
Abstract
Background
Twin-ribozyme introns represent a complex class of mobile group I introns that harbour a lariat capping (LC) ribozyme and a homing endonuclease gene embedded in a conventional self-splicing group I ribozyme (GIR2). Twin-ribozyme introns have so far been confined to nucleolar DNA in Naegleria amoeboflagellates and the myxomycete Didymium iridis.
Results
We characterize structural organization, catalytic properties and molecular evolution of a new twin-ribozyme intron in Allovahlkampfia (Heterolobosea). The intron contains two ribozyme domains with different functions in ribosomal RNA splicing and homing endonuclease mRNA maturation. We found Allovahlkampfia GIR2 to be a typical group IC1 splicing ribozyme responsible for addition of the exogenous guanosine cofactor (exoG), exon ligation and circularization of intron RNA. The Allovahlkampfia LC ribozyme, by contrast, represents an efficient self-cleaving ribozyme that generates a small 2′,5′ lariat cap at the 5′ end of the homing endonuclease mRNA, and thus contributes to intron mobility.
Conclusions
The discovery of a twin-ribozyme intron in a member of Heterolobosea expands the distribution pattern of LC ribozymes. We identify a putative regulatory RNA element (AP2.1) in the Allovahlkampfia LC ribozyme that involves homing endonuclease mRNA coding sequences as an important structural component.
Similar content being viewed by others

Background
Group I introns are commonly found as insertion elements in the nucleolar ribosomal DNA (rDNA) of eukaryotic microorganisms, where they interrupt highly conserved sites, both in small subunit (SSU) and large subunit (LSU) ribosomal RNA (rRNA) genes [1]. Group I introns are inherited vertically or horizontally, and are often referred to as selfish genetic elements. Most nucleolar group I introns are ribozymes (catalytic RNAs) that perform self-splicing as naked RNA in vitro by a common mechanism based on two coupled transesterification reactions [2, 3]. Splicing is initiated by nucleophilic attack at the 5′ splice site (SS) by the 3′OH group of an exogenous guanosine cofactor (exoG). This exoG becomes covalently bound at the 5′ end of the intron RNA, and subsequently, in a second reaction, the free 3′OH group of the 5′ exon attacks the phosphodiester bond at the 3′SS. This process results in the release of the intron RNA as a linear molecule and the accurate ligation of the flanking exons. Additional and alternative transesterification reactions occur during intron RNA processing and result in full-length or truncated circular forms of the intron [1, 4, 5].
A well-defined and highly conserved RNA core-structure is responsible for the catalysis. A group I ribozyme core is organized into three helical stacks, named the catalytic domain (P3 and P7, proximal P8 and P9), the substrate domain (P1 and proximal P2), and the scaffold domain (P4, P5 and P6) [1, 6]. About 5% to 10% of all known nucleolar group I introns harbour homing endonuclease gene (HEG) insertions. Homing endonuclease genes, which encode homing endonucleases (HEs) involved in intron mobility [7, 8], are located as large insertions within the peripheral parts of the helices P1, P2, P6, P8 or P9. Here, unconventional expression strategies are utilized for these protein-coding genes embedded in nucleolar rDNA [9].
The most complex nucleolar group I introns known are the twin-ribozyme introns [3, 10]. These introns consist of a regular group I splicing ribozyme (GIR2) with an insertion in P2 or P6 that contains a HEG as well as a lariat capping ribozyme (LC ribozyme). Whereas GIR2 is responsible for intron forward and reverse splicing and intron RNA circularization [5, 8, 11–13], the LC ribozyme performs the 2′,5′ branching reaction at an internal intron site and is directly involved in 5′ capping of the HE mRNA, a prerequisite for intron mobility [14–17].
Two natural variants of twin-ribozyme introns have been reported: the myxomycete Didymium iridis intron Dir.S956-1 [11, 18], and introns (Nae.S516) in various species and isolates of the Naegleria amoeboflagellate [12, 19]. Whereas both intron variants self-splice as naked RNA, express functional HEs and have a similar overall structural composition, a number of differences in distribution, inheritance and structural organization have been reported [10]. The Didymium LC ribozyme (DirLC; formerly DiGIR1) has been investigated in more detail and recent reports include studies of the catalytic reaction, RNA conformational changes, RNA:RNA interactions, 3D models and high-resolution X-ray crystal structures [10, 16, 20–22]. Similarly, structural and functional properties of Naegleria LC ribozymes (NaeLC; formerly NaGIR1) have been studied [12, 15, 17, 19, 23]. Importantly, both DirLC and NaeLC perform the branching reaction but contain different sequence motifs and structures in the flanking regulatory domains [17].
Here we report an expansion of the distribution pattern of twin-ribozyme introns. Allovahlkampfia sp., a species that belongs to the Heterolobosea amoeba, contains a twin-ribozyme intron (Asp.S516). Asp.S516 resembles the Naegleria introns in sequence, organization and insertion site. We have characterized the catalytic properties of the Asp.S516 RNA transcript in vitro. Allovahlkampfia GIR2 performs both self-splicing and complex intron RNA circle formations. We also show that the Allovahlkampfia LC ribozyme (AspLC) is an efficient branching ribozyme that generates a small 2′,5′ lariat cap at the 5′ end of the HE mRNA. Finally, by comparative sequence analysis we find supporting evidence that a putative regulatory RNA element known from the DirLC crystal structure is conserved in AspLC, but in a surprising context.
Results
Allovahlkampfia contains an optional group I intron at position S516 in the SSU rRNA gene
A BLAST search in the National Center for Biotechnology Information (NCBI) database using the Naegleria jamiesoni twin-ribozyme group I intron [12] as a query sequence identified several homologous introns in various Naegleria isolates [3, 19], as well as a significant hit towards a nuclear SSU rDNA intron of Heterolobosea sp. BA (accession number DQ388519 [24]). The Heterolobosea sequence corresponds to a 1310 bp group I intron at position S516. Position 516 in the SSU rRNA gene is a common group I intron insertion site among eukaryotic microorganisms [19, 25] and hosts the Naegleria twin-ribozyme intron.
Phylogenetic analysis has previously identified the Heterolobosea sp. BA as a very close relative to Allovahlkampfia spelaea, owing to almost identical SSU rRNA gene sequences, and as a sister species to the Acrasid slime moulds, but clearly distinct from the free-living Naegleria amoeboflagellates [26, 27]. Thus, we renamed the Heterolobosea sp. BA isolate Allovahlkampfia sp. and its corresponding intron Asp.S516, according to the current nomenclature of group I introns in rDNA [28]. Asp.S516 appears optional in Allovahlkampfia sp. since only the BA isolate harbours an intron at the S516 site [27].
Asp.S516 is organized as a twin-ribozyme intron composed of two distinct ribozyme domains and a homing endonuclease gene
A secondary structure diagram of Asp.S516 is presented in Figure 1A and compared with the consensus structure diagram of the Naegleria Nae.S516 twin-ribozyme intron (Figure 1B) [19]. Asp.S516 and Nae.S516 are similar in the overall structural features, and consist of three domains representing distinct structures and functions. Here, the GIR2 splicing ribozyme contains a large insertion sequence in P6 that harbours the HEG and the LC ribozyme. We previously reported that Nae.S516 was inherited vertically during evolution, and that NaeLC and HEG are evolutionary linked [19]. To further investigate the evolutionary relationship between Asp.S516 and Nae.S516, we performed phylogenetic tree analysis and found an apparent basal position of Asp.S516 compared with the Naegleria twin-ribozyme introns (Additional file 1: Figure S1).

Schematic presentation of the twin-ribozyme introns in Allovahlkampfia sp. (Asp.S516) and Naegleria (Nae.S516). (A) Secondary structure diagram of Asp.S516 folded according to previously reported models [19] with some modifications. Asp.S516 contains two ribozyme domains, AspLC and AspGIR2, and a homing endonuclease gene (I-Asp I HEG). Paired RNA segments are denoted P3 to P15 in AspLC and P1 to P13 in AspGIR2. Nucleotide positions invariant among the Naegleria isolates but different in Allovahlkampfia are indicated in blue. Regions in Asp.S516 that deviate from Nae.S516 are boxed (LC-GIR2 junction; P13). (B) Consensus secondary structure diagram of Nae.S516 based on 13 Naegleria introns [19]. Red circles represent variable positions based on at least one deviating Naegleria intron. Invariant positions are presented as uppercase letters. (C) Alignment of the His-Cys homing endonucleases encoded by Asp.S516 and Nae.S516. Identical residues are boxed, and deletions are indicated by dashes. Functionally important C, H and N residues involved in zinc binding and catalysis are indicated (yellow boxes). The I-Nae I sequence represents a consensus of nine Naegleria HEs (see Additional file 2: Figure S2). Uppercase letter, invariant amino acid position; lowercase letter, conserved amino acid position in at least five of nine sequences; X, non-conserved amino acid position.
The Asp.S516 HEG corresponds to a 252 amino acid His-Cys box HE with only limited sequence similarity to the 245 amino acid Naegleria HEs (Additional file 2: Figure S2). Whereas the amino acid identity among different Naegleria HEs varies from 81% to 100% [19], the Allovahlkampfia HE (hereafter named I-Asp I) contains only 46% identical positions, as compared with the consensus Naegleria HE (Figure 1C). Four of the Naegleria HEs have previously been tested and shown to cleave the intron-less rDNA allele [15, 29, 30]. Since I-Asp I shares all residues known to be essential for HE active site definition, catalysis and zinc coordination (Additional file 2: Figure S2) [19], it is likely that I-Asp I represents a functional HE.
AspGIR2 represents a typical group IC1 splicing ribozyme similar in sequence and structure to the Naegleria S516 GIR2 (Figure 1A,B) and with structural resemblance to the well-studied Tetrahymena intron [31]. Consistent with a more distant relationship of AspGIR2 to NaeGIR2, we detected 73 nucleotide substitutions present in Allovahlkampfia at invariant positions among Naegleria isolates (Figure 1A). The L5b-P6 GNRA-tetraloop interaction proposed to be a more recently derived structural feature in Naegleria[19] is missing in AspGIR2, and represents further support of a more basal position. Indels in AspGIR2 compared with NaeGIR2 are practically lacking, except for one notable example in the sequences flanking the P13 kissing-loop structure. Whereas NaeGIR2 has a 3-nt insertion, 5′ of P13 (P9 domain), AspGIR2 has a corresponding 5-nt insertion at the 3′ side (boxed in Figure 1A,B).
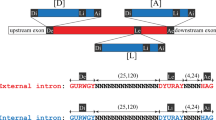
The AspLC ribozyme represents the Naegleria-type of LC ribozymes and contains the hallmark P3/P15 pseudoknot found in all LC ribozymes described to date [11, 12, 32]. AspLC contains 33 positions that deviate from the corresponding invariant positions in NaeLC ribozymes, but all are consistent with the secondary structure model (Figure 1A). The changes are mostly located in loop regions or found as compensatory changes in stems, including four compensatory base-pair changes in P15. In addition, unique nucleotide variants in P5 and J5/4 regions not seen among the Naegleria LC ribozymes are observed. Of most interest is the LC-GIR2 junction, which is only 20 nt in Asp.S516 but 31 nt in Nae.S516 (Figure 1A,B). This region is supposed to carry regulatory sequences responsible for conformation switching in LC ribozymes [10]. A closer inspection of LC-GIR2 junction sequences in Asp.S516 and all Naegleria S516 twin-ribozyme introns revealed highly conserved base-pairing features (named pHEG-2 and P2.1) that involve the 5′ part of the HE mRNA (Figure 2A). This structure is further supported by compensatory base pairings between the Allovahlkampfia and Naegleria introns, and among the different Naegleria intron species. Furthermore, the larger size of the LC-GIR2 junction in Nae.S516 compared with Asp.S516 results from the presence of a paired extension in NP2.1 and by pHEG-3 (Figure 2B).

Proposed base-pairing interactions between the LC ribozyme and HEG. (A) Schematic box presentation of pHEG-1, pHEG-2 and AP2.1 in Allovahlkampfia and pHEG-1, pHEG-2, pHEG-3, and NP2.1 in N. pringsheimi twin-ribozyme introns, respectively. P6 is from the GIR2 ribozyme. 5′ (n′) and 3′ (n″) sequence boxes are indicated, as well as the start (AUG) and stop (UGA) codons of the HEG. (B) Secondary structure diagram of the LC-GIR2 junctions in Asp.S516 and Npr.S516. Corresponding HEGs (red letters) are indicated by start codon (AUG), polyA signal (AAUAAA), and stop codon (UGA). pHEG-1, pHEG-2 and P2.1 are present in both introns and indicate base pairings between the LC ribozyme and HEG. AspGIR2 and NprGIR2, group I splicing ribozymes; AspLC and NprLC, lariat capping ribozymes; BP, branch point nucleotide; IPS, internal processing site.
AspGIR2 performs intron self-splicing and intron RNA circularization
Asp.S516 DNA including parts of the flanking exons, but lacking most of the HEG was synthesized in vitro and cloned into a plasmid vector (Figure 3A). Corresponding in vitro transcribed RNA (Asp.S516Δ742) was made from a PCR product generated by amplification using an upstream exon primer containing the T7 promoter (C716) and a downstream exon primer (C717; Additional file 3: Table S1), and subjected to self-splicing, exoG-labelling, or intron circularization conditions. Figure 3B presents a time-course gel analysis of the RNA processing and self-splicing products generated after incubation at self-splicing conditions with or without exoG. Several important features are noted: (1) The ligated exon was stimulated in the presence of exoG (RNA#9); (2) the excised full-length intron was accumulated more efficiently at splicing conditions with exoG than without (RNA#4); (3) circular intron RNA species were generated both with and without exoG, but with different patterns during the time-course reaction (RNA#1); and (4) internal intron processing independent of exoG was observed (RNA#7 and RNA#8).

RNA processing activity by AspGIR2. (A) Schematic organization of Asp.S516 intron and corresponding splicing RNA construct (Asp.S516Δ742 RNA) and lariat capping RNA construct (AspLC-191.28 RNA). SSU; small subunit ribosomal RNA exons. (B) Time-course gel analysis of Asp.S516 self-splicing products. In-vitro transcribed 32P-labelled RNA from Asp.S516Δ742 was incubated at self-splicing with and without exoG. RNA species generated are indicated to the right. RNA#1, circular intron; RNA#2, precursor transcript; RNA#3, 5′SS or 3′SS processed precursor transcript; RNA#4, excised linear intron; RNA#5, IPS processed 5′ precursor; RNA#6, IPS processed 5′ intron; RNA#7, IPS processed 3′ precursor; RNA#8, IPS processed 3′ intron; RNA#9, ligated exon; RNA#10, free 5′ or 3′ exons. Exons, AspLC and AspGIR2 are indicated as dark grey, black and light grey boxes, respectively. (C) ExoG-labelling of Asp.S516Δ742 RNA compared with uniformly labelled time-course experiment of the same RNA. RNA#4 (excised linear intron) is the main labelled RNA species. (D) Ligation junction sequence of ligated exons generated in self-splicing reaction with exoG. Sequences were determined from RT-PCR generated products of ligated exons (RNA#9, Figure 3B) using primer combinations C716/C736 in amplification and C736 in sequencing reactions. DNA sequence reads from the reactions (right) and is complementary to that of the RNA sequences presented. Arrow marks intron insertion site or ligated exon site. (E) Full-length circle-junction sequences of Asp.S516 generated in self-splicing reaction without exoG. Sequences were determined from RT-PCR generated products of spliced Asp.S516Δ742 RNA using primer combination C734/C735 in amplification and C734 in sequencing reactions. The signal one nucleotide above the branch point is due to a capping-independent primer extension stop. Note that the sequencing ladder is generated from the opposite strand. (F) Dominating circle junction generated with exoG in the self-splicing reaction. The sequence corresponds to truncated circles lacking the first two nucleotides of the intron.
The RNA corresponding to ligated exons (RNA#9) was further assessed by RT-PCR amplification and sequencing following elution from the gel. As expected, the unique sequence resulting from exons ligation was identified (Figure 3D). In group I intron self-splicing, exoG becomes covalently attached to the 5′ end of the excised intron RNA. This reaction can be monitored in a radioactive exoG-labelling experiment. In the exoG-labelling experiment of the Asp.S516Δ742 RNA (Figure 3C), one of the RNA species (RNA#4) became strongly labelled during splicing, and corresponds to the excised linear intron. We infer that Asp.S516 performs exoG-dependent self-splicing catalyzed by the AspGIR2 ribozyme.
Circularization of intron RNA during self-splicing and subsequent processing is a common feature among group I introns [5, 33], and three distinct pathways of generating intron RNA circles have been described and reviewed [3]. Each pathway has a unique sequence hallmark at the circularization junction and can be easily distinguished by RT-PCR sequencing. To evaluate circle identities, RNA#1 (Figure 3B) was eluted from the gel and subjected to RT-PCR sequencing. Full-length intron circle RNA was generated at reaction conditions lacking exoG (Figure 3E), an observation consistent with the splicing-independent circularization pathway initiated by 3′ SS hydrolysis of precursors [5]. A different result was observed from the reaction containing exoG. Here, a truncated circle lacking the first two nucleotides at the 5′ end of the intron was found (Figure 3F). Truncated circles are generated from excised intron RNAs [34]. A third circularization pathway incorporates exoG in the circular RNA and is also generated from the excised introns during splicing [35]. However, no support of exoG-containing intron circles was obtained since the exoG-labelling experiment did not detect any labelled circular RNAs (Figure 3C) and the RT-PCR sequencing approach did not reveal a circle junction with an additional G nucleotide. We conclude that Asp.S516 performs two types of intron circularization reactions, one dependent and one independent of exoG.
AspLC performs 5′ lariat capping at the intron homing endonuclease mRNA
The observed exoG-independent cleavage at the internal processing site (IPS; Figure 3C) was likely to be catalyzed by the AspLC ribozyme. Based on recent structural and functional characterization of the N. pringsheimi LC ribozyme (NprLC) [17], we designed an AspLC construct with 169 nt upstream from and 28 nt downstream from the IPS (AspLC-169.28) for kinetic cleavage analysis (Figure 4A). This construct was made from a template generated by amplification of the Asp.S516Δ742 intron using the upstream primer C718 containing a T7 promoter and a downstream primer (C719; Additional file 3: Table S1). The complete AspLC-169.28 and NprLC-191.28 ribozymes (Figure 4A,B) were subjected to LC conditions incubated at different time points from 0 to 240 min, and subsequently separated by denaturing polyacrylamide electrophoresis (Figure 4C). AspLC was found to be a very efficient self-cleaving ribozyme (kobs 0.08; endpoint 0.08), almost as active as the corresponding NprLC (Npr191.28; kobs 0.12; endpoint 0.08) (Figure 4D).

Structure and cleavage activity of AspLC. Secondary structure diagrams of the active forms of (A) Allovahlkampfia AspLC-169.28 and (B) Naegleria pringsheimi NprLC-191.28. Substitutions in AspLC compared with NprLC are indicated in red. Helical indels are boxed. (C) Time-course cleavage analysis of AspLC-169.28 and NprLC-191.28 transcripts separated on 5% urea polyacrylamide gels. Pre, LC precursor RNA; 5′LC, 5′ cleavage product containing the LC ribozyme sequence. (D) Kinetic cleavage analysis of AspLC-169.28 and NprLC-191.28 transcripts presented as fraction of uncleaved precursor versus time. (E) Primer extension analysis of the same sample of AspLC-169.28 analyzed in C and D, but tracking the 3′ cleavage product. BP, branch point nucleotide; IPS, internal processing site. A sequencing ladder made by sequencing of the AspLC-169.28 plasmid construct with the same primer (C720) as used for primer extension is shown. Interpretations of IPS and branch point are presented below the primer extension analysis. (F) Schematic presentation of the lariat capped 3′ cleavage product (I-Asp I mRNA) and released 5′ cleavage product (AspLC ribozyme sequence).
Naegleria LC ribozymes generate a 3-nt lariat cap structure at the IPS that constitutes the 5′ end of the downstream HE mRNA [17]. The lariat cap structure can be detected experimentally by several approaches, including a primer extension assay [16, 17]. Primer extension generates a stop at the branch point nucleotide when the cap is present, or at the IPS nucleotide when cleaved by hydrolysis. Figure 4E presents a primer extension experiment of the cleavage product after AspLC catalysis at six different time points. A strong signal was observed at the branch point nucleotide located only 10 nt upstream from the AUG start codon (Figure 4E). This result is consistent with a lariat cap structure present at the majority of 5′ ends. We interpret that AspLC is an active LC ribozyme able to generate a 3-nt 2′,5′ cap at the 5′ end of the downstream HE mRNA (Figure 4F).
Discussion
Expanded distribution of twin-ribozyme group I introns
We showed that Asp.S516, present in the Heterolobosea amoeba species Allovahlkampfia sp., is a twin-ribozyme intron. This finding expands the distribution pattern of mobile twin-ribozyme introns beyond Naegleria amoeboflagellate and Didymium myxomycete genera. Asp.S516 is related to the Naegleria introns and consists of two ribozymes with distinct functions in splicing and mRNA maturation. AspGIR2 represents a typical group IC1 ribozyme that catalyzes exoG-dependent intron excision and exon ligation, as well as the formation of full-length and truncated intron RNA circles. The AspLC ribozyme, on the other hand, is a new member of the LC ribozymes, and is responsible for forming a 5′ cap at the intron HE mRNA.
In Myxomycetes, only one isolate (D. iridis Panama 2) has been reported to harbour a mobile twin-ribozyme intron [8, 11, 18], despite extensive searches for nucleolar group I introns in Myxomycetes [36]. Naegleria twin-ribozyme introns appear much more frequent in nature, and 29 out of 70 isolates investigated harbour the Nae.S516 intron [10, 19]. Nae.S516 is strictly vertically inherited, a feature that includes both ribozyme domains (NaeLC and NaeGIR2) and the HEG [19]. Furthermore, structural characterizations suggested that the intron was gained as a pre-organized genetic element early in the evolution of the Naegleria genus. Our recent finding of a similar twin-ribozyme intron in the more distantly related Allovahlkampfia indicates an even earlier occurrence.
Role of circular intron RNAs
A biological role of group I introns beyond splicing has been debated and recently reviewed [1, 3, 37]. The fact that Asp.S516 and Nae.S516 are optional in Allovahlkampfia and Naegleria genera supports the notion of group I introns as selfish genetic elements, but a more complex picture appears when considering RNA processing data. Intron splicing catalyzed by AspGIR2 results in ligation of the essential host SSU rRNA. Thus, the host is dependent on a functional ribozyme in order to survive. Furthermore, the excised intron RNA has the potential of performing reverse splicing into cognate or non-cognate RNA sequence sites [13, 38], and the generation of truncated intron circles from excised group I introns has been suggested to reduce or eliminate the reverse splicing reaction [5]. Interestingly, very similar truncated circles are generated both from Asp.S516 and Nae.S516 during self-splicing in the presence of exoG (Figure 3C) [12], and this suggests that truncated circles represent a conserved RNA processing feature.
The full-length intron circles, however, are generated from a different pathway and initiated by hydrolytic cleavage at the 3′SS of precursor RNAs [5]. AspGIR2 catalyzes the formation of full-length intron circles in the absence of exoG, and subsequently generates non-ligated and presumably non-functional SSU rRNAs. Why Allovahlkampfia, Naegleria and Didymium twin-ribozyme introns generate full-length intron circles is not clear. One explanation could be that the full-length intron circles, which contain all the genetic information in the intron, are involved in intron mobility at the RNA level [5, 13, 18]. The generation of non-ligated SSU rRNA exons is likely to result in an imbalance in ribosomal components and generate nucleolar stress. Thus, intron circularization is expected to impact host physiology. Interestingly, all Heterolobosea amoeba studied to date have an unusual nucleolar structure consisting of several thousand identical copies of circular rDNA plasmids [39]. Loss of mature SSU rRNA due to full-length intron circularization could be compensated either by an increasing fraction of rDNA plasmids committed to transcription, or by increasing transcription initiation efficiency at each rDNA plasmid. These possibilities have to be experimentally investigated and evaluated.
P2.1 is a conserved feature of lariat capping ribozymes
AspLC was found to be an efficient branching ribozyme in vitro with a number of conserved structural features in common with NaeLC (Figures 1A,B and 4A,B). Several compensatory changes in helices P3, P5, P6, P8, P9 and P15 were noted that further supported the general secondary structure diagram of the Naegleria-type LC ribozyme [17]. The most dramatic difference was found at the LC-GIR2 junction (boxed in Figures 1A,B and 2B), which contains almost no conserved nucleotides between AspLC and NaeLC. Here we propose that the LC-GIR2 junctions in Allovahlkampfia and Naegleria are involved in base-pairing interactions with the HE mRNA (Figure 2). This finding adds additional structural support to the observed mutual relationships between the LC ribozyme and the corresponding HE mRNA sequence in the Naegleria-type twin-ribozyme introns [15].
The Didymium LC ribozyme contains an RNA structure element (DP2.1) shown to be essential in catalysis [40], but a corresponding P2.1 appears to be missing in the NaeLC ribozyme [10]. DP2.1 is supported by structural probing and mutational experiments [40], and the recent 2.5 Å DirLC crystal structure shows that DP2.1 bridges the catalytic core segments P10 and L5, thus participating in ribozyme activation [22]. Here we identified structures homologous to P2.1 in Allovahlkampfia (AP2.1) and Naegleria (NP2.1) LC ribozymes (Figure 5A). AP2.1 and NP2.1 are supported by sequence conservation to DP2.1, by the proximity to P10 and L9 interactions and by compensatory changes in the Allovahlkampfia and Naegleria helices (Figure 5A). The structural model of AspLC (Figure 5B) further suggests an integrated role of AP2.1, with a location that apparently corresponds to that of DP2.1 in the Didymium LC ribozyme [22]. We conclude that the peripheral helix P2.1 is a conserved feature of all LC ribozymes. P2.1 in Allovahlkampfia and Naegleria LC ribozymes appear unusual, owing to involvements of protein-coding sequences from the HEG region. However, it remains to be explored if AP2.1 and NP2.1 participate in conformational switch regulation of the branching reaction, and fold back onto the catalytic core, as seen for the DP2.1 [22, 32].

P2.1 in LC ribozymes. (A) Secondary structure diagrams of P2.1 in Allovahlkampfia (AP2.1), Naegleria (NP2.1) and Didymium (DP2.1) LC ribozymes. Yellow boxes: identical nucleotide positions in P2.1 and P10 of all three LC ribozyme variants. Red boxes: compensatory base-pair changes in P2.1 observed between the AspLC and NprLC ribozymes. Note that pHEG-1 in AspLC and NprLC has a similar location to that of the L9-tetraloop DP2 receptor interaction in DirLC [21], and that DP2.1 is a regular hairpin structure, while AP2.1/NP2.1 is made by a LC:HE mRNA interaction. (B) Structure model of the pre-cleaved AspLC ribozyme. Coloured sequence segments in the secondary structure (left) correspond to those in the 3D structure model (right). The HEG sequence (red line, dot) is involved in AP2.1 (yellow box).
Conclusions
Our study expands the distribution pattern of nuclear twin-ribozyme introns among eukaryote microorganisms. Here we show that an intron in Allovahlkampfia sp. ribosomal DNA consists of a HEG and two ribozyme domains with distinct functions in splicing and mRNA maturation. One of the ribozymes is a typical group I intron ribozyme that catalyzes the intron splicing and intron circularization reactions. The other ribozyme represents a new member of the LC ribozyme class. The Allovahlkampfia LC ribozyme is responsible for forming a cap structure at the homing endonuclease mRNA, and thus contributes to intron mobility. Comparative analysis and 3D modelling support the hypothesis that mRNA coding sequences are directly involved in base parings with sequences of the LC ribozyme, forming a regulatory RNA element (P2.1) conserved among all member of the LC ribozyme class. This observation adds support to a mutual relationship between the LC ribozyme and corresponding homing endonuclease mRNA.
Methods
Templates and in-vitro transcription
The DNA fragment of Asp.S516 twin-ribozyme intron (lacking most (742 bp) of the HEG sequence) and parts of flanking exons were synthesized by Eurofins MWG Operon based on sequence information available in the accession number DQ388519 from the sequence database and cloned using the standard vector pEX-A (Eurofins MWG Operon). The authentic Asp.S516 sequence was confirmed by dideoxy sequencing analysis. Templates for in-vitro transcription were made by PCR using Pfu DNA polymerase. Primer sequences are compiled in Additional file 3: Table S1. All upstream primers contain the T7 polymerase promoter sequence. PCR products were purified with GeneJET PCR purification kit (Fermentas) and in-vitro transcribed by T7 RNA polymerase (Fermentas). Labelled transcript was transcribed using trace amount of [α-32P] uridine triphosphate, as previously described [41], and purified through Illustra MicroSpin S-200 HR columns. All transcripts were excised after gel electrophoresis on 5% denaturing (urea) polyacrylamide gels (UPAGs), eluted by diffusion into 0.25 M sodium acetate pH 6.0, 1 mM EDTA), followed by ethanol precipitation and resuspension in diethylpyrocarbonate-treated water.
Cleavage analysis
Twin-ribozyme transcript was incubated at 45°C in splicing buffer (40 mM Tris-HCl pH 7.5, 25 mM MgCl2, 0.5 M KCl, 2 mM spermidine, 5 mM dithiothreitol) with or without 0.2 mM guanosine triphosphate. LC ribozyme RNA was pre-incubated at 45°C for 5 min in renaturation buffer (10 mM acetate buffer pH 5.5, 25 mM MgCl2, 1 M KCl) to promote correct folding. For kinetic cleavage experiments, the reactions were initiated by the addition of four volumes of start buffer (47.5 mM HEPES-KOH pH 7.5, 25 mM MgCl2, 1 M KCl) to the folded RNA. Aliquots of reaction were removed at various time points and terminated by addition of an equal volume of UBB (50% urea, 10 mM EDTA, 1 mg/ml bromophenol blue, 1 mg/ml xylene cyanol). RNA samples were run on 5% UPAG and exposed to storage phosphor screens. Interpretations of individual RNA species and processing reactions were made according to [42].
Sequencing of circle junction and ligated exon junction
The twin-ribozyme intron transcript was incubated in splicing buffer for 5 min and 15 min with or without 0.2 mM guanosine triphosphate, respectively. Circularization RNA products were excised after gel electrophoresis on 5% UPAG, eluted by diffusion into 0.25 M sodium acetate pH 6.0, 1 mM EDTA, ethanol precipitated, and resuspended in diethylpyrocarbonate-treated water. Reverse transcription was performed using M-MuLV reverse transcriptase (Fermentas) and primer C734. cDNA was amplified by primers C734 and C735. The PCR product was then dideoxy sequenced with labelled primer C734 using a sequencing kit (USB) and separated on 8% UPAG. Ligated exon RNA was isolated after gel electrophoresis, subjected to reverse transcription using the primer C736, and amplified by PCR with C716 and C736. The PCR product was sequenced using labelled primer C736.
ExoG-labelling
Unlabelled twin-ribozyme precursor transcript was incubated with [α-32P]guanosine triphosphate in splicing buffer at 45°C for 5 min. The reaction was terminated by adding equal amount of UBB and separated on 5% UPAG, together with kinetic cleavage reaction of labelled twin-ribozyme transcript.
Data analysis of LC ribozyme cleavage reaction
RNA bands were quantified using ImageQuant 5.2 software and the kinetic data were fitted using one phase exponential decay with an endpoint correction:
to obtain values for kobs and endpoints of the reaction [41]. All cleavage reactions were performed three times in parallel and the results were highly reproducible.
Primer extension
Primer extension analysis was used to map the 5′ end of the downstream LC RNA cleavage product (3′ RNA). The 3′ RNA was generated by self-cleavage reaction of the precursor LC RNA at various time points and annealed to end-labelled complementary primer C720. Reverse transcription was performed using M-MuLV reverse transcriptase (Fermentas), as previously described [40]. A dideoxy sequencing ladder was prepared from the corresponding PCR product using the same primer and run adjacent to the primer extension products as markers. The reactions were run on 8% UPAG.
Molecular modelling
The AspLC ribozyme was built by homology modelling using Assemble software [43], taking the recent wild-type DirLC crystal structure (PDB ID 4p8z [22]) as a template. Differences in secondary structure and sequence were manually accommodated in the same software. One additional nucleotide was added in L8 and in J5/4. The extension of P6 was modelled as a loop E found between residues 216 and 235 of the Deinococcus radiodurans large ribosomal subunit in the 3PIP entry [44] of the pdb, except for the structures of L8 and J5/4. These were kept identical to DirLC, because the segments left unchanged are not expected to interfere with AP2.1. Figure 5B (right) was prepared using the PyMOL program [45].
Abbreviations
- AspLC:
-
Allovahlkampfia LC ribozyme
- DirLC:
-
Didymium LC ribozyme
- EDTA:
-
ethylenediaminetetraacetic acid
- exoG:
-
exogenous guanosine cofactor
- HE:
-
homing endonuclease
- HEG:
-
homing endonuclease gene
- IPS:
-
internal processing site
- LC:
-
lariat capping
- LSU:
-
large subunit
- NCBI:
-
National Center for Biotechnology Information
- NaeLC:
-
Naegleria LC ribozyme
- NprLC:
-
N. pringsheimi LC ribozyme
- PCR:
-
polymerase chain reaction
- RT-PCR:
-
reverse transcription polymerase chain reaction
- SS:
-
splice site
- SSU:
-
small subunit
- UBB:
-
50% urea, 10 mM EDTA, 1 mg/ml bromophenol blue, 1 mg/ml xylene cyanol
- UPAG:
-
urea polyacrylamide gel.
References
Hedberg A, Johansen SD: Nuclear group I introns in self-splicing and beyond. Mob DNA 2013, 4: 17. 10.1186/1759-8753-4-17
Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR: Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena . Cell 1982, 31: 147-157. 10.1016/0092-8674(82)90414-7
Nielsen H, Johansen SD: Group I introns, moving in new directions. RNA Biol 2009, 6: 375-383. 10.4161/rna.6.4.9334
Brehm SL, Cech TR: Fate of an intervening sequence ribonucleic acid: excision and cyclization of the Tetrahymena ribosomal ribonucleic acid intervening sequence in vivo . Biochem 1983, 22: 2390-2397. 10.1021/bi00279a014
Nielsen H, Fiskaa T, Birgisdottir AB, Haugen P, Einvik C, Johansen S: The ability to form full-length intron RNA circles is a general property of nuclear group I introns. RNA 2003, 9: 1464-1475. 10.1261/rna.5290903
Vicens Q, Cech TR: Atomic level architecture of group I introns revealed. Trends Biochem Sci 2006, 31: 41-51. 10.1016/j.tibs.2005.11.008
Muscarella DE, Vogt VM: A mobile group I intron in the nuclear rDNA of Physarum polycephalum . Cell 1989, 56: 443-454. 10.1016/0092-8674(89)90247-X
Johansen S, Elde M, Vader A, Haugen P, Haugli K, Haugli F: In vivo mobility of a group I twintron in nuclear ribosomal DNA of the myxomycete Didymium iridis . Mol Microbiol 1997, 24: 737-745. 10.1046/j.1365-2958.1997.3921743.x
Johansen SD, Haugen P, Nielsen H: Expression of protein-coding genes embedded in ribosomal DNA. Biol Chem 2007, 388: 679-686.
Nielsen H, Beckert B, Masquida B, Johansen SD: The GIR1 branching ribozyme. In Ribozymes and RNA Catalysis. Cambridge: The Royal Society of Chemistry Publishing; 2008:229-252.
Decatur WA, Einvik C, Johansen S, Vogt VM: Two group-I ribozymes with different functions in a nuclear rDNA intron. EMBO J 1995, 14: 4558-4568.
Einvik C, Decatur WA, Embley TM, Vogt VM, Johansen S: Naegleria nucleolar introns contain two group I ribozymes with different functions in RNA splicing and processing. RNA 1997, 3: 710-720.
Birgisdottir AB, Johansen S: Site-specific reverse splicing of a HEG-containing group I intron in ribosomal RNA. Nucleic Acids Res 2005, 33: 2042-2051. 10.1093/nar/gki341
Vader A, Nielsen H, Johansen S: In vivo expression of the nucleolar group I intron-encoded I- Dir I homing endonuclease involves the removal of a spliceosomal intron. EMBO J 1999, 18: 1003-1013. 10.1093/emboj/18.4.1003
Decatur WA, Johansen S, Vogt VM: Expression of the Naegleria intron endonuclease is dependent on a functional group I self-cleaving ribozyme. RNA 2000, 6: 616-627. 10.1017/S1355838200992203
Nielsen H, Westhof E, Johansen S: An mRNA is capped by a 2′, 5′ lariat catalyzed by a group I-like ribozyme. Science 2005, 309: 1584-1587. 10.1126/science.1113645
Tang Y, Nielsen H, Birgisdottir ÅB, Johansen SD: A natural fast-cleaving branching ribozyme from the amoeboflagellate Naegleria pringsheimi . RNA Biol 2011, 8: 1-8. 10.4161/rna.8.1.15302
Johansen S, Vogt VM: An intron in the nuclear ribosomal DNA of Didymium iridis codes for a group I ribozyme and a novel ribozyme that cooperate in self-splicing. Cell 1994, 76: 725-734. 10.1016/0092-8674(94)90511-8
Wikmark OG, Einvik C, De Jonckheere JF, Johansen SD: Short-term sequence evolution and vertical inheritance of the Naegleria twin-ribozyme group I intron. BMC Evol Biol 2006, 6: 39. 10.1186/1471-2148-6-39
Beckert B, Nielsen H, Einvik C, Johansen SD, Westhof E, Masquida B: Molecular modelling of the GIR1 branching ribozyme gives new insight into evolution of structurally related ribozymes. EMBO J 2008, 27: 667-678. 10.1038/emboj.2008.4
Birgisdottir AB, Nielsen H, Beckert B, Masquida B, Johansen SD: Intermolecular interaction between a branching ribozyme and associated homing endonuclease mRNA. Biol Chem 2011, 392: 491-499.
Meyer M, Nielsen H, Olieric V, Roblin P, Johansen SD, Westhof E, Masquida B: Speciation of a group I intron into a lariat capping ribozyme. Proc Natl Acad Sci USA 2014, 111: 7659-7664. 10.1073/pnas.1322248111
Jabri E, Aigner S, Cech TR: Kinetic and secondary structure analysis of Naegleria andersoni GIR1, a group I ribozyme whose putative biological function is site-specific hydrolysis. Biochemistry 1997, 36: 16345-16354. 10.1021/bi9718595
Shutt TE, Gray MW: Environmental Heterolobosean isolates. GenBank 2006. DQ388519 [http://www.ncbi.nlm.nih.gov/nuccore/DQ388519] DQ388519 []
Haugen P, Coucheron DH, Rønning B, Haugli K, Johansen S: The molecular evolution and structural organization of self-splicing group I introns at position 516 in nuclear SSU rDNA of myxomycetes. J Euk Microbiol 2003, 50: 283-292. 10.1111/j.1550-7408.2003.tb00135.x
Walochnik J, Mulec J: Free-living amoebae in carbonate precipitating microhabitats of Karst Caves and a new vahlkampfiid amoeba, Allovahlkampfia spelaea gen. nov., sp. nov. Acta Protozool 2009, 48: 25-33.
Brown MW, Silberman JD, Spiegel FW: A morphologically simple species of Acrasis (Heterolobosea, Excavata), Acrasis helenhemmesae n. sp. J Eukaryot Microbiol 2010, 57: 346-353. 10.1111/j.1550-7408.2010.00481.x
Johansen S, Haugen P: A new nomenclature of group I introns in ribosomal DNA. RNA 2001, 7: 935-936. 10.1017/S1355838201010500
Elde M, Haugen P, Willassen NP, Johansen S: I- Nja I, a nuclear intron-encoded homing endonuclease from Naegleria , generates a pentanucleotide 3′ cleavage-overhang within a 19 base-pair partially symmetric DNA recognition site. Eur J Biochem 1999, 259: 281-288. 10.1046/j.1432-1327.1999.00035.x
Elde M, Willassen NP, Johansen S: Functional characterization of isoschizomeric His-Cys box homing endonuclease from Naegleria . Eur J Biochem 2000, 267: 7257-7266. 10.1046/j.1432-1327.2000.01862.x
Guo F, Gooding AR, Cech TR: Structure of the Tetrahymena ribozyme: base triple sandwich and metal ion at the active site. Mol Cell 2004, 16: 351-362.
Nielsen H, Einvik C, Lentz TE, Hedegaard MM, Johansen SD: A conformational switch in the DiGIR1 ribozyme involved in release and folding of the downstream I- Dir I mRNA. RNA 2009, 15: 958-967. 10.1261/rna.669209
Haugen P, De Jonckheere JF, Johansen S: Characterization of the self-splicing products of two complex Naegleria LSU rDNA group I introns containing homing endonuclease genes. Eur J Biochem 2002, 269: 1641-1649. 10.1046/j.1432-1327.2002.02802.x
Zaug AJ, Grabowski PJ, Cech TR: Autocatalytic cyclization of an excised intervening sequence RNA is a cleavage-ligation reaction. Nature 1983, 301: 578-583. 10.1038/301578a0
Vicens Q, Cech TR: A natural ribozyme with 3′,5′ RNA ligase activity. Nature Chem Biol 2009, 5: 97-99. 10.1038/nchembio.136
Nandipati SCR, Haugli K, Coucheron DH, Haskins EF, Johansen SD: Polyphyletic origin of the genus Physarum (Physarales, Myxomycetes) revealed by nuclear rDNA mini-chromosome analysis and group I intron synapomorphy. BMC Evol Biol 2012, 12: 166. 10.1186/1471-2148-12-166
Edgell DR, Chalamcharla VR, Belfort M: Learning to live together: mutualism between self-splicing introns and their hosts. BMC Biol 2011, 9: 22. 10.1186/1741-7007-9-22
Roman J, Woodson SA: Integration of the Tetrahymena group I intron into bacterial rRNA by reverse splicing in vivo . Proc Natl Acad Sci USA 1998, 95: 2134-2139. 10.1073/pnas.95.5.2134
Clark CG, Cross GAM: Circular ribosomal RNA genes are a general feature of schizopyrenid amoebae. J Protozool 1988, 35: 326-329. 10.1111/j.1550-7408.1988.tb04352.x
Einvik C, Nielsen H, Nour R, Johansen S: Flanking sequences with an essential role in hydrolysis of a self-cleaving group I-like ribozyme. Nucleic Acids Res 2000, 28: 2194-2200. 10.1093/nar/28.10.2194
Einvik C, Nielsen H, Westhof E, Michel F, Johansen S: Group I-like ribozymes with a novel core organization perform obligate sequential hydrolytic cleavages at two processing sites. RNA 1998, 4: 530-541. 10.1017/S1355838298971758
Nielsen H: Group I ribozymes. Methods Mol Biol 2012, 848: 73-89. 10.1007/978-1-61779-545-9_6
Jossinet F, Ludwig TE, Westhof E: Assemble: an interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics 2010, 26: 2057-2059. 10.1093/bioinformatics/btq321
Belousoff MJ, Shapira T, Bashan A, Zimmerman E, Rozenberg H, Arakawa K, Kinashi H, Yonath A: Crystall structure of the synergistic antibiotic pair, lankamycin and lankacidin, in complex with the large ribosomal subunit. Proc Natl Acad Sci USA 2011, 108: 2717-2722. 10.1073/pnas.1019406108
The PyMOL Molecular Graphics System http://www.pymol.org
Acknowledgements
This work was supported by grants from The Lundbeck Foundation (HN), the Danish Council for Independent Research, Natural Sciences (HN) and the Norwegian Research Council (SDJ).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YT performed the experimental work and participated in analyses and discussions of the results. BM performed 3D RNA modelling and participated in discussions of the results. PPG participated in bioinformatic discovery and discussion of the results. SDJ and HN directed the research. SDJ, HN and YT wrote the manuscript in collaboration with all authors. All authors read and approved the final manuscript.
Electronic supplementary material
13100_2014_110_MOESM1_ESM.pdf
Additional file 1: Figure S1: Unrooted neighbour-joining analysis based on twin-ribozyme intron sequences from Naegleria and Allovahlkampfia isolates. (PDF 497 KB)
13100_2014_110_MOESM2_ESM.pdf
Additional file 2: Figure S2: Amino acid alignment of nine Naegleria homing endonuclease sequences and that from Allovahlkampfia. (PDF 485 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Tang, Y., Nielsen, H., Masquida, B. et al. Molecular characterization of a new member of the lariat capping twin-ribozyme introns. Mobile DNA 5, 25 (2014). https://doi.org/10.1186/1759-8753-5-25
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1759-8753-5-25