases may be due to upregulation of SAA2 (sera amyloid A-2 protein) and downregulation of YLPM1 (YLP motif-containing protein 1, Additional file 1: Fig. S1), revealing an acute-phase response and an enhanced repair of inflammation-induced telomere shortening [47]. The network degree changes of some proteins were associated with cytokine storm. Immunoglobulin heavy variable 3-73 (IGHV3-73) participates in antigen recognition [48]. MTTP stimulates phosphatidylcholine transport [49]. Alpha-2-macroglobulin (A2M) influences cytokines signaling [50], and SIRT7 suppresses inflammation [51]. Since network degree suggests the co-expression associations with other proteins, the network degree changes of these proteins also uncorvered systematic molecular changes in severe cases. Our study showed that the predictive result of the model with five different features was better than that of the model with one single feature (Fig. 3B), suggesting the benefits of integrating multiple protein context in disease prediction and stratification.
Some limitations of this study should be noted. There were missing features in the two test sets. Seven features were not included in the TMT data, and nine features were excluded in the German cohort data. Median value of all the valued features were used to impute these missing features. The sample size of the training set is limited. Nevertheless, the model achieved satisfactory AUCs in these independent tests. Neither these limitations compromise the major conclusion of this study that integrating protein context information improves COVID-19 severity classification. Moreover, the protein complex information was obtained from cellular complexes, meaning that not all the complexes are necessarily formed in the serum, which needs to be verified by future research. Finally, building ratios may create an overfitting danger, but this can be avoided by building models with other types of features together.
Conclusion
Protein complexes, stoichiometric ratios, pathways and network degrees could be used as biomarkers to identify severe cases. Our present study confirms some of the previously reported molecular changes and identifies some new features that may contribute to understand the pathogenesis of COVID-19.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- APPs:
-
Acute phase proteins
- GC:
-
Vitamin D-binding protein
- TTR:
-
Transthyretin
- RBP4:
-
Retinol-binding protein 4
- TDRD1:
-
Tudor domain-containing protein 1
- SAA2:
-
Sera amyloid A-2 protein
- A2M:
-
Alpha-2-2 macroglobulin
- IGHV3-73:
-
Immunoglobulin heavy variable 3-73
References
Hui DS, Esam IA, Madani TA, Ntoumi F, Kock R, Dar O, Ippolito G, McHugh TD, Memish ZA, Drosten C, Zumla A, Petersen E. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—the latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis. 2020;91:264–6.
Gao YD, Ding M, Dong X, Zhang JJ, Kursat Azkur A, Azkur D, Gan H, Sun YL, Fu W, Li W, Liang HL, Cao YY, Yan Q, Cao C, Gao HY, Bruggen MC, van de Veen W, Sokolowska M, Akdis M, Akdis CA. Risk factors for severe and critically ill COVID-19 patients: a review. Allergy. 2021;76(2):428–55.
Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, Quan S, Zhang F, Sun R, Qian L, Ge W, Liu W, Liang S, Chen H, Zhang Y, Li J, Xu J, He Z, Chen B, Wang J, Yan H, Zheng Y, Wang D, Zhu J, Kong Z, Kang Z, Liang X, Ding X, Ruan G, Xiang N, Cai X, Gao H, Li L, Li S, Xiao Q, Lu T, Zhu Y, Liu H, Chen H, Guo T. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell. 2020. https://doi.org/10.1016/j.cell.2020.05.032.
Messner CB, Demichev V, Wendisch D, Michalick L, White M, Freiwald A, Textoris-Taube K, Vernardis SI, Egger AS, Kreidl M, Ludwig D, Kilian C, Agostini F, Zelezniak A, Thibeault C, Pfeiffer M, Hippenstiel S, Hocke A, von Kalle C, Campbell A, Hayward C, Porteous DJ, Marioni RE, Langenberg C, Lilley KS, Kuebler WM, Mulleder M, Drosten C, Suttorp N, Witzenrath M, Kurth F, Sander LE, Ralser M. Ultra-high-throughput clinical proteomics reveals classifiers of COVID-19 infection. Cell Syst. 2020;11(1):11-24 e4.
Gutmann C, Takov K, Burnap SA, Singh B, Ali H, Theofilatos K, Reed E, Hasman M, Nabeebaccus A, Fish M, McPhail MJ, O’Gallagher K, Schmidt LE, Cassel C, Rienks M, Yin X, Auzinger G, Napoli S, Mujib SF, Trovato F, Sanderson B, Merrick B, Niazi U, Saqi M, Dimitrakopoulou K, Fernandez-Leiro R, Braun S, Kronstein-Wiedemann R, Doores KJ, Edgeworth JD, Shah AM, Bornstein SR, Tonn T, Hayday AC, Giacca M, Shankar-Hari M, Mayr M. SARS-CoV-2 RNAemia and proteomic trajectories inform prognostication in COVID-19 patients admitted to intensive care. Nat Commun. 2021;12(1):3406.
Laudanski K, Jihane H, Antalosky B, Ghani D, Phan U, Hernandez R, Okeke T, Wu J, Rader D, Susztak K. Unbiased analysis of temporal changes in immune serum markers in acute COVID-19 infection with emphasis on organ failure, anti-viral treatment, and demographic characteristics. Front Immunol. 2021;12: 650465.
Demichev V, Tober-Lau P, Lemke O, Nazarenko T, Thibeault C, Whitwell H, Rohl A, Freiwald A, Szyrwiel L, Ludwig D, Correia-Melo C, Aulakh SK, Helbig ET, Stubbemann P, Lippert LJ, Gruning NM, Blyuss O, Vernardis S, White M, Messner CB, Joannidis M, Sonnweber T, Klein SJ, Pizzini A, Wohlfarter Y, Sahanic S, Hilbe R, Schaefer B, Wagner S, Mittermaier M, Machleidt F, Garcia C, Ruwwe-Glosenkamp C, Lingscheid T, de Jarcy LB, Stegemann MS, Pfeiffer M, Jurgens L, Denker S, Zickler D, Enghard P, Zelezniak A, Campbell A, Hayward C, Porteous DJ, Marioni RE, Uhrig A, Muller-Redetzky H, Zoller H, Loffler-Ragg J, Keller MA, Tancevski I, Timms JF, Zaikin A, Hippenstiel S, Ramharter M, Witzenrath M, Suttorp N, Lilley K, Mulleder M, Sander LE, PA-COVID-19 Study group, Ralser M, Kurth F. A time-resolved proteomic and prognostic map of COVID-19. Cell Syst. 2021;12(8):780-794 e7.
Demichev V, Tober-Lau P, Nazarenko T, Aulakh SK, Whitwell H, Lemke O, Röhl A, Freiwald A, Mittermaier M, Szyrwiel L, Ludwig D, Correia-Melo C, Lippert LJ, Helbig ET, Stubbemann P, Olk N, Thibeault C, Grüning N-M, Blyuss O, Vernardis S, White M, Messner CB, Joannidis M, Sonnweber T, Klein SJ, Pizzini A, Wohlfarter Y, Sahanic S, Hilbe R, Schaefer B, Wagner S, Machleidt F, Garcia C, Ruwwe-Glösenkamp C, Lingscheid T, de Jarcy LB, Stegemann MS, Pfeiffer M, Jürgens L, Denker S, Zickler D, Spies C, Edel A, Müller NB, Enghard P, Zelezniak A, Bellmann-Weiler R, Weiss G, Campbell A, Hayward C, Porteous DJ, Marioni RE, Uhrig A, Zoller H, Löffler-Ragg J, Keller MA, Tancevski I, Timms JF, Zaikin A, Hippenstiel S, Ramharter M, Müller-Redetzky H, Witzenrath M, Suttorp N, Lilley K, Mülleder M, Sander LE, PA-COVID-19 Study group, Kurth F, Ralser M. A proteomic survival predictor for COVID-19 patients in intensive care. PLOS Digit Health. 2022. https://doi.org/10.1371/journal.pdig.0000007.
Mao K, Tan Q, Ma Y, Wang S, Zhong H, Liao Y, Huang Q, Xiao W, Xia H, Tan X, Luo P, Xu J, Long D, Jin Y. Proteomics of extracellular vesicles in plasma reveals the characteristics and residual traces of COVID-19 patients without underlying diseases after 3 months of recovery. Cell Death Dis. 2021;12(6):541.
Zhang Y, Cai X, Ge W, Wang D, Zhu G, Qian L, Xiang N, Yue L, Liang S, Zhang F, Wang J, Zhou K, Zheng Y, Lin M, Sun T, Lu R, Zhang C, Xu L, Sun Y, Zhou X, Yu J, Lyu M, Shen B, Zhu H, Xu J, Zhu Y, Guo T. Potential use of serum proteomics for monitoring COVID-19 progression to complement RT-PCR detection. J Proteome Res. 2022;21(1):90–100.
Ori A, Iskar M, Buczak K, Kastritis P, Parca L, Andres-Pons A, Singer S, Bork P, Beck M. Spatiotemporal variation of mammalian protein complex stoichiometries. Genome Biol. 2016;17:47.
Goh WWB, Wong L. NetProt: complex-based feature selection. J Proteome Res. 2017;16(8):3102–12.
Goh WW. Fuzzy-FishNET: a highly reproducible protein complex-based approach for feature selection in comparative proteomics. BMC Med Genomics. 2016;9(Suppl 3):67.
Fossati A, Li C, Uliana F, Wendt F, Frommelt F, Sykacek P, Heusel M, Hallal M, Bludau I, Capraz T, Xue P, Song J, Wollscheid B, Purcell AW, Gstaiger M, Aebersold R. PCprophet: a framework for protein complex prediction and differential analysis using proteomic data. Nat Methods. 2021. https://doi.org/10.1038/s41592-021-01107-5.
Leake MC, Chandler JH, Wadhams GH, Bai F, Berry RM, Armitage JP. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature. 2006;443(7109):355–8.
Wang J, Peng X, Peng W, Wu FX. Dynamic protein interaction network construction and applications. Proteomics. 2014;14(4–5):338–52.
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL, Tummino TA, Huttenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Meyer B, Roesch F, Vallet T, Mac Kain A, Miorin L, Moreno E, Naing ZZC, Zhou Y, Peng S, Shi Y, Zhang Z, Shen W, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, Lyu J, Mathy CJP, Perica T, Pilla KB, Ganesan SJ, Saltzberg DJ, Rakesh R, Liu X, Rosenthal SB, Calviello L, Venkataramanan S, Liboy-Lugo J, Lin Y, Huang XP, Liu Y, Wankowicz SA, Bohn M, Safari M, Ugur FS, Koh C, Savar NS, Tran QD, Shengjuler D, Fletcher SJ, O’Neal MC, Cai Y, Chang JCJ, Broadhurst DJ, Klippsten S, Sharp PP, Wenzell NA, Kuzuoglu-Ozturk D, Wang HY, Trenker R, Young JM, Cavero DA, Hiatt J, Roth TL, Rathore U, Subramanian A, Noack J, Hubert M, Stroud RM, Frankel AD, Rosenberg OS, Verba KA, Agard DA, Ott M, Emerman M, Jura N, von Zastrow M, Verdin E, Ashworth A, Schwartz O, d’Enfert C, Mukherjee S, Jacobson M, Malik HS, Fujimori DG, Ideker T, Craik CS, Floor SN, Fraser JS, Gross JD, Sali A, Roth BL, Ruggero D, Taunton J, Kortemme T, Beltrao P, Vignuzzi M, Garcia-Sastre A, Shokat KM, Shoichet BK, Krogan NJ. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459–68.
Ruepp A, Brauner B, Dunger-Kaltenbach I, Frishman G, Montrone C, Stransky M, Waegele B, Schmidt T, Doudieu ON, Stumpflen V, Mewes HW. CORUM: the comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 2008;36(Database issue):D646-50.
Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, Vaites LP, Ordureau A, Rad R, Erickson BK, Wuhr M, Chick J, Zhai B, Kolippakkam D, Mintseris J, Obar RA, Harris T, Artavanis-Tsakonas S, Sowa ME, De Camilli P, Paulo JA, Harper JW, Gygi SP. The BioPlex network: a systematic exploration of the human interactome. Cell. 2015;162(2):425–40.
Dai H, Li L, Zeng T, Chen L. Cell-specific network constructed by single-cell RNA sequencing data. Nucleic Acids Res. 2019;47(11): e62.
Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019;47(W1):W191–8.
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, HLH Across Speciality Collaboration, UK. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–4.
Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340(6):448–54.
Polycarpou A, Howard M, Farrar CA, Greenlaw R, Fanelli G, Wallis R, Klavinskis LS, Sacks S. Rationale for targeting complement in COVID-19. EMBO Mol Med. 2020;12(8): e12642.
Georg P, Astaburuaga-Garcia R, Bonaguro L, Brumhard S, Michalick L, Lippert LJ, Kostevc T, Gabel C, Schneider M, Streitz M, Demichev V, Gemund I, Barone M, Tober-Lau P, Helbig ET, Hillus D, Petrov L, Stein J, Dey HP, Paclik D, Iwert C, Mulleder M, Aulakh SK, Djudjaj S, Bulow RD, Mei HE, Schulz AR, Thiel A, Hippenstiel S, Saliba AE, Eils R, Lehmann I, Mall MA, Stricker S, Rohmel J, Corman VM, Beule D, Wyler E, Landthaler M, Obermayer B, von Stillfried S, Boor P, Demir M, Wesselmann H, Suttorp N, Uhrig A, Muller-Redetzky H, Nattermann J, Kuebler WM, Meisel C, Ralser M, Schultze JL, Aschenbrenner AC, Thibeault C, Kurth F, Sander LE, Bluthgen N, Sawitzki B, PA-COVID-19 Study Group. Complement activation induces excessive T cell cytotoxicity in severe COVID-19. Cell. 2021. https://doi.org/10.1016/j.cell.2021.12.040.
Nie X, Qian L, Sun R, Huang B, Dong X, Xiao Q, Zhang Q, Lu T, Yue L, Chen S, Li X, Sun Y, Li L, Xu L, Li Y, Yang M, Xue Z, Liang S, Ding X, Yuan C, Peng L, Liu W, Yi X, Lyu M, Xiao G, Xu X, Ge W, He J, Fan J, Wu J, Luo M, Chang X, Pan H, Cai X, Zhou J, Yu J, Gao H, Xie M, Wang S, Ruan G, Chen H, Su H, Mei H, Luo D, Zhao D, Xu F, Li Y, Zhu Y, Xia J, Hu Y, Guo T. Multi-organ proteomic landscape of COVID-19 autopsies. Cell. 2021;184(3):775-791 e14.
Wang C, Xie J, Zhao L, Fei X, Zhang H, Tan Y, Nie X, Zhou L, Liu Z, Ren Y, Yuan L, Zhang Y, Zhang J, Liang L, Chen X, Liu X, Wang P, Han X, Weng X, Chen Y, Yu T, Zhang X, Cai J, Chen R, Shi ZL, Bian XW. Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. EBioMedicine. 2020;57: 102833.
Vollmy F, van den Toorn H, Chiozzi RZ, Zucchetti O, Papi A, Volta CA, Marracino L, Vieceli Dalla Sega F, Fortini F, Demichev V, Tober-Lau P, Campo G, Contoli M, Ralser M, Kurth F, Spadaro S, Rizzo P, Heck AJ. A serum proteome signature to predict mortality in severe COVID-19 patients. Life Sci Alliance. 2021. https://doi.org/10.26508/lsa.202101099.
Geyer PE, Arend FM, Doll S, Louiset ML, Virreira Winter S, Muller-Reif JB, Torun FM, Weigand M, Eichhorn P, Bruegel M, Strauss MT, Holdt LM, Mann M, Teupser D. High-resolution serum proteome trajectories in COVID-19 reveal patient-specific seroconversion. EMBO Mol Med. 2021;13(8): e14167.
Chevrier S, Zurbuchen Y, Cervia C, Adamo S, Raeber ME, de Souza N, Sivapatham S, Jacobs A, Bachli E, Rudiger A, Stussi-Helbling M, Huber LC, Schaer DJ, Nilsson J, Boyman O, Bodenmiller B. A distinct innate immune signature marks progression from mild to severe COVID-19. Cell Rep Med. 2021;2(1): 100166.
Altay O, Arif M, Li X, Yang H, Aydin M, Alkurt G, Kim W, Akyol D, Zhang C, Dinler-Doganay G, Turkez H, Shoaie S, Nielsen J, Boren J, Olmuscelik O, Doganay L, Uhlen M, Mardinoglu A. Combined metabolic activators accelerates recovery in mild-to-moderate COVID-19. Adv Sci. 2021;8(17): e2101222.
D’Alessandro A, Thomas T, Dzieciatkowska M, Hill RC, Francis RO, Hudson KE, Zimring JC, Hod EA, Spitalnik SL, Hansen KC. Serum proteomics in COVID-19 patients: altered coagulation and complement status as a function of IL-6 level. J Proteome Res. 2020;19(11):4417–27.
Zhong W, Altay O, Arif M, Edfors F, Doganay L, Mardinoglu A, Uhlen M, Fagerberg L. Next generation plasma proteome profiling of COVID-19 patients with mild to moderate symptoms. EBioMedicine. 2021;74: 103723.
Su Y, Chen D, Yuan D, Lausted C, Choi J, Dai CL, Voillet V, Duvvuri VR, Scherler K, Troisch P, Baloni P, Qin G, Smith B, Kornilov SA, Rostomily C, Xu A, Li J, Dong S, Rothchild A, Zhou J, Murray K, Edmark R, Hong S, Heath JE, Earls J, Zhang R, Xie J, Li S, Roper R, Jones L, Zhou Y, Rowen L, Liu R, Mackay S, O’Mahony DS, Dale CR, Wallick JA, Algren HA, Zager MA, Unit IS-SCB, Wei W, Price ND, Huang S, Subramanian N, Wang K, Magis AT, Hadlock JJ, Hood L, Aderem A, Bluestone JA, Lanier LL, Greenberg PD, Gottardo R, Davis MM, Goldman JD, Heath JR. Multi-omics resolves a sharp disease-state shift between mild and moderate COVID-19. Cell. 2020;183(6):1479-1495 e20.
Zhou S, Butler-Laporte G, Nakanishi T, Morrison DR, Afilalo J, Afilalo M, Laurent L, Pietzner M, Kerrison N, Zhao K, Brunet-Ratnasingham E, Henry D, Kimchi N, Afrasiabi Z, Rezk N, Bouab M, Petitjean L, Guzman C, Xue X, Tselios C, Vulesevic B, Adeleye O, Abdullah T, Almamlouk N, Chen Y, Chasse M, Durand M, Paterson C, Normark J, Frithiof R, Lipcsey M, Hultstrom M, Greenwood CMT, Zeberg H, Langenberg C, Thysell E, Pollak M, Mooser V, Forgetta V, Kaufmann DE, Richards JB. A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID-19 susceptibility and severity. Nat Med. 2021;27(4):659–67.
Shah AB, DiMartino SJ, Trujillo G, Kew RR. Selective inhibition of the C5a chemotactic cofactor function of the vitamin D binding protein by 1,25(OH)2 vitamin D3. Mol Immunol. 2006;43(8):1109–15.
El-Amraoui A, Schonn JS, Kussel-Andermann P, Blanchard S, Desnos C, Henry JP, Wolfrum U, Darchen F, Petit C. MyRIP, a novel Rab effector, enables myosin VIIa recruitment to retinal melanosomes. EMBO Rep. 2002;3(5):463–70.
Lu C, Hou N. Skin hyperpigmentation in coronavirus disease 2019 patients: is polymyxin B the culprit? Front Pharmacol. 2020;11:01304.
Myron Johnson A, Merlini G, Sheldon J, Ichihara K, Scientific Division Committee on Plasma Proteins (C-PP), International Federation of Clinical Chemistry and Laboratory Medicine (IFCC). Clinical indications for plasma protein assays: transthyretin (prealbumin) in inflammation and malnutrition. Clin Chem Lab Med. 2007;45(3):419–26.
Rubin LP, Ross AC, Stephensen CB, Bohn T, Tanumihardjo SA. Metabolic effects of inflammation on vitamin A and carotenoids in humans and animal models. Adv Nutr. 2017;8(2):197–212.
Marcos-Jimenez A, Sanchez-Alonso S, Alcaraz-Serna A, Esparcia L, Lopez-Sanz C, Sampedro-Nunez M, Mateu-Albero T, Sanchez-Cerrillo I, Martinez-Fleta P, Gabrie L, Del Campo Guerola L, Rodriguez-Frade JM, Casasnovas JM, Reyburn HT, Vales-Gomez M, Lopez-Trascasa M, Martin-Gayo E, Calzada MJ, Castaneda S, de la Fuente H, Gonzalez-Alvaro I, Sanchez-Madrid F, Munoz-Calleja C, Alfranca A. Deregulated cellular circuits driving immunoglobulins and complement consumption associate with the severity of COVID-19 patients. Eur J Immunol. 2021;51(3):634–47.
Carvelli J, Demaria O, Vely F, Batista L, Chouaki Benmansour N, Fares J, Carpentier S, Thibult ML, Morel A, Remark R, Andre P, Represa A, Piperoglou C, Cordier PY, Le Dault E, Guervilly C, Simeone P, Gainnier M, Morel Y, Ebbo M, Schleinitz N, Vivier E, Explore COVID-19 IPH group, Explore COVID-19 Marseille Immunopole group. Association of COVID-19 inflammation with activation of the C5a-C5aR1 axis. Nature. 2020;588(7836):146–50.
Zhao P, Wu J, Lu F, Peng X, Liu C, Zhou N, Ying M. The imbalance in the complement system and its possible physiological mechanisms in patients with lung cancer. BMC Cancer. 2019;19(1):201.
Zhao J, Wang B, Yu H, Wang Y, Liu X, Zhang Q. tdrd1 is a germline-specific and sexually dimorphically expressed gene in Paralichthys olivaceus. Gene. 2018;673:61–9.
Bergmann CC, Lane TE, Stohlman SA. Coronavirus infection of the central nervous system: host-virus stand-off. Nat Rev Microbiol. 2006;4(2):121–32.
Ciavarella C, Motta I, Valente S, Pasquinelli G. Pharmacological (or synthetic) and nutritional agonists of PPAR-gamma as candidates for cytokine storm modulation in COVID-19 disease. Molecules. 2020. https://doi.org/10.3390/molecules25092076.
Taghizadeh S, Vazehan R, Beheshtian M, Sadeghinia F, Fattahi Z, Mohseni M, Arzhangi S, Nafissi S, Kariminejad A, Najmabadi H, Kahrizi K. Molecular diagnosis of hereditary neuropathies by whole exome sequencing and expanding the phenotype spectrum. Arch Iran Med. 2020;23(7):426–33.
Lefranc MP. Immunoglobulin and T cell receptor genes: IMGT((R)) and the birth and rise of immunoinformatics. Front Immunol. 2014;5:22.
Sirwi A, Hussain MM. Lipid transfer proteins in the assembly of apoB-containing lipoproteins. J Lipid Res. 2018;59(7):1094–102.
Cater JH, Wilson MR, Wyatt AR. Alpha-2-macroglobulin, a hypochlorite-regulated chaperone and immune system modulator. Oxid Med Cell Longev. 2019;2019:5410657.
Chen KL, Li L, Li CM, Wang YR, Yang FX, Kuang MQ, Wang GL. SIRT7 regulates lipopolysaccharide-induced inflammatory injury by suppressing the NF-kappaB signaling pathway. Oxid Med Cell Longev. 2019;2019:3187972.
Acknowledgements
We thank the Supercomputer Center of Westlake University for its assistance in data storage and computation.
Funding
This work is supported by Grants from the National Key R&D Program of China (No. 2020YFE0202200), the National Science Fund for Young Scholars (21904107), the National Natural Science Foundation of China (81972492), Zhejiang Provincial Natural Science Foundation for Distinguished Young Scholars (LR19C050001), Hangzhou Agriculture and Society Advancement Program (20190101A04), and Westlake Education Foundation, Tencent Foundation. Work in the MR lab is supported by the ERC (SyG-2020 951475) and the Wellcome Trust (IA 200829/Z/16/Z), and the specific work received funding from the Ministry of Education and Research (BMBF), as part of the National Research Node ‘Mass spectrometry in Systems Medicine (MSCoresys), under Grant agreements 031L0220, and Research (NaFoUniMedCOVID-19—NUM-NAPKON, FKZ: 01KX2021).
Author information
Authors and Affiliations
Contributions
TNG and YZ designed and supervised the project. JLG summarized the pathological mechanism of severe cases and wrote the manuscript. TNG and YZ guided the writing of manuscript. JLH and FFZ organized and analyzed data. SQZ designed the algorithm. QX provided suggestion for figures and manuscript. YZ, DLW, GJZ, JW and BS provided clinical guidance and sample. MR provided the test set 2 data. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethics committee of Wenzhou Medical University. All included participants provided written informed consent for the study.
Consent for publication
Not applicable.
Competing interests
TG and YZ are shareholders of Westlake Omics, Inc. The remaining authors declare no conflicts of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Expression of the 25 features in COVID-19 sera. (A) log2-scaled protein intensity; (B) Protein complex features indicated by z score and stoichiometric ratio of SAA2/YLPM1; (C) z scores of pathways; (D) z scores of network degree features.
Additional file 2: Table S1.
Information of 40 patients in the training set and all total patients.
Additional file 3: Table S2.
The total 868 features based on 331 proteins.
Additional file 4: Table S3.
The complexs, differential proteins and enriched pathways used as features.
Additional file 5: Table S4.
The total 192 differential features between severe and non-severe patients.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gao, J., He, J., Zhang, F. et al. Integration of protein context improves protein-based COVID-19 patient stratification. Clin Proteom 19, 31 (2022). https://doi.org/10.1186/s12014-022-09370-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12014-022-09370-0