
Abstract
Objective
It is important for psychiatrists to be aware of certain inborn errors of metabolism (IEMs) as these rare disorders can present as psychosis, and because definitive treatments may be available for treating the underlying metabolic cause. A systematic review was conducted to examine IEMs that often present with schizophrenia-like symptoms.
Data sources
Published literature on MEDLINE was assessed regarding diseases of homocysteine metabolism (DHM; cystathionine beta-synthase deficiency [CbS-D] and homocysteinemia due to methyltetrahydrofolate reductase deficiency [MTHFR-D]), urea cycle disorders (UCD), acute porphyria (POR), Wilson disease (WD), cerebrotendinous-xanthomatosis (CTX) and Niemann-Pick disease type C (NP-C).
Study selection
Case reports, case series or reviews with original data regarding psychiatric manifestations and cognitive impairment published between January 1967 and June 2012 were included based on a standardized four-step selection process.
Data extraction
All selected articles were evaluated for descriptions of psychiatric signs (type, severity, natural history and treatment) in addition to key disease features.
Results
A total of 611 records were identified. Information from CbS-D (n = 2), MTHFR-D (n = 3), UCD (n = 8), POR (n = 12), WD (n = 11), CTX (n = 14) and NP-C publications (n = 9) were evaluated. Six non-systematic literature review publications were also included. In general, published reports did not provide explicit descriptions of psychiatric symptoms. The literature search findings are presented with a didactic perspective, showing key features for each disease and psychiatric signs that should trigger psychiatrists to suspect that psychotic symptoms may be secondary to an IEM.
Conclusion
IEMs with a psychiatric presentation and a lack of, or sub-clinical, neurological signs are rare, but should be considered in patients with atypical psychiatric symptoms.
Similar content being viewed by others

Introduction
A range of medical conditions may be associated with schizophrenia-like psychosis [1]. The landmark review of psychosis associated with organic disorders by Davison and Bagley, which utilized the 1957 WHO operational criteria for schizophrenia, highlighted a number of disorders where the association with psychosis significantly exceeded chance [2]. Many of the disorders identified showed pathology in the temporal lobe and diencephalon.
A large study of 268 consecutive patients with first-episode psychosis found that 6% had organic cerebral disease that was potentially causally linked with psychiatric symptoms [3], which emphasizes the importance of a thorough diagnostic evaluation to exclude underlying medical illness at first presentation. Recently, more than 60 different congenital conditions associated with psychosis were reviewed. Interestingly, some of them are not associated with dysmorphia, mental retardation or prominent neurological features that may otherwise trigger a search for an organic cause of illness [4].
The diagnosis of medical or neurological illnesses underlying psychosis is of great importance as many of these conditions are progressive or fatal, associated with significant additional medical comorbidity, and may be partially or entirely reversible with definitive treatment. Inborn errors of metabolism (IEMs) represent a particular focus for research as they are frequently under-detected or misdiagnosed, a number are treatable, and new diagnostic methods and therapies have become available.
IEMs are a group of diseases that generally result from the absence or deficiency of an intracellular component of a metabolic pathway (usually, but not exclusively, an enzyme), which may lead to altered intracellular synthesis and catabolism [5]. There are hundreds of IEMs, and many remain poorly characterized. Most result in clinical disease due to the accumulation of substances that are toxic to, or interfere with, normal cellular function, or which may be due to the effects of a reduced ability to synthesize essential compounds.
The overall incidence of IEMs has been estimated to be approximately 40 cases per 100 000 live births [6]. However, this rate may be an underestimation, as new disorders continue to be discovered and characterized and because diagnostic techniques continue to improve in sensitivity and accuracy.
Up to 80% of IEMs are diagnosed during childhood, but an increasing recognition of late-onset presentations has recently raised awareness and diagnoses of adult-onset forms [7]. A number of adult-onset IEMs are associated with schizophrenia-like symptoms, including diseases of homocysteine metabolism (DHM), urea cycle disorders (UCD), porphyria (POR), Wilson disease (WD), cerebrotendinous xanthomatosis (CTX) and Niemann-Pick disease type C (NP-C) [7].
This article reports findings from a systematic literature review and provides a guide for the diagnosis of treatable IEMs associated with schizophrenia-like symptoms based on the review findings. The major features of IEMs that can be associated with psychosis are summarized, and a diagnostic algorithm to assist psychiatrists in the detection of atypical symptoms is proposed that may be related to underlying IEMs.
Methods
Review scope
A meeting was held in mid-2012 to decide which IEMs associated with psychosis are currently treatable, with the aim of conducting a systematic bibliographic search to address the clinical challenges associated with these conditions. Based on a consensus reached during that meeting, the following seven IEMs were chosen as a focus for this review: homocysteinemia due to methyltetrahydrofolate reductase deficiency (MTHFR-D), cystathionine beta-synthase deficiency (CbS-D), UCD, POR, WD, CTX and NP-C.
Literature search methodology and data sources
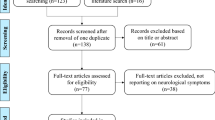
The public MEDLINE database was searched according to a standard four-step protocol, as described in the following sections and summarized in Figure 1.

Summary of literature review process.
Identification
All terms, including complete names and abbreviations for MTHFR-D, CbS-D, UCD, POR, WD, CTX and NP-C were searched alongside the generic tag, ‘psy*’ using EndNote X5 software (Thomson Reuters), which enabled the identification and deletion of any duplicates. In total, 708 potentially relevant records published between January 1967 and June 2012 were identified, from which 97 duplicate records were removed. Seven separate EndNote databases were created – one for each IEM. The numbers of articles for each IEM database were: MTHFR-D (n = 12); CbS-D (n = 6); UCD (n = 15); POR (n = 75); WD (n = 451); CTX (n = 15); and NP-C (n = 31). Six non-systematic literature reviews were also identified and included [7–12]. A total of 611 records were collated for screening.
Screening
Two groups worked separately in screening abstracts from relevant articles from the literature review (Group 1: M. Walterfang and H.H. Kluenemann. Group 2: O. Bonnot, D. Cohen, Sylvie Tordjman and F. Sedel). Case reports, case series with original data regarding psychiatric manifestations and cognitive impairments, and previous reviews containing relevant data were selected. Articles were excluded from full text analysis (see ‘Eligibility’ stage) according to the following exclusion criteria: 1) the article mentioned psychiatric manifestations without data pertaining to any of the seven chosen treatable diseases; 2) an unrelated article, mentioning an IEM without describing psychiatric presentations; 3) literature reviews not containing any new data and; 4) data already reported elsewhere. Screening excluded 26 of the initial records from the POR database and 400 records from the WD database. In cases where the two analysis groups did not agree, records were kept and included in the next step.
Eligibility
The same two analysis groups accessed the full texts of all remaining articles (n = 185) and checked them further for eligibility according to the same exclusion criteria used in the abstract screening stage. The numbers of articles considered eligible after this process were: MTHFR-D (n = 12), CbS-D (n = 6), UCD (n = 15), POR (n = 49), WD (n = 51), CTX (n = 15) and NP-C (n = 31). The six previous reviews were also kept.
Inclusion
Among the eligible records, information on key IEM disease features as well as psychiatric manifestations was included from the following numbers of publications, per database: MTHFR-D (n = 3); CbS-D (n = 2); UCD (n = 8); POR (n = 12); WD (n = 11); CTX (n = 14) and; NP-C (n = 9). Including the six previous reviews, this brought the final total of source articles to 59.
Results
To understand the metabolic pathways implicated in these disorders, we will provide an explanation in the text referring directly to the cited diseases. For figures and complete presentation, we refer the reader to the KFGG website which illustrates theses pathways in detail [13].
Disorders of homocysteine metabolism (DHMs)
CbS-D and MTHFR-D are two key DHMs that commonly feature psychiatric signs.
Key features of CbS-D
Homocysteinuria due to CbS-D is characterized by the involvement of the ocular, skeletal, central nervous and vascular systems. Prevalence is estimated around 1/344 000 birth in countries were systematic search of CbS deficiency is provided for every newborn, however recent data from systematic search for CbS mutation show important prevalence up to 1/20 000 [14]. Two articles that addressed psychiatric symptoms and psychosis were identified [15, 16]. The disease is an autosomal recessive disorder of methionine metabolism, caused by mutations in the CbS gene (21q22.3). CbS normally converts homocysteine to cystathionine in the trans-sulfuration pathway of the methionine cycle, and requires pyridoxal 5-phosphate as a cofactor. The other two cofactors involved in methionine remethylation include vitamin B12 and folic acid. Clinical diagnosis of CbS-D is confirmed by blood amino acid analysis (including total homocysteine measurement), assays of CbS enzyme activity, or screening for CbS mutations.
Patients appear normal at birth but display a progressive disease course if left untreated. Eye anomalies include ectopia lentis (in 85% of cases) and high myopia. Skeletal changes include genu valgum and pes cavus, followed by dolichostenomelia, pectus excavatum or carinatum, kyphoscoliosis and osteoporosis. A Marfan-like body habitus may occur, with tall stature and arachnodactyly. Thromboembolism affecting both large and small arteries and veins is the most striking cause of morbidity and mortality, and affects 25% of individuals by the age of 15 years. While some individuals have a normal IQ, mental retardation is common and, when present, may progress if the disorder is left untreated. Brittle hair and livedo reticularis have also been reported.
Psychiatric signs associated with CbS-D
In one of the few studies in this field, psychiatric illness was found in 51% of cases overall, with symptoms falling into four diagnostic categories: episodic depression (10%), chronic disorders of behavior (17%), obsessive-compulsive disorder (5%), and personality disorder (19%) [15]. In the same study, aggressive behavior and other conduct disorders were particularly common among patients with mental retardation and those who were nonresponsive to vitamin B6. In some cases, psychiatric symptoms may be the initial presenting symptom with no neurological signs [16].
Key features of MTHFR-D
MTHFR-D is another autonomic recessive trait, and is caused by mutations in the 5,10-methylenetetrahydrofolate reductase (MTHFR) gene (1p36.3). MTHFR deficiency results in abnormal intracellular folic acid metabolism, and prevents reduction of 5–10 methylenetetrahydrofolate to 5-methyltetrahydrofolate – the methyl donor for the remethylation of homocysteine into methionine. As a result, the disorder leads to MTHFR-D and consequently to homocysteinuria and hypomethioninemia. To our knowledge prevalence is unknown.
The onset of MTHFR-D usually occurs during the first year of life, characterized by severe neurological signs, recurrent apnea, microcephaly and convulsions without megaloblastic anemia. However, there are some forms with onset during childhood, adolescence or adulthood that present with mental regression, ataxia and schizophrenia-like psychosis. Other symptoms such as sub-acute degeneration of the spinal cord have been reported.
Diagnoses of DHM are made through analysis of amino acids by chromatography and total plasma homocysteine measurement; an elevated level is defined at >100 micromol/L [17]. Methionine levels may be useful, as they are decreased in MTHFR-D and increased in CbS-D.
There are currently three recognized treatment modalities for DHM. For pyridoxine-responsive patients, treatment with pharmacological doses of pyridoxine combined with folic acid and vitamin B12 supplements is recommended. In pyridoxine non-responsive patients, the treatment should comprise a methionine-restricted, cysteine-supplemented diet in combination with the pyridoxine, folic acid and vitamin B12 supplementation. Betaine anhydrous is a methyl donor that may lead to lowering of homocysteine levels in MTHFR-D patients, and can be used as an adjunct to such a diet.
Homocysteine is cleared by transulfuration to cysteine and glutathione, an important antioxidant. Transulfuration requires vitamins B6 and B12. Treatment with vitamin B6, a precursor of homocysteine, can be effective in treating psychiatric symptoms if instituted early [7].
Psychiatric signs associated with MTHFR-D
Three articles were identified from the systematic literature review [18–20]. Psychiatric symptoms are not uncommon in MTHFR-D, and may be the presenting symptom [18]. Their onset may be acute or may follow a more insidious course. Acute manifestations occur mainly after surgery, and present with visual and/or auditory hallucinations, thought disorder and delusions [18]. Roze and colleagues describe two siblings (16- and 24-year-old women) with presumed late-onset MTHFR-D. Three years after the diagnosis of her older sister, the 16-year-old sister was initially seen with a 3-month history of dissociative symptoms and delusions of persecution with visual and auditory hallucinations. One month prior to hospital admission, she had also developed an unsteady gait and urinary incontinence. She was previously healthy and had been an average student. Physical examination on admission showed only an areflexic paraparesis with an extensor plantar response on the right side and impaired vibration and position sense in the lower limbs. Diagnosis was made because her sister was known to have the disease. Combined daily treatment with intravenous hydroxocobalamin (2 mg), oral betaine (9 g), L-carnitine (3 g), and folinic acid (10 mg) was started, and a dramatic clinical improvement, with the recovery of arm function and the disappearance of psychotic features and lethargy, was observed after 6 weeks.
In a different approach, a recent meta-analysis examining the association between MTHFR gene polymorphisms and psychiatric disorders demonstrated a strong association between the MTHFR C677T gene variant and unipolar depression, schizophrenia and bipolar disorder, with odds ratios of 1.36, 1.44 and 1.82, respectively [20]. Notably, the metabolic syndrome secondary to antipsychotic medication may be more frequent in patients with reduced MTHFR activity associated with schizophrenia-like psychosis [21].
It is well documented that aberrant methylated compounds are linked to mental state and behavior. A recent review of the ‘one-carbon metabolism hypothesis’ described a range of factors that can contribute to folate and/or vitamin B12 deficiency [22]. Moreover, folic acid is a water-soluble B vitamin involved in the synthesis, repair and methylation of DNA, leading to epigenetic regulation of crucial developmental genes implicated in the pathogenesis of schizophrenia [23, 24]. Deficiency of B vitamins leads to an increased level of homocysteine, which is a highly toxic metabolite to neural and vascular development [25]. Elevated serum levels of homocysteine have also been shown to be associated with schizophrenia, although the evidence is far from conclusive [26, 27].
Urea cycle disorders (UCDs)
Key features
The urea cycle is the metabolic process by which the body eliminates nitrogen. Six enzymes take part in this process; a deficiency of any one of them disrupts this pathway and results in excess nitrogen accumulating in the body in the form of ammonia. The six UCDs include deficiency of: 1) carbamyl phosphate synthetase; 2) n-acetylglutamate synthetase; 3) ornithine transcarbamylase; 4) argininosuccinic acid synthetase (also called citrullinemia); 5) argininosuccinase acid lyase and; 6) arginase. UCD has an estimated incidence of 1:8.000 [28].
If the enzyme deficiency is severe, symptoms will be present at birth and can present as irritability, nausea and vomiting followed by lethargy, seizures and poor muscle tone. If left untreated, patients can develop respiratory distress or fall victim to coma or premature death due to pathological levels of ammonia in the blood.
If the enzyme deficiency is partial, symptom onset may not occur until childhood or adulthood. In such cases, symptoms may include nausea and vomiting associated with headache and a clouded sensorium in the context of infection or a high-protein diet. Medications may worsen or trigger the disease, particularly corticosteroids and sodium valproate.
There is no cure for UCDs, although prompt diagnosis allows measures to be taken that can reduce the consequences of hyperammonemia. Measurement of plasma ammonemia is key to the diagnosis of UCDs, and treatment consisting of a protein-restricted diet and special supplements is essential [29]. In addition, several medications including sodium benzoate, sodium phenylacetate and sodium phenylbutyrate can bind with ammonia and remove it from the circulation. Hemodialysis may represent an alternative treatment, especially in emergency situations [30].
Psychiatric signs
Eight relevant articles were identified in the literature review [31–38]. Arn and colleagues [31] reported a 21-year-old white woman who presented 8 days postpartum with headache and confusion, and became uncommunicative. She was admitted to a psychiatric hospital and diagnosed with postpartum depression. Within 24 hours of admission (11 days postpartum) she became comatose and had generalized tonic–clonic seizures, decorticate posturing, and papilledema. She was treated successfully with hemodialysis and received intravenous sodium benzoate and arginine hydrochloride. Enns and colleagues reported a similar case, suggesting that UCD may present initially with postpartum psychiatric symptoms and may represent an under-recognized cause of ‘postpartum psychosis.’ [32]
Interestingly, late-onset (between 13 and 48 years of age in our review) UCD may present with behavioral and hallucinatory psychiatric and organic signs, often featuring vomiting, which is clearly a key trigger sign for the consideration of UCD in psychiatric situations [35–37]. Patients may have a history of anorexia and atypical depression [35] or psychosis [36], often with associated confusion [37].
Acute porphyria (POR)
Key features
The PORs comprise a group of eight hereditary metabolic diseases characterized by intermittent neurovisceral manifestations, cutaneous lesions, or the combination of both. All porphyrias are caused by a deficiency in one of the enzymes of the heme biosynthesis pathway. These deficiencies result in an accumulation of porphyrins and/or their precursors – delta-aminolevulinic acid (ALA) and porphobilinogen (PBG) – in the liver or bone marrow. Neurological manifestations are caused by the neurotoxic effects of these precursors, particularly ALA. Prevalence is estimated around 0.54:100 000 [39].
Enzyme deficiencies in the porphyrias result from mutations of the correspondingly coded genes, and transmission of hereditary porphyrias occurs in either an autosomal dominant fashion with weak penetrance, or in a recessive manner with complete penetrance.
Clinical signs of disease usually appear in adulthood, although some porphyrias affect children. Porphyrias are classified into two groups – hepatic and erythropoietic – according to the main location of the metabolic anomaly. Chronic hepatic porphyrias and erythropoietic porphyrias manifest with bullous cutaneous lesions or acute pain in areas exposed to the sun, without neurological symptoms. However, neuro-visceral attacks occur in patients with acute hepatic porphyrias, manifesting as intense abdominal pain (often associated with nausea, vomiting and constipation), and neurological and psychological symptoms. Two acute hepatic porphyrias (variegate porphyria and hereditary coproporphyria) may also present with cutaneous photosensitivity.
Diagnosis is mainly based on the measurement of porphyrins and their precursors in biological samples such as urine, stools and blood. The key diagnostic procedure is the measurement of ALA and PBG in urine. Genetic counseling should be offered to affected families to identify individuals susceptible to developing and transmitting the disease. Acute attacks should be treated urgently with an injection of human hemin and/or perfusion of carbohydrates.
Psychiatric signs
Twelve relevant articles were included from the systematic literature review. Psychiatric manifestations are widely known and well documented [40–43], occurring in 24–70% of patients in acute porphyria series [44, 45]. The most common manifestations reported are delirium, psychosis and depression, with some authors suggesting that 40% of acute porphyrias present with delirium and hallucinations [46].
Some cases are spectacular. For example, Santosh and Malhotra reported the case of a 13- year-old boy with six episodes of psychosis with various presentations, including delusions, hallucinations, hypomania and catatonia, but with no obvious organic signs [43]. Crimlisk described three noteworthy cases. The first was that of a 53-year-old woman with a history of cognitive decline since the age of 20, and episodes of visual hallucinations, ataxia, abdominal pain and weight loss; diagnosis was achieved after an acute vomiting episode at 53 years of age. A 52-year-old woman with a history of episodic psychiatric disturbance with associated ideas of reference, auditory hallucinations, emotional lability and abdominal pain was also described, with episodes tending to occur pre-menstrually. The third case was a young boy with a history of generalized pain, vesicular rash, fever and nausea, in whom a diagnosis was made when psychiatric symptoms (paranoid schizophrenia) appeared.
Finally, Mandoki and Summer reported a sub-acute psychiatric dysphoric presentation with emotional lability and aggression, as well as headaches and abdominal pain, in a 9-year-old girl who had a history of aggressive behavior and early anorexia [47]. The patient was diagnosed with coproporphyria – a subtype of porphyria in which psychiatric signs (labile mood and psychosis) have occasionally been described among affected children and young adults [47–51].
Wilson disease (WD)
Key features
WD is an autosomal recessive disorder with a prevalence of 6 per 100 000 of the general population [39]. A mutation in the ATP7B gene coding for a key copper transport protein leads to copper accumulation in the liver, brain, kidney and skeletal system, caused by reduced excretion in the bile [52].
In approximately half of patients, computerized tomography reveals characteristic hypodensities in the basal ganglia [53]. Virtually all patients show magnetic resonance imaging (MRI) abnormalities, including T2-weighted hyperintensities in the thalamus, brainstem and lenticular nuclei [54]. Functional imaging generally shows significant hypometabolism in the lenticular nuclei [55].
Classically, symptoms of WD appear between the ages of 6 and 20 years. Approximately one-third of patients initially present with hepatic disease, one-third with neurological symptoms, and one-third with psychiatric symptomatology. Kayser-Fleischer ring is seen in some patients during ophthalmological examinations. Between one- and two-thirds of patients report psychiatric symptoms at initial presentation [56–58]. Psychiatric signs are present in almost 50% of patients at any one time [59], and present before motor signs in 20% of cases; up to half of patients may be seen initially by a psychiatrist [60].
Psychiatric signs
Eleven relevant case reports or case series (psychiatric or general including psychiatric patients) were identified, and data from most of these were included in a large review [57]. Four main psychiatric symptom clusters have been identified: mood and affective change; behavior and personality change; psychosis and; cognitive impairment [61]. Personality changes are very common, particularly irritability and aggression [56, 62]. Mood disturbance, including both depression and mania, is the most common formal neuropsychiatric illness [63–67]. Psychosis, delusional states and catatonia, while less frequent in WD, can be extremely disabling [56, 57, 60, 61, 68]. Schizophrenia-like symptoms were reported to be present in up to 10% of patients [63], but were less prevalent in one case series (2.4%) [69]. Whilst delusions in WD have been reported to be uncommon [60], a number of psychotic presentations meeting criteria for delusional disorder have recently been described [70–73].
Deteriorating academic performance or work function is another key neurological feature of WD. Neurologically symptomatic patients display a range of cognitive difficulties including impairments of executive function, aspects of memory and visuospatial processing [59, 74, 75]. In contrast, no such deficits are found in neurologically asymptomatic patients [76]. Lesions within the basal ganglia seem to be of central importance in cognitive change due to their interruption of frontal-subcortical circuits [76, 77].
After initiating treatment with chelation therapy, the disease often stabilizes or improves, but disease progression during treatment is more likely for neuropsychiatric symptoms than for hepatic symptoms [78]. Resolution of neuropsychiatric illness following chelation has been reported [67, 73, 79, 80].
The use of neuroleptic medication may be problematic due to the increased risk of movement disorder side effects in the setting of degenerative basal ganglia disease [81–84]. However, some reports suggest the relatively safe use of atypical medications such as olanzapine, risperidone, quetiapine and clozapine, which each have a lower propensity to cause movement disorders [72, 83, 85, 86]. Nevertheless, these agents should be used with caution because of the increased risk of agranulocytosis in the presence of hypersplenism or penicillamine treatment.
Treatment of mania with mood stabilizers can be difficult because valproate or carbamazepine may be contraindicated in the presence of significant hepatic impairment [84]. Lithium may also be contraindicated in the presence of renal tubular acidosis [84], although successful lithium treatment without metabolic compromise has been reported [86, 87].
Electroconvulsive therapy (ECT) has been successfully used in cases of catatonia [88], psychosis [89] and depression [90–92].
Depression has been reported as responding to both tricyclic antidepressants and selective serotonin reuptake inhibitors [91–93], although treatment-resistance to traditional antidepressants has also been described [91]. A manic switch in response to antidepressant therapy has also been described in one patient [93].
Cerebrotendinous xanthomatosis (CTX)
Key features
CTX is an autosomal recessive disease of bile acid synthesis. It is caused by mutations in the CYP27A1 gene, which is localized on the long arm of chromosome 2 and codes for the mitochondrial enzyme, sterol-27-hydroxylase. This enzyme is involved in the synthesis of chenodeoxycholic and cholic acids from cholesterol. The metabolic block resulting from the mutant gene causes a progressive storage of cholesterol and its poorly soluble by-product, cholestanol, which is deposited in many tissues including the brain and tendons [94]. A recent review found more than 300 patients with CTX reported worldwide, and identified 50 different mutations in the CYP27A1 gene associated with this disease [95]. Prevalence is estimated around 2:100 000 [96].
Clinical presentations of CTX are quite variable. The initial symptoms typically begin in childhood with non-specific mild mental retardation, juvenile cataract, chronic diarrhea or epilepsy. Progressive neurological deterioration follows in adolescence or adulthood with acute psychiatric signs [10, 97], progressive spastic paraparesis, cerebellar ataxia, polyneuropathy, epilepsy and cognitive deficits leading to severe handicap or death. These neurological signs can be accompanied by the appearance of tendon xanthomata, which are usually visible at the level of the Achilles’ tendons. An MRI of the brain typically shows a specific pattern with high signals in the dentate nuclei of the cerebellum on T2-weighted sequences [98].
Chenodeoxycholic acid is the primary treatment for CTX. This agent blocks the accumulation of cholestanol by replenishing the pool of bile acid in the liver and hepatic circulation, and shuts down the abnormal hepatic bile acid synthesis pathway. Although it is efficient at normalizing circulating levels of cholestanol, and clearly stabilizes disease progression, it does not improve already existing neurological signs. In addition, xanthomata do not decrease in size.
Psychiatric signs
Fourteen articles were identified in the systematic literature review [10, 97]. Psychiatric manifestations in CTX have only been described in sporadic reports and two patient series [97, 99–111]. Unfortunately, many of these cases are poorly documented and do not contain a systematic psychiatric evaluation.
Acute psychotic episodes have been described, but most psychiatric symptoms are non-specific and occur during childhood and/or adolescence [10, 97]. Hyperactivity is the most common syndrome seen during youth, and is associated with cognitive impairments in speech and comprehension [112].
The Dotti et al. series described 11/13 patients (85%) with psychiatric symptoms [Note: please confirm that 11 patients out of the 13 studied had psychiatric symptoms, as queried]: five with behavioural changes, four with psychosis and two with depression [107], suggesting an over-representation of psychiatric disorders in this population. This contrasts with the documented rarity of psychiatric signs in CTX (around 10%). In the only small series specifically focusing on the psychiatric spectrum of CTX, Berginer et al. reported four patients with disparate psychiatric syndromes, including irritability and personality changes with hypersexuality, atypical psychosis and paranoid delusions, and severe catatonia [97]. Diagnoses of CTX were made on the basis of pes cavus and Achilles xanthomata in all patients, and caratacts and cognitive impairment in two cases.
Two siblings were recently described with an early psychiatric presentation comprising attention deficit hyperactivity disorder (ADHD) and oppositional defiant disorder (ODD) associated with mild intellectual disability [112]. In both patients, treatment with chenodeoxycholic acid improved externalizing symptoms, and a partial recovery of cognitive impairment was observed.
Niemann Pick disease type C (NP-C)
Key features
NP-C is a pan-ethnic, autosomal recessive neurodegenerative disease with an incidence estimated between 1 case per 150 000 and 1 case per 120 000 live births [113, 114]. The disease is characterized by a variety of progressive, disabling neurological symptoms including clumsiness, limb and gait ataxia, dysarthria, dysphagia and cognitive deterioration [113, 115].
NP-C is associated with mutations of the NPC1 and NPC2 genes, with no primary defect in catabolic enzymes. NPC1 gene mutations are present in 95% of cases and NPC2 mutations are present in approximately 4%. At the cellular level, these mutations give rise to characteristic abnormalities in the intracellular transport of cholesterol, glycosphingolipids and sphingosine. Impaired function of the NPC1 and NPC2 gene products, which normally function cooperatively in intracellular lipid transport, leads to the accumulation of these lipids in the late-endosomal/lysosomal intracellular compartment, and excess build up in various tissues. Unesterified cholesterol, sphingomyelin, bis(monoacylglycero)-phosphate, glycosphingolipids and sphingosine are stored in excess in the liver and spleen, while levels of glucosylceramide, lactosylceramide and, above all, GM2 and GM3 gangliosides are markedly increased in the brain [116].
NP-C has an extremely heterogeneous clinical presentation characterized by a wide range of symptoms that are not specific to the disease, and which arise and progress over varied periods of time [113, 117]. This complicates diagnosis, and is likely an important factor in the under-detection of the NP-C and, in some cases, its misdiagnosis. In the first decade of life, the most common presentations are neurological, although early-onset patients are often diagnosed based on isolated systemic manifestations (e.g. neonatal jaundice, splenomegaly). Many cases are also diagnosed in adulthood, sometimes even up to the seventh decade of life [118].
The age at onset of neurological symptoms has a major influence on disease progression; [119] if neurological symptoms arise early in life the rate of deterioration is generally faster and premature death occurs sooner. Patients with the perinatal-onset form present during the first 3 months of life with an enlarged liver and spleen, prolonged cholestasis, hydrops fetalis and/or respiratory failure [113, 114], usually without presenting neurological signs. Infantile, juvenile and adolescent/adult forms usually present with neurological signs including progressive ataxia, dystonia, dysarthria, dysphagia, deafness, cataplexy or, more rarely, epilepsy. Most notably, vertical supranuclear gaze palsy (VSGP) – particularly paresis of downgaze – is a highly specific and highly prevalent sign that may be present at an early stage of the disease [113, 115]. VSGP or discrete slowing of saccades is present in almost all cases at some point during the disease course.
Diagnosis of NP-C requires a skin biopsy and a fibroblast culture in a specialized center, with the resulting cultured cells stained with filipin (which binds excess cholesterol) and tested for cholesterol esterification. However, data suggest that plasma oxysterol measurements may represent a simpler screening and/or diagnostic method in the coming years [120].
Therapy for NP-C has, until recently, been limited to supportive measures, including pharmacotherapy to alleviate neurological and psychiatric symptoms [113, 115, 121]. Miglustat, an iminosugar compound that reversibly inhibits glucosyl ceramide synthetase and thus inhibits the formation of excess gangliosides, is a substrate-reduction therapy that has been shown to stabilize neurological manifestations in children and adults [122–124].
Psychiatric signs
Numerous cases of NP-C presenting with schizophrenia-like symptoms have been reported in adolescent and adult patients, and nine case series and reports were identified and included in this literature analysis [125–133]. Definitive diagnoses are commonly delayed in patients with adult psychiatric presentations of NP-C, sometimes by up to 10 years [134].
Psychotic presentations among children and adolescents with NP-C have been reported, and may be comorbid with a pervasive developmental disorder (PDD). Sandu et al. reported a case of an 8-year-old with PDD who presented with auditory hallucinations and 7 years later developed a typical paranoid schizophrenic illness that was partially responsive to risperidone [132]. One notable report described two siblings with psychosis [131]. The male sibling presented at 16 years old with visual and auditory hallucinations, and later developed dysarthria and ataxia leading to a definitive diagnosis at the age of 24 years when vertical supranuclear ophthalmoplegia was discovered. His sister developed schizophrenia-like symptoms a decade later, but diagnosis was made rapidly when she was examined for vertical supranuclear ophthalmoplegia based on the family history. Notably, her later onset and lower antipsychotic dosage required to effect symptom resolution mirrored the gender dimorphism seen in typical schizophrenia, which raises the possibility of a gender effect in presentation and progression of NP-C [131].
The onset of developmental delay is commonly seen between 6 and 15 years of age in NP-C, and may result in a learning disorder and/or impaired school performance [117, 135, 136]. Patients commonly display cognitive impairments involving logical thinking and abstraction, impaired attentional processes, poor working memory, word retrieval difficulties, and a lack of interpersonal ‘distance’ [121, 136]. The typical cognitive profile in adult patients is one of significant executive dysfunction and impaired working memory [115, 136].
Discussion
This article is the first systematic review in this field. Widely unknown and neglected by psychiatrists, IEMs represent a growing field in research that interfaces with clinical psychiatry due to the fact that a number of disorders may initially present to psychiatrists. New treatments are available for a number of these diseases [137].
One key finding of this literature review is that the clinical signs of IEMs are poorly documented in terms of both quantity (only 63 articles with original data for seven different disorders) and quality (non-systematic clinical evaluations, lack of standardized clinical scales, sparse clinical description). A second clear finding is that many cases of WD, MTHFR-D and NP-C are strongly associated with psychotic illness.
It is not realistic, and probably unnecessary due to the rarity of the association, to consider IEMs in all psychiatric patients. It is also not feasible to train psychiatrists to become metabolic specialists. However, it is crucial for all professionals working in psychiatry to be aware of the large variety of organic disorders that may be associated with psychiatric diseases, particularly treatable IEMs such as those addressed in this review, and to be aware of clinical features that may herald an underpinning organic disorder for the patient’s psychiatric presentation.
One major difficulty in considering the association of IEMs with psychosis is in ascribing causality. If prevalence rates across a number of populations are higher than 0.8–1.0% (the general population prevalence for schizophrenia) [138, 139] it may be difficult to consider an association with IEMs as significant as their prevalence is more likely around 1 in 10 000. In addition, when treatment of an organic disorder leads to an improvement in psychotic symptoms, this association is strengthened, and the Bradford-Hill criteria of strength, consistency and temporality are met [140]. These associations may also aid in shedding light on the potential neurobiological origins of schizophrenia. There is a wide consensus regarding the neurodevelopmental hypothesis of schizophrenia [141, 142], as well as the role of complex genetic determinism and gene–environment interactions [143]. The historical association of a range of organic disorders with schizophrenia-like psychosis has shed light on the role that the medial temporal lobe and diencephalon play in the origin of psychotic symptoms [2], and similarly the recent advancement in our understanding of the neurobiology of various IEMs has shed light on the role of anatomical disconnection and disruption to a range of neurotransmitter systems in the genesis of psychotic illness [130].
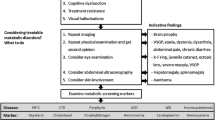
The second major difficulty is to recognize IEMs and to think about an organic etiology in clinical practice. In order to help psychiatrists, it could be clinically useful to identify psychiatric features that may trigger for the search of organic disorder in patients with schizophrenia. Unfortunately, data regarding organic psychosis and its specific associated symptoms are scarce. One study, which was not specific for IEMs, analyzed the phenomenology of 74 patients with ‘organic schizophrenia’ compared with ‘non-organic schizophrenia’ [144]. Visual hallucinations and confusion were seen more often among patients with organic schizophrenia, and comparable features have been observed in elderly schizophrenia patients [145, 146]. A handful of inborn errors of metabolism may cause elementary hallucination and visual hallucinations which are associated with various organic and psychiatric conditions [147]. Hallucinations are a core symptom of schizophrenia and are more often auditory or at least, auditory hallucinations are more important than visual hallucinations. We suggest therefore that predominant visual hallucinations are highly suggestive of organic disorders such as IEM. An acute onset of psychiatric symptoms may also raise suspicion of IEMs (e.g. UCDs, porphyria or homocysteinemia with CbS-D). It is also notable that data indicate a high degree of association of catatonia with organic disorders, especially if it occurs during childhood or adolescence [148]. An unusually high proportion of patients with organic disorders has been reported in large series of patients with early-onset schizophrenia, which suggests that an early-onset of schizophrenia-like symptoms is another indicator for possible organic origin of disease, especially if associated with progressive cognitive decline, which is a common feature in IEMs [149]. Finally, treatment resistance is frequently associated with IEMs [150], again suggesting its possible use as an indicator for possible organic disease. In summary, we may suggest six readily recognizable features that should trigger the suspicion of organicity associated with schizophrenia-like symptoms: 1) acute confusion; 2) visual hallucinations more important than auditory hallucinations; 3) catatonia; 4) progressive cognitive decline; 5) early or acute onset and; 6) treatment resistance (see Table 1). As the validity and specificity of these atypical psychiatric signs have not yet been evaluated, they are presented to raise awareness and suggest clinical and neurological exams prior to further progressive screening. Both atypical psychiatric signs and main clinical/biomarker features of IEM lead us to propose an algorithm (see Figure 2). This algorithm is based on clinical practice of OB, HK and MW and DC. We plan to study his validity and reliance in further study in population of patients with psychiatric signs and IEM. Further research is needed to develop a real suspicion index from our group of atypical psychiatric signs associated with this algorithm.

Diagnostic algorithm for diagnosing inborn errors of metabolism in patients with schizophrenia-like symptoms. Negative: If exams are negative and suspicion is high. Positive: Could lead to diagnoses or high suspicion of specific disease. MRI = magnetic resonance imaging; MTHFR-CbS = methylenetetrahydrofolate reductase-cystathionine beta-synthase; NP-C = Niemann-Pick disease type C; UCDs = urea cycle disorders, WD = Wilson disease.
Biological screening is not recommended for all patients but only in case of high suspicion. It is also important to know that the cost of screening for most of these disorders in standard metabolic reference laboratories is usually modest, generally consisting of serum or urine tests for various metabolites, comparable or less than the cost of basic neuroimaging. The costs of treating these IEMs varies widely; from B-vitamin replacement and/or dietary modification in a number of disorders, to more expensive long-term treatments in disorders such as NPC, and potential high-cost hospitalizations during metabolic crises in UCDs and porphyria. This should however be referenced against the cost of treating true schizophrenia, at a cost of at least $USD 50000 per year [151], with at least one third of this borne in direct treatment.
Toward atypical psychiatric signs, IEMs presents specificities and Table 2 provides a summary that encompasses the main clinical signs of six treatable IEMs, and highlights the main clinical symptoms, biomarkers and context as well as ophthalmologic signs, which occur in at least four of the treatable IEMs.
It is important for psychiatrists and other associated professionals to be specifically aware of potential IEMs when patients present with such indicators (organic signs and atypical psychiatric symptoms) of possible organic psychosis.
Conclusions
Based on published evidence this review highlights the role of a range of IEMs as possible underlying organic causes of schizophrenia or schizophrenia-like syndromes. It is important to identify such cases as some IEMs are treatable (sometimes simply with vitamin replacement or supplementation) and new treatments continue to appear. Clinical studies suggest that some IEM-specific treatments may be most effective during the early stages of disease when psychiatric symptoms may be evident. Efficient recognition and identification of the underlying organic disease could therefore allow earlier initiation of specific therapy and, possibly, improve outcomes.Although the literature base from which to draw conclusions is limited, clinicians managing patients presenting with new-onset psychosis should pay particular attention to IEMs as a possible underlying cause in patients with atypical symptoms, and in the presence of specific clinical contexts. It is hoped that this review summarizing six easily assessed features that might trigger suspicion of organicity in patients with psychosis will help to detect patients with treatable IEMs as early as possible during their disease course. While not intended to replace specialized psychiatric or neurological examination and measurements, our proposed algorithm (Figure 2) is a pragmatic tool that can be used to reduce the risk of mistaken diagnoses among patients with atypical psychiatric signs and treatable IEMs.
References
Price G, Ron MA: Schizophrenia and schizophrenia-like psychosis. Neurology and Clinical Neuroscience. Edited by: Schapira AHV. 2003, St Louis: Elsevier-Mosby, 223-233. 1
Davison K, Bagley CR: Schizophrenia-like psychoses associated with organic cerebral disorders: a review. Psychiatr Dev. 1983, 1 (1): 1-33.
Johnstone EC, Macmillan JF, Crow TJ: The occurrence of organic disease of possible or probable aetiological significance in a population of 268 cases of first episode schizophrenia. Psychol Med. 1987, 17 (2): 371-379.
Lauterbach MD, Stanislawski-Zygaj AL, Benjamin S: The differential diagnosis of childhood and young adult onset disorders that include psychosis. J Neuropsychiatry Clin Neurosci. 2008, 20 (4): 409-418.
Saudubray JM: Neurometabolic disorders. J Inherit Metab Dis. 2009, 32 (5): 595-596.
Applegarth DA, Toone JR, Lowry RB: Incidence of inborn errors of metabolism in British Columbia, 1969–1996. Pediatrics. 2000, 105 (1): e10-
Sedel F, Baumann N, Turpin JC, Lyon-Caen O, Saudubray JM, Cohen D: Psychiatric manifestations revealing inborn errors of metabolism in adolescents and adults. J Inherit Metab Dis. 2007, 30 (5): 631-641.
Wilson WP, Nashold BS: Psychiatric considerations of certain neurological diseases treated neurosurgically. Int Psychiatry Clin. 1967, 4 (2): 189-204.
Bonnot O, Cohen D: [Psychiatric and cognitive signs associated with inborn errors of metabolism]. Rev Neurol. 2011, 167 (12): 881-885.
Estrov Y, Scaglia F, Bodamer OA: Psychiatric symptoms of inherited metabolic disease. J Inherit Metab Dis. 2000, 23 (1): 2-6.
Martins AM: Inborn errors of metabolism: a clinical overview. Sao Paulo medical journal =. Rev Paul Med. 1999, 117 (6): 251-265.
Trifiletti RR, Packard AM: Metabolic disorders presenting with behavioral symptoms in the school-aged child. Child Adolesc Psychiatr Clin N Am. 1999, 8 (4): 791-806.
KEGG: Reference Metabolic Pathways. 2013, [cited 2014 January]; Available from: http://www.genome.jp/kegg/pathway.html
Yap S: 2007, [cited January, 2014]; Available from: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=FR&Expert=394
Abbott MH, Folstein SE, Abbey H, Pyeritz RE: Psychiatric manifestations of homocystinuria due to cystathionine beta-synthase deficiency: prevalence, natural history, and relationship to neurologic impairment and vitamin B6-responsiveness. Am J Med Genet. 1987, 26 (4): 959-969.
Li SC, Stewart PM: Homocystinuria and psychiatric disorder: a case report. Pathology. 1999, 31 (3): 221-224.
Baric I: Inherited disorders in the conversion of methionine to homocysteine. J Inherit Metab Dis. 2009, 32 (4): 459-471.
Mattson MP, Shea TB: Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 2003, 26 (3): 137-146.
Roze E, Gervais D, Demeret S, Ogier de Baulny H, Zittoun J, Benoist JF, Said G, Pierrot-Deseilligny C, Bolgert F: Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch Neurol. 2003, 60 (10): 1457-1462.
Gilbody S, Lewis S, Lightfoot T: Methylenetetrahydrofolate reductase (MTHFR) genetic polymorphisms and psychiatric disorders: a HuGE review. Am J Epidemiol. 2007, 165 (1): 1-13.
Ellingrod VL, Miller DD, Taylor SF, Moline J, Holman T, Kerr J: Metabolic syndrome and insulin resistance in schizophrenia patients receiving antipsychotics genotyped for the methylenetetrahydrofolate reductase (MTHFR) 677C/T and 1298A/C variants. Schizophr Res. 2008, 98 (1–3): 47-54.
Frankenburg FR: The role of one-carbon metabolism in schizophrenia and depression. Harv Rev Psychiatry. 2007, 15 (4): 146-160.
Friso S, Choi SW: Gene-nutrient interactions in one-carbon metabolism. Curr Drug Metab. 2005, 6 (1): 37-46.
Sharma RP: Schizophrenia, epigenetics and ligand-activated nuclear receptors: a framework for chromatin therapeutics. Schizophr Res. 2005, 72 (2–3): 79-90.
Regland B: Schizophrenia and single-carbon metabolism. Prog Neuro-Psychopharmacol Biol Psychiatry. 2005, 29 (7): 1124-1132.
O'Donnell C, Stephens T: The significance of homocysteine levels in schizophrenia. Am J Psychiatry. 2005, 162 (7): 1387-1388. author reply 1388–1389
Muntjewerff JW, Kahn RS, Blom HJ, den Heijer M: Homocysteine, methylenetetrahydrofolate reductase and risk of schizophrenia: a meta-analysis. Mol Psychiatry. 2006, 11 (2): 143-149.
Haberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, Karall D, Martinelli D, Crespo PS, Santer R, Servais A, Valayannopoulos V, Lindner M, Rubio V, Dionisi-Vici C: Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012, 7: 32-
Singh RH: Nutritional management of patients with urea cycle disorders. J Inherit Metab Dis. 2007, 30 (6): 880-887.
Smith W, Kishnani PS, Lee B, Singh RH, Rhead WJ, Sniderman King L, Smith M, Summar M: Urea cycle disorders: clinical presentation outside the newborn period. Crit Care Clin. 2005, 21 (4 Suppl): S9-17.
Arn PH, Hauser ER, Thomas GH, Herman G, Hess D, Brusilow SW: Hyperammonemia in women with a mutation at the ornithine carbamoyltransferase locus. A cause of postpartum coma. N Engl J Med. 1990, 322 (23): 1652-1655.
Enns GM, O'Brien WE, Kobayashi K, Shinzawa H, Pellegrino JE: Postpartum “psychosis” in mild argininosuccinate synthetase deficiency. Obstet Gynecol. 2005, 105 (5 Pt 2): 1244-1246.
Bachmann C: Outcome and survival of 88 patients with urea cycle disorders: a retrospective evaluation. Eur J Pediatr. 2003, 162 (6): 410-416.
Krivitzky L, Babikian T, Lee HS, Thomas NH, Burk-Paull KL, Batshaw ML: Intellectual, adaptive, and behavioral functioning in children with urea cycle disorders. Pediatr Res. 2009, 66 (1): 96-101.
Legras A, Labarthe F, Maillot F, Garrigue MA, Kouatchet A, Ogier de Baulny H: Late diagnosis of ornithine transcarbamylase defect in three related female patients: polymorphic presentations. Crit Care Med. 2002, 30 (1): 241-244.
Myers JH, Shook JE: Vomiting, ataxia, and altered mental status in an adolescent: late-onset ornithine transcarbamylase deficiency. Am J Emerg Med. 1996, 14 (6): 553-557.
Panlaqui OM, Tran K, Johns A, McGill J, White H: Acute hyperammonemic encephalopathy in adult onset ornithine transcarbamylase deficiency. Intensive Care Med. 2008, 34 (10): 1922-1924.
Thurlow VR, Asafu-Adjaye M, Agalou S, Rahman Y: Fatal ammonia toxicity in an adult due to an undiagnosed urea cycle defect: under-recognition of ornithine transcarbamylase deficiency. Ann Clin Biochem. 2010, 47 (Pt 3): 279-281.
Orphanet: Prevalence of rare diseases by alphabetical list. 2013, [cited January, 2014]; Available from: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf
Cashman MD: Psychiatric aspects of acute porphyria. Lancet. 1961, 1 (7168): 115-116.
Tishler PV, Woodward B, O'Connor J, Holbrook DA, Seidman LJ, Hallett M, Knighton DJ: High prevalence of intermittent acute porphyria in a psychiatric patient population. Am J Psychiatry. 1985, 142 (12): 1430-1436.
Boon FF, Ellis C: Acute intermittent porphyria in a children’s psychiatric hospital. J Am Acad Child Adolesc Psychiatry. 1989, 28 (4): 606-609.
Santosh PJ, Malhotra S: Varied psychiatric manifestations of acute intermittent porphyria. Biol Psychiatry. 1994, 36 (11): 744-747.
Goldberg A: Acute intermittent porphyria: a study of 50 cases. Q J Med. 1959, 28 (110): 183-209.
Stein JA, Tschudy DP: Acute intermittent porphyria. A clinical and biochemical study of 46 patients. Medicine. 1970, 49 (1): 1-16.
Bonkowsky HL, Schady W: Neurologic manifestations of acute porphyria. Semin Liver Dis. 1982, 2 (2): 108-124.
Mandoki MW, Sumner GS: Psychiatric manifestations of hereditary coproporphyria in a child. J Nerv Ment Dis. 1994, 182 (2): 117-118.
Brodie MJ, Thompson GG, Moore MR, Beattie AD, Goldberg A: Hereditary coproporphyria. Demonstration of the abnormalities in haem biosynthesis in peripheral blood. Q J Med. 1977, 46 (182): 229-241.
Crimlisk HL: The little imitator–porphyria: a neuropsychiatric disorder. J Neurol Neurosurg Psychiatry. 1997, 62 (4): 319-328.
Kuhnel A, Gross U, Doss MO: Hereditary coproporphyria in Germany: clinical-biochemical studies in 53 patients. Clin Biochem. 2000, 33 (6): 465-473.
Gross U, Puy H, Meissauer U, Lamoril J, Deybach JC, Doss M, Nordmann Y, Doss MO: A molecular, enzymatic and clinical study in a family with hereditary coproporphyria. J Inherit Metab Dis. 2002, 25 (4): 279-286.
Pfeffer RF: Wilson’s disease. Semin Neurol. 2007, 27 (2): 123-132.
Williams J, Walshe J: Wilson’s disease: an analysis of the cranial computerized tomographic appearances found in 60 patients and the changes in response to treatment with chelating agents. Brain. 1981, 104: 735-752.
Roh JK, Lee TG, Wie BA, Lee SB, Park SH, Chang KH: Initial and follow-up brain MRI findings and correlation with the clinical course in Wilson’s disease. Neurology. 1994, 44 (6): 1064-1068.
Hawkins RA, Mazziotta JC, Phelps ME: Wilson’s disease studied with FDG and positron emission tomography. Neurology. 1987, 37 (11): 1707-1711.
Barthel W, Markwardt F: Aggregation of blood platelets by adrenaline and its uptake. Biochem Pharmacol. 1975, 24 (20): 1903-1904.
Dening TR: The neuropsychiatry of Wilson’s disease: a review. Int J Psychiatry Med. 1991, 21 (2): 135-148.
Schwartz M, Fuchs S, Polak H, Sharf B: [Psychiatric manifestations in Wilson’s disease]. Harefuah. 1993, 124 (2): 75-77. 120
Rathbun JK: Neuropsychological aspects of Wilson’s disease. Int J Neurosci. 1996, 85 (3–4): 221-229.
Dening TR, Berrios GE: Wilson’s disease. Psychiatric symptoms in 195 cases. Arch Gen Psychiatry. 1989, 46 (12): 1126-1134.
Dening TR: Psychiatric aspects of Wilson’s disease. Br J Psychiatry. 1985, 147: 677-682.
Coscia L, Causa P, Giuliani E, Nunziata A: Pharmacological properties of new neuroleptic compounds. Arzneimittelforschung. 1975, 25 (9): 1436-1442.
Akil M, Brewer GJ: Psychiatric and behavioral abnormalities in Wilson’s disease. Adv Neurol. 1995, 65: 171-178.
Akil M, Schwartz JA, Dutchak D, Yuzbasiyan-Gurkan V, Brewer GJ: The psychiatric presentations of Wilson’s disease. J Neuropsychiatry Clin Neurosci. 1991, 3 (4): 377-382.
Dening TR, Berrios GE: Wilson’s disease: a longitudinal study of psychiatric symptoms. Biol Psychiatry. 1990, 28 (3): 255-265.
Medalia A, Scheinberg IH: Psychopathology in patients with Wilson’s disease. Am J Psychiatry. 1989, 146 (5): 662-664.
Srinivas K, Sinha S, Taly AB, Prashanth LK, Arunodaya GR, Janardhana Reddy YC, Khanna S: Dominant psychiatric manifestations in Wilson’s disease: a diagnostic and therapeutic challenge!. J Neurol Sci. 2008, 266 (1–2): 104-108.
Renaud B, Buda M, Lewis BD, Pujol JF: Effects of 5,6-dihydroxytryptamine on tyrosine-hydroxylase activity in central catecholaminergic neurons of the rat. Biochem Pharmacol. 1975, 24 (18): 1739-1742.
Taly AB, Meenakshi-Sundaram S, Sinha S, Swamy HS, Arunodaya GR: Wilson disease: description of 282 patients evaluated over 3 decades. Medicine. 2007, 86 (2): 112-121.
Sagawa M, Takao M, Nogawa S, Mizuno M, Murata M, Amano T, Koto A: [Wilson’s disease associated with olfactory paranoid syndrome and idiopathic thrombocytopenic purpura]. No to shinkei = Brain and nerve. 2003, 55 (10): 899-902.
Wichowicz H, Cubala W, Slawek J: Wilson’s disease asociated with delusional disorder. Psychiatry Clin Neurosci. 2006, 60: 758-760.
Spyridi S, Diakogiannis I, Michaelides M, Sokolaki S, Iacovides A, Kaprinis G: Delusional disorder and alcohol abuse in a patient with Wilson’s disease. Gen Hosp Psychiatry. 2008, 30 (6): 585-586.
Stiller P, Kassubek J, Schonfeldt-Leucona C, Connemann B: Wilson’s disease in psychiatric patients. Psychiatry Clin Neurosci. 2008, 56: 649-
Medalia A, Isaacs-Glaberman K, Scheinberg IH: Neuropsychological impairment in Wilson’s disease. Arch Neurol. 1988, 45 (5): 502-504.
Isaacs-Glaberman K, Medalia A, Scheinberg IH: Verbal recall and recognition abilities in patients with Wilson’s disease. Cortex. 1989, 25 (3): 353-361.
Ardenne M, Reitnauer PG: Demonstration of tumor inhibiting properties of a strongly immunostimulating low-molecular weight substance. Comparative studies with ifosfamide on the immuno-labile DS carcinosarcoma. Stimulation of the autoimmune activity for approx. 20 days by BA 1, a N-(2-cyanoethylene)-urea. Novel prophylactic possibilities. Arzneimittelforschung. 1975, 25 (9): 1369-1379.
Kroger H, Donner I, Skiello G: Influence of a new virostatic compound on the induction of enzymes in rat liver. Arzneimittelforschung. 1975, 25 (9): 1426-1429.
Smith RJ, Bryant RG: Metal substitutions incarbonic anhydrase: a halide ion probe study. Biochem Biophys Res Commun. 1975, 66 (4): 1281-1286.
Walter G, Lyndon B: Depression in hepatolenticular degeneration (Wilson’s disease). Aust N Z J Psychiatry. 1997, 31 (6): 880-882.
Machado A, Deguti M, Caixeta L, Spitz M, Lucato L, Barbosa E: Mania as the first manifestation of Wilson’s disease. Bipolar Disord. 2008, 10: 447-450.
Tu J: The inadvisability of neuroleptic medication in Wilson’s disease. Biol Psychiatry. 1981, 16 (10): 963-968.
Hoogenraad T: Wilson’s Disease. 1996, London: Saunders
Chroni E, Lekka NP, Tsibri E, Economou A, Paschalis C: Acute, progressive akinetic-rigid syndrome induced by neuroleptics in a case of Wilson’s disease. J Neuropsychiatry Clin Neurosci. 2001, 13 (4): 531-532.
Varghese ST, Narayanan D, Dinesh D: Mania in a patient with Wilson’s disease awaiting liver transplant. J Neuropsychiatry Clin Neurosci. 2008, 20 (4): 501-502.
Krim E, Barroso B: [Psychiatric disorders treated with clozapine in a patient with Wilson’s disease]. Presse Med. 2001, 30 (15): 738-
Kulaksizoglu IB, Polat A: Quetiapine for mania with Wilson’s disease. Psychosomatics. 2003, 44 (5): 438-439.
Longanathan S, Nayak R, Sinha S, Taly A, Math S, Varghese M: Treating mania in Wilson’s disease with lithium. J Neuropsychiatry Clin Neurosci. 2008, 20: 487-489.
Rodrigues AC, Dalgalarrondo P, Banzato CE: Successful ECT in a patient with a psychiatric presentation of Wilson’s disease. J ECT. 2004, 20 (1): 55-
Shah N, Kumar D: Wilson’s disease, psychosis, and ECT. Convuls Ther. 1997, 13 (4): 278-279.
Negro P, Louza Neto M: Results of ECT for a case of depression in Wilson disease. J Neuropsychiatry Clin Neurosci. 1995, 7: 384-
Sechi G, Antonio Cocco G, Errigo A, Deiana L, Rosati G, Agnetti V, Stephen Paulus K, Mario Pes G: Three sisters with very-late-onset major depression and parkinsonism. Parkinsonism Relat Disord. 2007, 13 (2): 122-125.
Chan K, Cheung R, Au-Yeung K, Mak W, Cheng T, Ho S: Wilson’s disease with depression and parkinsonism. J Clin Neurosci. 2004, 12: 303-305.
Keller R, Torta R, Lagget M, Crasto S, Bergamasco B: Psychiatric symptoms as late onset of Wilson’s disease: neuroradiological findings, clinical features and treatment. Ital J Neurol Sci. 1999, 20: 49-54.
Moghadasian MH, Salen G, Frohlich JJ, Scudamore CH: Cerebrotendinous xanthomatosis: a rare disease with diverse manifestations. Arch Neurol. 2002, 59 (4): 527-529.
Gallus GN, Dotti MT, Federico A: Clinical and molecular diagnosis of cerebrotendinous xanthomatosis with a review of the mutations in the CYP27A1 gene. Neurol Sci. 2006, 27 (2): 143-149.
Orphanet: Prevalence of Rare Diseases. 2013, [cited 2014 January]; Available from: http://www.orpha.net/orphacom/cahiers/docs/FR/Prevalence_des_maladies_rares_par_prevalence_decroissante_ou_cas.pdf
Berginer VM, Foster NL, Sadowsky M, Townsend JA, Siegel GJ, Salen G: Psychiatric disorders in patients with cerebrotendinous xanthomatosis. Am J Psychiatry. 1988, 145 (3): 354-357.
Barkhof F, Verrips A, Wesseling P, van Der Knaap MS, van Engelen BG, Gabreels FJ, Keyser A, Wevers RA, Valk J: Cerebrotendinous xanthomatosis: the spectrum of imaging findings and the correlation with neuropathologic findings. Radiology. 2000, 217 (3): 869-876.
Philippart M, Van Bogaert L: Cholestanolosis (cerebrotendinous xanthomatosis). A follow-up study on the original family. Arch Neurol. 1969, 21 (6): 603-610.
Shapiro S: Depression in a patient with dementia secondary to cerebrotendinous xanthomatosis. J Nerv Ment Dis. 1983, 171 (9): 568-571.
Burnstein M, Buckwalter KA, Martel W, McClatchey KD, Quint D: Case report 427: Cerebrotendinous xanthomatosis. Skelet Radiol. 1987, 16 (4): 346-349.
Laurent A, Dairou F, Luc G, Truffert J, Lapresle J, de Gennes JL: Van Bogaert’s cerebrotendinous xanthomatosis. A study of 3 cases. Ann Med Interne. 1988, 139 (6): 395-402.
Wevers RA, Cruysberg JR, Van Heijst AF, Janssen-Zijlstra FS, Renier WO, Van Engelen BG, Tolboom JJ: Paediatric cerebrotendinous xanthomatosis. J Inherit Metab Dis. 1992, 15 (3): 374-376.
Soffer D, Benharroch D, Berginer V: The neuropathology of cerebrotendinous xanthomatosis revisited: a case report and review of the literature. Acta Neuropathol. 1995, 90 (2): 213-220.
Verrips A, Steenbergen-Spanjers GC, Luyten JA, van den Heuvel LP, Keyser A, Gabreels FJ, Wevers RA: Two new mutations in the sterol 27-hydroxylase gene in two families lead to cerebrotendinous xanthomatosis. Hum Genet. 1996, 98 (6): 735-737.
Sperhake JP, Matschke J, Orth U, Gal A, Puschel K: Sudden death due to cerebrotendinous xanthomatosis confirmed by mutation analysis. Int J Legal Med. 2000, 113 (2): 110-113.
Dotti MT, Rufa A, Federico A: Cerebrotendinous xanthomatosis: heterogeneity of clinical phenotype with evidence of previously undescribed ophthalmological findings. J Inherit Metab Dis. 2001, 24 (7): 696-706.
Lee Y, Lin PY, Chiu NM, Chang WN, Wen JK: Cerebrotendinous xanthomatosis with psychiatric disorders: report of three siblings and literature review. Chang Gung Med J. 2002, 25 (5): 334-340.
Guyant-Marechal L, Verrips A, Girard C, Wevers RA, Zijlstra F, Sistermans E, Vera P, Campion D, Hannequin D: Unusual cerebrotendinous xanthomatosis with fronto-temporal dementia phenotype. Am J Med Genet A. 2005, 139A (2): 114-117.
Price Evans DA, Salah KA, Mobrad MA, Mitchell WD, Olin M, Eggertsen G: Cerebrotendinous xanthomatosis in a Saudi Arabian family-genotyping and long-term follow-up. Saudi Med J. 2007, 28 (7): 1113-1118.
Gonzalez-Cuyar LF, Hunter B, Harris PL, Perry G, Smith MA, Castellani RJ: Cerebrotendinous xanthomatosis: case report with evidence of oxidative stress. Redox Rep. 2007, 12 (3): 119-124.
Bonnot O, Fraidakis MJ, Lucanto R, Chauvin D, Kelley N, Plaza M, Dubourg O, Lyon-Caen O, Sedel F, Cohen D: Cerebrotendinous xanthomatosis presenting with severe externalized disorder: improvement after one year of treatment with chenodeoxycholic Acid. CNS Spectr. 2010, 15 (4): 231-236.
Wraith JE, Baumgartner MR, Bembi B, Covanis A, Levade T, Mengel E, Pineda M, Sedel F, Topcu M, Vanier MT, Widner H, Wijburg FA, Patterson MC, Group N-CGW: Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol Genet Metab. 2009, 98 (1–2): 152-165.
Vanier MT: Niemann-Pick disease type C. Orphanet J Rare Dis. 2010, 5: 16-
Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F, on behalf of the NPCGWG: Recommendations for the diagnosis and management of Niemann-Pick disease type C: An update. Mol Genet Metab. 2012, 106 (3): 330-344.
Vanier MT: Lipid changes in Niemann-Pick disease type C brain: personal experience and review of the literature. Neurochem Res. 1999, 24 (4): 481-489.
Sevin M, Lesca G, Baumann N, Millat G, Lyon-Caen O, Vanier MT, Sedel F: The adult form of Niemann-Pick disease type C. Brain. 2007, 130 (Pt 1): 120-133.
Trendelenburg G, Vanier MT, Maza S, Millat G, Bohner G, Munz DL, Zschenderlein R: Niemann-Pick type C disease in a 68-year-old patient. J Neurol Neurosurg Psychiatry. 2006, 77 (8): 997-998.
Wraith JE, Guffon N, Rohrbach M, Hwu WL, Korenke GC, Bembi B, Luzy C, Giorgino R, Sedel F: Natural history of Niemann-Pick disease type C in a multicentre observational retrospective cohort study. Mol Genet Metab. 2009, 98 (3): 250-254.
Porter FD, Scherrer DE, Lanier MH, Langmade SJ, Molugu V, Gale SE, Olzeski D, Sidhu R, Dietzen DJ, Fu R, Wassif CA, Yanjanin NM, Marso SP, House J, Vite C, Schaffer JE, Ory DS: Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci Transl Med. 2010, 2 (56): 56ra81-
Patterson MC, Platt F: Therapy of Niemann-Pick disease, type C. Biochim Biophys Acta. 2004, 1685 (1–3): 77-82.
Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE: Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 2007, 6 (9): 765-772.
Wraith JE, Vecchio D, Jacklin E, Abel L, Chadha-Boreham H, Luzy C, Giorgino R, Patterson MC: Miglustat in adult and juvenile patients with Niemann-Pick disease type C: long-term data from a clinical trial. Mol Genet Metab. 2010, 99 (4): 351-357.
Patterson MC, Vecchio D, Jacklin E, Abel L, Chadha-Boreham H, Luzy C, Giorgino R, Wraith JE: Long-term miglustat therapy in children with Niemann-Pick disease type C. J Child Neurol. 2010, 25 (3): 300-305.
Shulman LM, Lang AE, Jankovic J, David NJ, Weiner WJ: Case 1, 1995: psychosis, dementia, chorea, ataxia, and supranuclear gaze dysfunction. Mov Disord. 1995, 10 (3): 257-262.
Campo JV, Stowe R, Slomka G, Byler D, Gracious B: Psychosis as a presentation of physical disease in adolescence: a case of Niemann-Pick disease, type C. Dev Med Child Neurol. 1998, 40 (2): 126-129.
Turpin JC, Baumann N: [Presenting psychiatric and cognitive disorders in adult neurolipidoses]. Rev Neurol. 2003, 159 (6–7 Pt 1): 637-647.
Josephs KA, Van Gerpen MW, Van Gerpen JA: Adult onset Niemann-Pick disease type C presenting with psychosis. J Neurol Neurosurg Psychiatry. 2003, 74 (4): 528-529.
Tyvaert L, Stojkovic T, Cuisset JM, Vanier MT, Turpin JC, De Seze J, Vermersch P: [Presentation of Niemann-Pick type C disease with psychiatric disturbance in an adult]. Rev Neurol. 2005, 161 (3): 318-322.
Walterfang M, Fietz M, Fahey M, Sullivan D, Leane P, Lubman DI, Velakoulis D: The neuropsychiatry of Niemann-Pick type C disease in adulthood. J Neuropsychiatry Clin Neurosci. 2006, 18 (2): 158-170.
Walterfang M, Fietz M, Abel L, Bowman E, Mocellin R, Velakoulis D: Gender dimorphism in siblings with schizophrenia-like psychosis due to Niemann-Pick disease type C. J Inherit Metab Dis. 2009, 221-226. Sup1
Sandu S, Jackowski-Dohrmann S, Ladner A, Haberhausen M, Bachmann C: Niemann-Pick disease type C1 presenting with psychosis in an adolescent male. Eur Child Adolesc Psychiatry. 2009, 18 (9): 583-585.
Walterfang M, Kornberg A, Adams S, Fietz M, Velakoulis D: Post-ictal psychosis in adolescent Niemann-Pick disease type C. J Inherit Metab Dis. 2010, 13 (Sup3): 63-65.
Klunemann HH, Santosh PJ, Sedel F: Treatable metabolic psychosis that go undetecd: What Niemann Pick type C can teach us. Int J Psychiatry Clin Pract. 2012, 16 (3): 1-8.
van de Vlasakker CJ, Gabreels FJ, Wijburg HC, Wevers RA: Clinical features of Niemann-Pick disease type C. An example of the delayed onset, slowly progressive phenotype and an overview of recent literature. Clin Neurol Neurosurg. 1994, 96 (2): 119-123.
Klarner B, Klunemann HH, Lurding R, Aslanidis C, Rupprecht R: Neuropsychological profile of adult patients with Niemann-Pick C1 (NPC1) mutations. J Inherit Metab Dis. 2007, 30 (1): 60-67.
Talele SS, Xu K, Pariser AR, Braun MM, Farag-El-Massah S, Phillips MI, Thompson BH, Cote TR: Therapies for inborn errors of metabolism: what has the orphan drug act delivered?. Pediatrics. 2010, 126 (1): 101-106.
Andreasen NC: The evolving concept of schizophrenia: from Kraepelin to the present and future. Schiz Res. 1997, 28 (2–3): 105-109.
van Os J, Driessen G, Gunther N, Delespaul P: Neighbourhood variation in incidence of schizophrenia. Evidence for person-environment interaction. Br J Psychiatry. 2000, 176: 243-258.
Hill AB: The Environment and Disease: Association or Causation?. Proc R Soc Med. 1965, 58: 295-300.
Weinberger DR: From neuropathology to neurodevelopment. Lancet. 1995, 346 (8974): 552-557.
Van Gorkom HJ, Pulles MP, Wessels JS: Light-induced changes of absorbance and electron spin resonance in small photosystem II particles. Biochim Biophys Acta. 1975, 408 (3): 331-339.
van Os J, Kapur S: Schizophrenia. Lancet. 2009, 374 (9690): 635-645.
Cutting J: The phenomenology of acute organic psychosis. Comparison with acute schizophrenia. Br J Psychiatry. 1987, 151: 324-332.
Horiguchi J, Miyaoka T, Shinno H: Pathogenesis and symptomatology of hallucinations (delusions) of organic brain disorder and schizophrenia. Psychogeriatrics. 2009, 9 (2): 73-76.
Barak Y, Aizenberg D, Mirecki I, Mazeh D, Achiron A: Very late-onset schizophrenia-like psychosis: clinical and imaging characteristics in comparison with elderly patients with schizophrenia. J Nerv Ment Dis. 2002, 190 (11): 733-736.
Teeple RC, Caplan JP, Stern TA: Visual hallucinations: differential diagnosis and treatment. Prim Care Companion J Clin Psychiatry. 2009, 11 (1): 26-32.
Cornic F, Consoli A, Tanguy ML, Bonnot O, Perisse D, Tordjman S, Laurent C, Cohen D: Association of adolescent catatonia with increased mortality and morbidity: evidence from a prospective follow-up study. Schizophr Res. 2009, 113 (2–3): 233-240.
Remschmidt H, Theisen F: Early-onset schizophrenia. Neuropsychobiology. 2012, 66 (1): 63-69.
Walterfang M, Bonnot O, Mocellin R, Velakoulis D: The neuropsychiatry of inborn errors of metabolism. J Inherit Metab Dis. 2013, 36 (4): 687-702.
McEvoy JP: The costs of schizophrenia. J Clin Psychiatry. 2007, 68 (Suppl 14): 4-7.
Acknowledgments
Matthew Reilly PhD, associated with InTouch Medical Ltd, provided editorial assistance in the preparation of the final draft of this manuscript, paid for by Actelion Pharmaceuticals Ltd.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
OB, MW, FS and HK declare past and present honorarium from Actelion Pharma. DC and ST have no competing interest.
Authors’ contribution
OB write the first draft of the article which was reviewed by all authors. ST, DC, MW and HK help OB for response to reviewers. Regarding specific systematic review work, two groups worked separately in screening abstracts from relevant articles from the literature review (Group 1: MW and HK. Group 2: OB, DC, ST and FS). All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Bonnot, O., Klünemann, H.H., Sedel, F. et al. Diagnostic and treatment implications of psychosis secondary to treatable metabolic disorders in adults: a systematic review. Orphanet J Rare Dis 9, 65 (2014). https://doi.org/10.1186/1750-1172-9-65
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1172-9-65