
Abstract
Epithelial ovarian cancer (EOC) is a significant cause of cancer-related mortality in women, and there has been no substantial decrease in the death rates due to EOC in the last three decades. Thus, basic knowledge regarding ovarian tumor cell biology is urgently needed to allow the development of innovative treatments for EOC. Traditionally, EOC has not been considered an immunogenic tumor, but there is evidence of an immune response to EOC in patients. Clinical data demonstrate that an antitumor immune response and immune evasion mechanisms are correlated with a better and lower survival, respectively, providing evidence for the immunoediting hypothesis in EOC. This review focuses on the immune response and immune suppression in EOC. The immunological roles of chemotherapy and surgery in EOC are also described. Finally, we detail pilot data supporting the efficiency of immunotherapy in the treatment of EOC and the emerging concept that immunomodulation aimed at counteracting the immunosuppressive microenvironment must be associated with immunotherapy strategies.
Similar content being viewed by others

Introduction
Epithelial ovarian cancer (EOC) is the fifth most common cancer among women and the fourth most common cause of cancer-related death among women in developing countries [1]. The prognosis is poor, with a 5-year survival rate of 30%. The majority of patients relapse within 16–18 months following the end of treatment and die from the disease despite response to first-line therapy consisting of debulking surgery and chemotherapy [2, 3]. 15% of patients die within the first year. No substantial decrease in the death rate occurred in the last three decades. Thus, there is an urgent need for basic knowledge of ovarian tumor biology for the development of innovative EOC treatments.
Unlike melanoma or renal and hematologic tumor diseases, EOC is not considered to be immunogenic. However, there is evidence of an immune response against EOC in patients [4]. Experimental data show that the inflammatory microenvironment of EOC prevents the maturation of myeloid cells, favors regulatory cell development and restrains the cytotoxic activity of effector lymphocytes, leading the tumor to escape from the immune system and triggering cancer progression [5]. Treatments such as chemotherapy with paclitaxel/carboplatin and debulking surgery are traditionally considered to negatively impact the immune system during EOC [6]. However, recent data challenge this concept and highlight the major role of immune response in EOC. Indeed, aforementioned treatments were shown to modulate the host response and to decrease the immunosuppression [7, 8]. Thus, immunotherapies aimed at increasing the host immune response or decreasing immunosuppression were tested in preclinical and clinical studies and are emerging as potential strategies to enhance classical EOC treatments.
In this article, we present an overview of the current understanding of the immune response and immune suppression in EOC. The immunological role of chemotherapy and surgery is highlighted, and pilot data supporting the efficiency of immunotherapy in EOC treatment are reviewed.
Evidence of an immune response in EOC
EOC expresses or overexpresses tumor-associated antigens (TAA), i.e. antigens (Ag) acquired by tumor cells in the process of neoplastic transformation that can elicit a specific T-cell immune response by the host. In 1993, EOC ascites were found to contain CD8+ T-cells capable of recognizing HER2/neu-positive tumor cells [9]. 5 to 66% of EOC exhibit this EGFR-related glycoprotein that activates signaling pathways involved in cellular proliferation [10, 11]. Many other TAA were described in EOC, such as folate receptor(FR)-α [12], epithelial cell adhesion molecule (EpCAM) [13], human epididymis protein 4 [14], p53 [15], mucin-like MUC16 (CA125) and MUC1 (CA15.3) [16] and TAA of the cancer-testis group [17, 18]. Tumor-reactive T-cells and antibodies (Ab) directed against TAA were detected in the peripheral blood of patients with advanced-stage disease at the time of diagnosis [15, 19], and tumor-reactive T-cells were isolated from tumors or ascites [20].
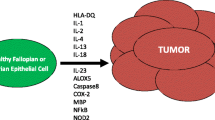
Furthermore, there is clinical evidence for the role of immunosurveillance against EOC. The detection of intraepithelial tumor-infiltrating lymphocytes (TIL) correlates with clinical outcome. Zhang et al. detected CD3+ TIL in 102/186 frozen specimens from patients with stage III/IV EOC [21]. The five-year progression-free survival rates were 31.0% and 8.7% for patients with and without TIL, respectively. The presence of TIL correlated with progression-free survival in multivariate analysis (p < 0.001) [21]. Recently, other studies confirmed that the CD3+ TIL count is a significant prognosis factor in EOC (Table 1) [22–32]. High frequencies and activity levels of immune effector cells such as CD8+ T-cells, Natural Killer(NK)-cells and Vγ9Vδ2T-cells are correlated with positive clinical outcomes for EOC patients [33, 34]. Thelper(Th)-17 cells, a recently discovered T-lymphocyte subset, were found in proportionally higher number in EOC microenvironment in comparison with other immune cells [35, 36]. In EOC patients, Th17 levels in the tumor correlated positively with Th1-cells, cytotoxic CD8+ T-cells and NK-cells and Th17 levels in ascites correlated positively with patient survival [35]. Intriguingly, Th17 were reported to promote either tumor cell growth or antitumor response and their role in cancer development is currently under debate [37]. Finally, in addition to TIL, the number of peripheral blood immune cells, e.g. NK-cells, is also correlated with survival in EOC [33]. All these results support the existence of immunosurveillance in EOC (Figure 1).

Immune network in EOC. EOC is immunogenic and expresses tumor-associated antigens such as HER2/neu, CA125 and Folate Receptor α. Various immune effectors such as CD8+ T-cells, NK-cells and Vγ9Vδ2T-cells can attack tumor cells, but immunosuppressive crosstalk counteracts the functions of these effector cells. Treg, tolerogenic DC, MDSC, B7-H4+ TAM and non-immune cells such as mesenchymal stem cells and tumor cells themselves halt antitumor activities through cell-cell contacts (CA125/siglec pathway, PD-1 and CTLA-4 immunosuppressive checkpoints) or the production of soluble factors (IL-10, TGF-β, PGE2, MIF, arginase-1, and IDO).
Immune escape in EOC
The tumor immunosurveillance concept was postulated in the 1960s by Burnet and Thomas, who proposed that the immune system patrols the body to recognize and destroy host cells that become cancerous and that the immune system is responsible for preventing cancer development [46]. This concept was then replaced by the cancer-immunoediting hypothesis, in which the immune system shapes tumor immunogenicity with three successive phases: elimination, equilibrium and escape [47].
Immune escape in EOC involves several mechanisms that implicate tumor, immune and stromal cells. Ovarian tumor cells escape immune recognition by downregulating surface molecules involved in Ag presentation, such as β2-microglobulin and Major Histocompatibility Complex (MHC) [28]. Similarly, the downregulation of MHC class I-related chain A (MICA) expression impedes the detection of tumor cells by innate cytotoxic effector cells through the engagement of the NKG2D-activating receptor [48, 49]. Additionally, ovarian tumor cells overexpress molecules that counteract the cytotoxic activities of immune cells: CA125 binds the NK-cell inhibitory receptor (KIR) siglec-9, thereby protecting themselves from NK-mediated lysis [50, 51]; the macrophage migration inhibitory factor (MIF) downregulates NKG2D-activating receptor expression on NK-cells [52]. Furthermore, engagement of programmed death-1 (PD-1) on CD8+ T-cells by programmed death-1 ligand-1 (PD-L1) expressed by ovarian tumor cells impairs the effector functions of these lymphocytes [24, 53]. Wide panel of cancers, including EOC, were also shown to express indolamine-2,3-dioxygenase (IDO), an intracellular enzyme that catalyzes the rate-limiting step in the metabolism of the essential amino acid tryptophan [54, 55]. IDO is a beneficial host mechanism regulating immune responses in various contexts such as pregnancy, transplantation or infection. It was proposed to elicit feedback process, therefore preventing deleterious consequences of excessive immune responses. However, this endogenous mechanism is hijacked by tumors to establish immunotolerance to tumor antigens [56, 57].
Immune cells also play a major role in the immune escape in EOC [58]. The EOC-specific recruitment of CD4+CD25+FoxP3+ regulatory T-cells (Treg), tolerogenic dendritic cells (DC), B7-H4+ tumor-associated macrophages (TAM) and myeloid-derived suppressor cells (MDSC) fosters immune privilege and predicts reduced survival in EOC (Table 1) [23, 36, 44, 59–62]. Accumulation of Treg is now well documented in various tumors including EOC [23, 36, 38–41]. CCR4 chemokine receptor expression confers to Treg higher capacity than effector T-cells to infiltrate the tumor in response to CCL22 chemokine produced by either tumor cells or TAM [36]. In addition, Treg could originate from in situ expansion. In that setting, ICOS-ligand costimulation provided by plasmacytoid DC (pDC) was recently highlighted as a prominent signal triggering in situ Treg expansion in some tumors, including EOC [63, 64]. At last, de novo conversion of FoxP3- cells into Treg was shown to occur in the tumor as a consequence of TGF-β stimulation or IDO induction [65, 66]. Treg mainly mediate immunosuppression through cell-cell contacts with DC or effector cells or by the secretion of immunosuppressive cytokines, including IL-10, IL-35 and TGF-β [67]. Treg notably contribute to DC tolerization, thereby further reducing the effector T-cell activation and proliferation. Interestingly, association of tumor regulatory T-cells with high hazard ratio for death and decreased survival times is currently well documented in EOC [23, 36, 42]. Besides Treg, DC are instrumental in establishing immunosupression in cancer. While DC were initially recognized as the primary orchestrators of the immune response, their role in the immunotolerance is now well established [68]. Importantly, both conventional myeloid DC (cDC) and pDC are characterized by high plasticity [69]. Consequently, their immune properties could be modulated by environmental stimuli and tumors may benefit from this Achille’s heel to induce DC tolerization and to reduce the adaptive immunity to tumor antigens. Accordingly, studies showed that the EOC microenvironment converts DC toward an immunosuppressive phenotype [70]. In a mouse model of EOC, Scarlett et al. showed that the DC phenotype controls EOC progression. Indeed, the switch of infiltrating-DC from activating to regulatory phenotype coincides with rapid tumor progression to terminal disease [62]. The role of pDC in EOC immunity was proposed by Zou et al. that evidenced the recruitment of pDC in response to stromal-derived factor-1 (SDF-1/CXCL-12) secretion by EOC [71]. The accumulation of pDC within the EOC was shown to be associated with shorter progression-free survival [44]. Tolerogenic DC may exert profound immunosuppressive effects on effector lymphocytes. Alteration of the IFN-α production by pDC was recently documented in EOC [44]. Moreover, through PD-L1/PD-L2 expression, DC can engage the PD-1 inhibitory pathway, thus inhibiting lymphocyte proliferation and effector functions [72, 73], inducing tumor-specific T-cell apoptosis [74] and promoting the differentiation of CD4+ T-cells into Treg [75]. Tolerogenic DC can also turn-down the immune response through the induction of IDO activity that inhibits CD8+ T-cell proliferation [76] and decreases NKG2D expression on NK-cells [77]. As aforementioned for DC, the tumor microenvironment also strongly polarizes the macrophage differentiation and gives rise to TAM [37]. B7-H4+ macrophages, a subset of TAM, was shown to suppress TAA-specific T-cell immunity [60]. An inverse correlation was evidenced between the intensity of B7-H4 expression on macrophages in EOC and patient survival [42]. Moreover, average 5-year survival rate was found significantly higher in EOC patients with low densities of TAM than in patients with increased TAM populations [78]. At last, MDSC are immature myeloid cells with immunosuppressive properties that were evidenced in both mouse model of EOC and EOC patients [61, 79, 80]. MDSC exhibit increased level of arginase-1 (ARG-1) and inductible Nitric Oxide Synthase (iNOS) activities. Deprivation of L-Arginine in the tumor microenvironment is emerging as a key immunosuppressive mechanism. It leads to CD3-zeta chain downregulation, thereby inhibiting effector T-cell activation [81]. Increased levels of NO, along with reactive oxygen and nitrogen species, disrupt signaling through the IL-2 receptor [82] and alter Ag recognition by nitrating the TCR [83]. Moreover, MDSC were shown to facilitate effector T-cell conversion into Treg [84] and to inhibit intratumoral migration of CD8+ effectors because of the nitration of CCL2 chemoattractant [85].
Third player in tumor escape is the stromal cell population. Overexpression of the endothelin-B receptor by tumor endothelial cells inhibits concurrent ICAM-1 expression, thereby impairing the ICAM-1/LFA-1-mediated transmigration of leukocytes [86]. Overexpression of the endothelin-B receptor is associated with the absence of TIL and short survival time in EOC patients [43]. Furthermore, stromal cells may provide chemoattractants for the immune cells e.g. SDF-1/CXCL12 that recruits pDC [71]. They are also able to secrete soluble immunosuppressive factors e.g. prostaglandin-E2 (PGE2) which is produced by mesenchymal stem cells (MSC).
Finally, the EOC microenvironment is characterized by the presence of numerous immunosuppressive soluble or cellular factors (IL-10, TGF-β, PGE2, MIF, HLA-G, IDO, arginase-1, PD-L1, B7-H4 and Fas-ligand), which can originate from various sources, including tumor, immune and stromal cells [87–91]. PGE2 can be secreted by both MSC and EOC tumor cells. Of note, overexpression of COX-2, an inducible enzyme that triggers PGE2 synthesis, by ovarian tumor cells correlates with resistance to chemotherapy and poor prognosis [92]. PGE2 inhibits NK and γδ T-cell cytotoxicity [45, 93, 94] and induces the differentiation of CD4+ T-cells into Treg [95]. Similarly, IDO is expressed in ovarian tumor cells and tumor-infiltrating DC [54, 55, 96]. IDO expression was reported in 43% of analyzed EOC tissues (83/192) [97]. Moreover, its expression was correlated with worse patient survival [54, 55] and with enhanced peritoneal tumor dissemination [55, 98]. IDO is currently thought as one of the main factors that contribute to tumor-induced immunosuppression by depleting tryptophan from the microenvironment and producing tryptophan metabolite kynurenine. Depletion of tryptophan is sensed by GCN2 kinase pathway driving effector T-cell anergy and apoptosis [99]. Effects of kynurenine are mediated by the aryl hydrocarbon receptor transcription factor that induces increased survival and motility in cancer cells while favoring Treg expansion and suppressive effects in effector T-cells [100, 101].
Thus, regulatory cells, along with soluble and cellular immunosuppressive factors, create a tolerogenic microenvironment in EOC that compromises the antitumoral immune response [89]. These EOC immunosuppressive networks characterize the “cancer immunoediting” concept, which emphasizes a dynamic process of interaction between cancer and the host immune system [47] (Figure 1).
Modulation of the immune response against EOC with debulking surgery or chemotherapy
Conventional EOC treatment uses debulking surgery and systemic chemotherapy. Surgery decreases the tumor burden and removes poorly vascularized tissues while cytotoxic drugs eradicate residual tumor cells [7, 102, 103]. Little information is available regarding the impact of surgery on the immunological status in EOC patients. Major surgery would induce immunosuppression because of the downregulation of T helper(Th)-1 response [6, 104]. However, there is some evidence that tumor debulking reduces tumor-induced immunosuppression in EOC [7, 105]. Napoletano et al. demonstrated that surgery significantly decreases the proportions of Treg and naive CD4+ T-cells while significantly increasing the ratio of CD8+ T-cells/Treg and the proportions of effector T-cells among the peripheral blood mononuclear cells [7]. Moreover, surgery significantly increases IFN-γ secretion by peripheral CD8+ T-cells and reduces the IL-10 immunosuppressive factor concentration in the serum [7]. Thus, cancer immunosuppression is partially reversible, and acquired immunity is enhanced by tumor debulking surgery in EOC [7].
Regarding chemotherapy, the frequent induction of lymphopenia suggests that this treatment may be immunosuppressive. However, recent data indicate that immunity plays a major role in the therapeutic mechanisms associated with chemotherapy [106, 107]. Accordingly, in advanced-EOC patients treated with platinum-based chemotherapy, an optimal tumor debulking outcome was more frequent when CD3+ TIL are present [21]. In addition, paclitaxel or cisplatin used in EOC cause the upregulation of mannose-6-phosphate receptor expression on murine tumor cells. This upregulation sensitizes tumor cells to granzyme-B protease released by cytotoxic T-lymphocytes [108]. Paclitaxel can also stimulate the proliferation of T-cells and enhance the cytolytic activity of NK-cells in models of breast cancer [106, 109]. Moreover, in advanced EOC, successful chemotherapy was shown to be associated with improved functions and increased proportions of CD8+ effector T-cells [7, 8]. Furthermore, chemotherapy decreases immunosuppression by reducing the number of circulating Treg observed after neoadjuvant chemotherapy in EOC [7]. Some antitumor agents can also trigger immunogenic tumor cell death, causing the cancer cells to expose or secrete immunogenic signals that trigger an anticancer immune response. Of note, not all types of chemotherapy, but oxaliplatin and 3/25 tested anthracyclines, elicit immunogenic cell death [110–112]. Altogether, these data provide evidence that debulking surgery and chemotherapy may restore, by direct and indirect effects, the equilibrium phase or the elimination phase in tumors that escaped initial immunosurveillance [106].
Immunotherapy in EOC: how to counteract immunosuppression?
Preclinical and preliminary clinical studies aimed at proving the immunotherapy concept in EOC were initiated by using monoclonal Ab (mAb), vaccinations or adoptive T-cell transferts [113–115]. The majority of these studies were uncontrolled phase I/II studies, with small sample sizes and heterogeneous inclusion criteria (recurrent or chemotherapy-refractory diseases) disrupting the comparisons and the identification of the best strategy.
Several mAb targeting TAA were tested in EOC [116]: anti-CA125 oregovomab and abagovomab [117–119]; anti-HER-2/neu trastuzumab and pertuzumab [10, 120]; anti-FR-α farletuzumab α [121], anti-EpCAM catumaxomab [122] and anti-Tag72 B72.3 [123]. All these mAb demonstrated adequate safety and tolerability but failed to demonstrate clear clinical benefits, even when an immunological response was evidenced [114, 115]. Active immunotherapy by vaccination based on peptides or cellular approaches were also evaluated. Clinically tested peptides include NY-ESO-1 [124, 125], p53 [126], HER2-neu [127] and multiple constructed-peptides (HER2-neu/MAGE-A1/FRα [128] and MUC-1/carcinoembryonic antigen [129]). In addition, cell vaccines include DC pulsed with ARNm (FRα [130]), peptides (HER2-neu, MUC1 [131]), autologous tumor Ag [132] or whole tumor cell lysate [133]. Vaccine therapies were well tolerated and demonstrated immunological responses, but provided only minor clinical benefits. Of note, these studies enrolled low numbers of patients and have generally not yet evolved past phase I/II studies. A third immunotherapy strategy is adoptive T-cell therapy, which uses cytotoxic lymphocytes with natural or engineered reactivity against cancer cells. Cytotoxic lymphocytes are generated in vitro and then transferred back into the patient to elicit cytotoxic responses against the patient’s own tumor cells. Only five phase I/II EOC studies, which enrolled few patients (<20), are available [134–138]. They used either TIL or peripheral autologous T-cells and were well tolerated; unfortunately, only modest clinical benefits were demonstrated. Thus, to date, results of these trials are disappointing, regardless of the strategy [115]. However, these trials may suffer from some pitfalls. First, they often enrolled patients with recurrent or refractory-chemotherapy diseases, i.e. patients at terminal stages of the disease, with strong immunodepression. It is likely that enrollment of patients at earlier stages of disease could be more successful. Secondly, all these trials focused on the recognition and killing of tumor cells and neglected to consider the immunosuppressive impact of the tumor microenvironment. Thus, improvement of theses immunotherapies is needed. For example, chimeric antigen receptor(CAR)-modified T cell therapy is highly promising [139, 140]. CAR T-cells could be engineered to only express the downstream pathway of activating receptors. This refined adoptive therapy skips inhibitory signals expressed by the tumor environment. In addition, use of adjuvant drugs targeting immunosuppressive cells or soluble/cellular immunomodulatory factors could be the key to fully unleash the potential of immunotherapy by breaking peripheral tolerance.
Below, we review some immunomodulatory tools already in clinical use or likely to be assessed in the near future, that interact with the immunosuppressive factors found in the EOC microenvironment.
First approach may consist in depleting the host of the regulatory cells or in limiting their recruitment within the tumor. Treg depletion may be achieved using low-dose cyclophosphamide which prevents, under incompletely understood mechanism, Treg development and functionality [141, 142]. An alternative strategy uses the expression by Treg of the IL-2 receptor alpha (CD25). Recombinant fusion protein of IL-2 and diphtheria toxin (Ontak®, Eisai) was tested in EOC patients and showed effective depletion of circulating Treg [143]. Moreover, in patients with metastatic breast cancer, the anti-CD25 mAb daclizumab (Zenapax®, Roche) demonstrated selective T-lymphocyte killing properties, allowing Treg depletion for several weeks [144]. However, it is unclear if Treg depletion occurs at EOC locations (solid tumor, malignant ascites) and results in tumor regression [143–145]. Moreover, as effector cells also express CD25, anti-CD25 mAb may also induce unwanted depletion of effector cells [146]. In addition, blocking the ICOS-pathway could inhibit the pDC-triggered proliferation of Treg within the tumor [64]. However, as ICOS pathway also favors the differentiation of T helper(Th)-17 cells which might either promote tumor growth or antitumor response [35, 37, 147–150] careful preclinical investigations of ICOS inhibitors (314.8 mAb) is needed [63].
The role of chemoattractants in the recruitment of immune cells also gives a great opportunity to reduce the infiltration of regulatory cells within the tumor [151]. First candidates are under investigation. CCR4 antagonists were shown to block the recruitment of Treg instructed by CCL22 and CCL17 and to favor the induction of antigen-specific CD8+ T cell response after vaccination [152]. Similarly, Bindarit® that inhibits CCL2 synthesis and therefore restricts the recruitment in the tumor of immature myeloid cells, was shown to induce tumor regression in prostate and breast cancer animal models [153]. Regulatory cell depletion could also be achieved by improving the maturation of immature myeloid cells [154] using all trans retinoic acid [155] or ultra-low non-cytotoxic doses of paclitaxel (chemo-immunomodulation) [156].
Another attractive approach is the use of either antagonists of immune-repressor molecules or agonists of immune-activating receptors [157]. Checkpoint blockade receptors comprise CTLA-4, PD-1 and NK inhibitory receptors (KIR) that, upon engagement, dampen the immune response. CTLA-4 predominantly regulates T-cells at the priming phase of activation by competing with CD28+ for binding of B7-1 and B7-2 on DC. CTLA-4 engagement prevents T-cells from achieving full activation. Accordingly, anti-CTLA-4 mAb were shown to activate CD4+ and CD8+ effector T-cells both directly by removing inhibitory checkpoints and indirectly via the inhibition of regulatory T-cell activity [158]. Eleven EOC patients, previously vaccinated with GM-CSF and irradiated autologous tumor cells, received anti-CTLA-4 ipilimumab (Yervoy®, Bristol-Myers-Squibb, BMS). Significant antitumor effects were observed in a minority of these patients and were correlated with increased CD8+ T-cells/Treg ratio [159]. In contrast to CTLA-4, PD-1 signaling occurs in the tumor, where PD-L1-expressing tumor cells can signal through PD-1 on TIL to turn-down the antitumor T-cell response. In EOC, the PD-1/PD-L1 pathway seems to be a dominant immunosuppression mechanism [73]. PD-L1 expression in EOC was demonstrated to be an independent unfavorable prognostic factor and to promote peritoneal dissemination [24, 160]. Several PD-1/PD-L1-pathway blocking agents were assessed in various cancer types and promising results were recently reported. Nivolumab (BMS-936558, Bristol-Myers-Squibb) was tested in 296 patients most harboring lung cancer, renal cell cancer and melanoma, with clinical benefits apparent in 20 to 25% of the patients [161]. Impressive durable responses were reported. 25/42 patients with PD-L1-positive tumor experienced an objective response while none of the 17/42 PD-L1-negative patients did. However, lack of prognostic association was reported elsewhere and the usefulness of PD-L1 as a biomarker need to be explored in larger prospective studies [162]. In addition, Bramher and colleagues reported that 1/17 EOC patients treated with anti-PD-L1 mAb (BMS-936559) experienced an objective response [163]. New trials enrolling patients with solid tumor of multiple origins are underway and informative data in EOC are expected [164]. Inhibition of the cytotoxic properties of NK-cells through KIR engagement may also contribute to the tumor escape. Some anti-KIR antibodies, such as lilirumab (Bristol-Myers-Squibb), recently entered clinical development phases. First data were obtained in hematological diseases and phase I studies recruiting patients with solid tumors are ongoing [165, 166]. As a corollary, agonistic agents of costimulatory molecules such as glucocorticoid-induced TNFR (GITR), OX40, CD137 are candidates to boost the antitumor immune response. A dose-escalation phase I clinical trial (NCT01239134) with agonist anti-GITR mAb (TRX518) was recently initiated.
Third possibility is to repress the activity of enzymes (IDO, ARG-1, iNOS) that were shown to inhibit the immune response. Data from first clinical trials using IDO inhibitors, notably the isomers of 1-methyl-tryptophan (1MT), were disappointing, but these studies may suffer from lack of potent and selective IDO inhibitors. New compounds recently entered clinical trials [167]. A phase II study of IDO inhibitor INCB024360 is currently recruiting patients with biochemical-recurrent-only EOC following complete remission with first-line chemotherapy (clinical trial: NCT01685255). In addition, inhibitors of phosphodiesterase(PDE)-5, e.g. sildenafil, were reported to increase intracellular concentrations of cGPM, resulting in the inhibition of both ARG-1 and iNOS. PDE-5 inhibitors along with nitroaspirin or specific ARG-1/iNOS inhibitors might provide new therapeutic strategy to recover potent antitumor immune response [154].
Lastly, PGE2 was shown to be a crucial immunosuppressive factor in EOC, as it impairs the cytotoxic properties of effector cells such as Vγ9Vδ2T-cells [45] and also induces the differentiation of MDSC from bone marrow stem cells in a mouse model [168]. PGE2 biosynthesis is regulated by the inducible COX-2 enzyme and could be inhibited by the COX-2-specific inhibitor celecoxib (Celebrex®, Pfizer). In a mouse model, celecoxib prevented the local and systemic expansion of MDSC, impaired the suppressive function of these cells, and significantly improved vaccine immunotherapy [169]. Thus, celecoxib, currently used in the prevention of colorectal adenomatous polyps [170], could be tested in combination with immunotherapy to reduce the immunosuppression by MDSC in EOC. Another possible strategy to counteract the immunosuppressive influence of PGE2 on Vγ9Vδ2T cells could be to restore the cytotoxic properties of these cells with a zoledronate perfusion [45]. In addition, zoledronate was shown to prevent the immunosuppressive polarization of TAM [171, 172] which is a major component of the leukocyte infiltrate in the tumor microenvironment and plays a dominant role in the production of immune suppressive cytokines in EOC [60]. Thus, zoledronate, which is currently used for the management of osteoporosis and bone metastasis, appears to be an attractive molecule to reinforce the immune response. Altogether, these data warrant further exploration of combinatorial therapies with immunotherapy and bisphosphonates.
In conclusion, accumulated evidences support the immunoediting hypothesis and the idea that EOC is immunogenic. Immunotherapeutic protocols aimed at modulating the immune system to strengthen the spontaneous antitumor immune response are under investigation. Targeting the immunosuppressive mechanisms could be the key to fully unleash the potential of immunotherapy. The combination of molecules endowed with immuno-modulatory properties with immunotherapy targeting the tumor cells will hopefully increase the survival of EOC patients. Careful preclinical evaluation will be necessary to screen optimal combinations before clinical trials.
References
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 2009, 59: 225-249. 10.3322/caac.20006.
Pfisterer J, Ledermann JA: Management of platinum-sensitive recurrent ovarian cancer. Semin Oncol. 2006, 33: S12-S16.
Hoskins P, Vergote I, Cervantes A, Tu D, Stuart G, Zola P, Poveda A, Provencher D, Katsaros D, Ojeda B: Advanced ovarian cancer: phase III randomized study of sequential cisplatin-topotecan and carboplatin-paclitaxel vs carboplatin-paclitaxel. J Natl Cancer Inst. 2010, 102: 1547-1556. 10.1093/jnci/djq362.
Knutson KL, Curiel TJ, Salazar L, Disis ML: Immunologic principles and immunotherapeutic approaches in ovarian cancer. Hematol Oncol Clin North Am. 2003, 17: 1051-1073. 10.1016/S0889-8588(03)00064-9.
Cubillos-Ruiz JR, Rutkowski M, Conejo-Garcia JR: Blocking ovarian cancer progression by targeting tumor microenvironmental leukocytes. Cell Cycle. 2010, 9: 260-268. 10.4161/cc.9.2.10430.
Brune IB, Wilke W, Hensler T, Holzmann B, Siewert JR: Downregulation of T helper type 1 immune response and altered pro-inflammatory and anti-inflammatory T cell cytokine balance following conventional but not laparoscopic surgery. Am J Surg. 1999, 177: 55-60. 10.1016/S0002-9610(98)00299-2.
Napoletano C, Bellati F, Landi R, Pauselli S, Marchetti C, Visconti V, Sale P, Liberati M, Rughetti A, Frati L: Ovarian cancer cytoreduction induces changes in T cell population subsets reducing immunosuppression. J Cell Mol Med. 2010, 14: 2748-2759. 10.1111/j.1582-4934.2009.00911.x.
Coleman S, Clayton A, Mason MD, Jasani B, Adams M, Tabi Z: Recovery of CD8+ T-cell function during systemic chemotherapy in advanced ovarian cancer. Cancer Res. 2005, 65: 7000-7006. 10.1158/0008-5472.CAN-04-3792.
Ioannides CG, Fisk B, Fan D, Biddison WE, Wharton JT, O'Brian CA: Cytotoxic T cells isolated from ovarian malignant ascites recognize a peptide derived from the HER-2/neu proto-oncogene. Cell Immunol. 1993, 151: 225-234. 10.1006/cimm.1993.1233.
Bookman MA, Darcy KM, Clarke-Pearson D, Boothby RA, Horowitz IR: Evaluation of monoclonal humanized anti-HER2 antibody, trastuzumab, in patients with recurrent or refractory ovarian or primary peritoneal carcinoma with overexpression of HER2: a phase II trial of the Gynecologic Oncology Group. J Clin Oncol. 2003, 21: 283-290. 10.1200/JCO.2003.10.104.
Camilleri-Broet S, Hardy-Bessard AC, Le Tourneau A, Paraiso D, Levrel O, Leduc B, Bain S, Orfeuvre H, Audouin J, Pujade-Lauraine E: HER-2 overexpression is an independent marker of poor prognosis of advanced primary ovarian carcinoma: a multicenter study of the GINECO group. Ann Oncol. 2004, 15: 104-112. 10.1093/annonc/mdh021.
Peoples GE, Anderson BW, Fisk B, Kudelka AP, Wharton JT, Ioannides CG: Ovarian cancer-associated lymphocyte recognition of folate binding protein peptides. Ann Surg Oncol. 1998, 5: 743-750. 10.1007/BF02303486.
Runz S, Keller S, Rupp C, Stoeck A, Issa Y, Koensgen D, Mustea A, Sehouli J, Kristiansen G, Altevogt P: Malignant ascites-derived exosomes of ovarian carcinoma patients contain CD24 and EpCAM. Gynecol Oncol. 2007, 107: 563-571. 10.1016/j.ygyno.2007.08.064.
Drapkin R, von Horsten HH, Lin Y, Mok SC, Crum CP, Welch WR, Hecht JL: Human epididymis protein 4 (HE4) is a secreted glycoprotein that is overexpressed by serous and endometrioid ovarian carcinomas. Cancer Res. 2005, 65: 2162-2169. 10.1158/0008-5472.CAN-04-3924.
Goodell V, Salazar LG, Urban N, Drescher CW, Gray H, Swensen RE, McIntosh MW, Disis ML: Antibody immunity to the p53 oncogenic protein is a prognostic indicator in ovarian cancer. J Clin Oncol. 2006, 24: 762-768. 10.1200/JCO.2005.03.2813.
Chauhan SC, Singh AP, Ruiz F, Johansson SL, Jain M, Smith LM, Moniaux N, Batra SK: Aberrant expression of MUC4 in ovarian carcinoma: diagnostic significance alone and in combination with MUC1 and MUC16 (CA125). Mod Pathol. 2006, 19: 1386-1394. 10.1038/modpathol.3800646.
Zhang S, Zhou X, Yu H, Yu Y: Expression of tumor-specific antigen MAGE, GAGE and BAGE in ovarian cancer tissues and cell lines. BMC Cancer. 2010, 10: 163-10.1186/1471-2407-10-163.
Chiriva-Internati M, Wang Z, Salati E, Timmins P, Lim SH: Tumor vaccine for ovarian carcinoma targeting sperm protein 17. Cancer. 2002, 94: 2447-2453. 10.1002/cncr.10506.
Schlienger K, Chu CS, Woo EY, Rivers PM, Toll AJ, Hudson B, Maus MV, Riley JL, Choi Y, Coukos G: TRANCE- and CD40 ligand-matured dendritic cells reveal MHC class I-restricted T cells specific for autologous tumor in late-stage ovarian cancer patients. Clin Cancer Res. 2003, 9: 1517-1527.
Peoples GE, Schoof DD, Andrews JV, Goedegebuure PS, Eberlein TJ: T-cell recognition of ovarian cancer. Surgery. 1993, 114: 227-234.
Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN: Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003, 348: 203-213. 10.1056/NEJMoa020177.
Raspollini MR, Castiglione F, Rossi Degl'innocenti D, Amunni G, Villanucci A, Garbini F, Baroni G, Taddei GL: Tumour-infiltrating gamma/delta T-lymphocytes are correlated with a brief disease-free interval in advanced ovarian serous carcinoma. Ann Oncol. 2005, 16: 590-596. 10.1093/annonc/mdi112.
Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C: Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005, 102: 18538-18543. 10.1073/pnas.0509182102.
Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, Higuchi T, Yagi H, Takakura K, Minato N: Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A. 2007, 104: 3360-3365. 10.1073/pnas.0611533104.
Clarke B, Tinker AV, Lee CH, Subramanian S, van de Rijn M, Turbin D, Kalloger S, Han G, Ceballos K, Cadungog MG: Intraepithelial T cells and prognosis in ovarian carcinoma: novel associations with stage, tumor type, and BRCA1 loss. Mod Pathol. 2009, 22: 393-402. 10.1038/modpathol.2008.191.
Shah CA, Allison KH, Garcia RL, Gray HJ, Goff BA, Swisher EM: Intratumoral T cells, tumor-associated macrophages, and regulatory T cells: association with p53 mutations, circulating tumor DNA and survival in women with ovarian cancer. Gynecol Oncol. 2008, 109: 215-219. 10.1016/j.ygyno.2008.01.010.
Tomsova M, Melichar B, Sedlakova I, Steiner I: Prognostic significance of CD3+ tumor-infiltrating lymphocytes in ovarian carcinoma. Gynecol Oncol. 2008, 108: 415-420. 10.1016/j.ygyno.2007.10.016.
Han LY, Fletcher MS, Urbauer DL, Mueller P, Landen CN, Kamat AA, Lin YG, Merritt WM, Spannuth WA, Deavers MT: HLA class I antigen processing machinery component expression and intratumoral T-Cell infiltrate as independent prognostic markers in ovarian carcinoma. Clin Cancer Res. 2008, 14: 3372-3379. 10.1158/1078-0432.CCR-07-4433.
Stumpf M, Hasenburg A, Riener MO, Jutting U, Wang C, Shen Y, Orlowska-Volk M, Fisch P, Wang Z, Gitsch G: Intraepithelial CD8-positive T lymphocytes predict survival for patients with serous stage III ovarian carcinomas: relevance of clonal selection of T lymphocytes. Br J Cancer. 2009, 101: 1513-1521. 10.1038/sj.bjc.6605274.
Leffers N, Gooden MJ, de Jong RA, Hoogeboom BN, ten Hoor KA, Hollema H, Boezen HM, van der Zee AG, Daemen T, Nijman HW: Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer Immunol Immunother. 2009, 58: 449-459. 10.1007/s00262-008-0583-5.
Milne K, Kobel M, Kalloger SE, Barnes RO, Gao D, Gilks CB, Watson PH, Nelson BH: Systematic analysis of immune infiltrates in high-grade serous ovarian cancer reveals CD20, FoxP3 and TIA-1 as positive prognostic factors. PLoS One. 2009, 4: e6412-10.1371/journal.pone.0006412.
Adams SF, Levine DA, Cadungog MG, Hammond R, Facciabene A, Olvera N, Rubin SC, Boyd J, Gimotty PA, Coukos G: Intraepithelial T cells and tumor proliferation: impact on the benefit from surgical cytoreduction in advanced serous ovarian cancer. Cancer. 2009, 115: 2891-2902. 10.1002/cncr.24317.
Garzetti GG, Cignitti M, Ciavattini A, Fabris N, Romanini C: Natural killer cell activity and progression-free survival in ovarian cancer. Gynecol Obstet Invest. 1993, 35: 118-120. 10.1159/000292678.
Thedrez A, Lavoué V, Dessarthe B, Daniel P, Henno S, Jaffre I, Levêque J, Catros V, Cabillic F: A quantitative deficiency in peripheral blood Vγ9Vδ2 cells is a negative prognostic biomarker in ovarian cancer patients. PLoS One. 2013, 8 (5): e63322-10.1371/journal.pone.0063322. Print 2013
Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, Huang E, Finlayson E, Simeone D, Welling TH: Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood. 2009, 114: 1141-1149. 10.1182/blood-2009-03-208249.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M: Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004, 10: 942-949. 10.1038/nm1093.
Wilke CM, Kryczek I, Zou W: Antigen-presenting cell (APC) subsets in ovarian cancer. Int Rev Immunol. 2011, 30: 120-126. 10.3109/08830185.2011.567362.
Callahan MJ, Nagymanyoki Z, Bonome T, Johnson ME, Litkouhi B, Sullivan EH, Hirsch MS, Matulonis UA, Liu J, Birrer MJ: Increased HLA-DMB expression in the tumor epithelium is associated with increased CTL infiltration and improved prognosis in advanced-stage serous ovarian cancer. Clin Cancer Res. 2008, 14: 7667-7673. 10.1158/1078-0432.CCR-08-0479.
Leffers N, Fehrmann RS, Gooden MJ, Schulze UR, Ten Hoor KA, Hollema H, Boezen HM, Daemen T, de Jong S, Nijman HW, van der Zee AG: Identification of genes and pathways associated with cytotoxic T lymphocyte infiltration of serous ovarian cancer. Br J Cancer. 2010, 103: 685-692. 10.1038/sj.bjc.6605820.
Wolf D, Wolf AM, Rumpold H, Fiegl H, Zeimet AG, Muller-Holzner E, Deibl M, Gastl G, Gunsilius E, Marth C: The expression of the regulatory T cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin Cancer Res. 2005, 11: 8326-8331. 10.1158/1078-0432.CCR-05-1244.
Dong HP, Elstrand MB, Holth A, Silins I, Berner A, Trope CG, Davidson B, Risberg B: NK- and B-cell infiltration correlates with worse outcome in metastatic ovarian carcinoma. Am J Clin Pathol. 2006, 125: 451-458.
Kryczek I, Wei S, Zhu G, Myers L, Mottram P, Cheng P, Chen L, Coukos G, Zou W: Relationship between B7-H4, regulatory T cells, and patient outcome in human ovarian carcinoma. Cancer Res. 2007, 67: 8900-8905. 10.1158/0008-5472.CAN-07-1866.
Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, Katsaros D, O'Brien-Jenkins A, Gimotty PA, Coukos G: Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008, 14: 28-36. 10.1038/nm1699.
Labidi-Galy SI, Sisirak V, Meeus P, Gobert M, Treilleux I, Bajard A, Combes JD, Faget J, Mithieux F, Cassignol A: Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res. 2011, 71: 5423-5434. 10.1158/0008-5472.CAN-11-0367.
Lavoué V, Cabillic F, Toutirais O, Thedrez A, Dessarthe B, de La Pintière CT, Daniel P, Foucher F, Bauville E, Henno S, Burtin F, Bansard JY, Levêque J, Catros V, Bouet-Toussaint F: Sensitization of ovarian carcinoma cells with zoledronate restores the cytotoxic capacity of Vγ9Vδ2 T cells impaired by the prostaglandin E2 immunosuppressive factor: implications for immunotherapy. Int J Cancer. 2012, 131 (4): E449-E462. 10.1002/ijc.27353. Epub 2011 Dec 21
Burnet FM: The concept of immunological surveillance. Prog Exp Tumor Res. 1970, 13: 1-27.
Schreiber RD, Old LJ, Smyth MJ: Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011, 331: 1565-1570. 10.1126/science.1203486.
Lu J, Aggarwal R, Kanji S, Das M, Joseph M, Pompili V, Das H: Human ovarian tumor cells escape gammadelta T cell recognition partly by down regulating surface expression of MICA and limiting cell cycle related molecules. PLoS One. 2011, 6: e23348-10.1371/journal.pone.0023348.
Thedrez A, Sabourin C, Gertner J, Devilder MC, Allain-Maillet S, Fournie JJ, Scotet E, Bonneville M: Self/non-self discrimination by human gammadelta T cells: simple solutions for a complex issue?. Immunol Rev. 2007, 215: 123-135. 10.1111/j.1600-065X.2006.00468.x.
Gubbels JA, Felder M, Horibata S, Belisle JA, Kapur A, Holden H, Petrie S, Migneault M, Rancourt C, Connor JP, Patankar MS: MUC16 provides immune protection by inhibiting synapse formation between NK and ovarian tumor cells. Mol Cancer. 2010, 9: 11-10.1186/1476-4598-9-11.
Belisle JA, Horibata S, Jennifer GA, Petrie S, Kapur A, Andre S, Gabius HJ, Rancourt C, Connor J, Paulson JC, Patankar MS: Identification of Siglec-9 as the receptor for MUC16 on human NK cells, B cells, and monocytes. Mol Cancer. 2010, 9: 118-10.1186/1476-4598-9-118.
Krockenberger M, Dombrowski Y, Weidler C, Ossadnik M, Honig A, Hausler S, Voigt H, Becker JC, Leng L, Steinle A: Macrophage migration inhibitory factor contributes to the immune escape of ovarian cancer by down-regulating NKG2D. J Immunol. 2008, 180: 7338-7348.
Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, Eppolito C, Qian F, Lele S, Shrikant P: Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A. 2010, 107: 7875-7880. 10.1073/pnas.1003345107.
Okamoto A, Nikaido T, Ochiai K, Takakura S, Saito M, Aoki Y, Ishii N, Yanaihara N, Yamada K, Takikawa O: Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin Cancer Res. 2005, 11: 6030-6039. 10.1158/1078-0432.CCR-04-2671.
Inaba T, Ino K, Kajiyama H, Yamamoto E, Shibata K, Nawa A, Nagasaka T, Akimoto H, Takikawa O, Kikkawa F: Role of the immunosuppressive enzyme indoleamine 2,3-dioxygenase in the progression of ovarian carcinoma. Gynecol Oncol. 2009, 115: 185-192. 10.1016/j.ygyno.2009.07.015.
Mellor AL, Munn DH: Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol. 2008, 8: 74-80. 10.1038/nri2233.
Johnson TS, Munn DH: Host indoleamine 2,3-dioxygenase: contribution to systemic acquired tumor tolerance. Immunol Invest. 2012, 41: 765-797. 10.3109/08820139.2012.689405.
Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK: Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol. 2012, 22: 275-281. 10.1016/j.semcancer.2012.01.011.
Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC: Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003, 9: 562-567. 10.1038/nm863.
Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, Brumlik M, Cheng P, Curiel T, Myers L: B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006, 203: 871-881. 10.1084/jem.20050930.
Yang R, Cai Z, Zhang Y, Yutzy WH, Roby KF, Roden RB: CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1 + CD11b + myeloid cells. Cancer Res. 2006, 66: 6807-6815. 10.1158/0008-5472.CAN-05-3755.
Scarlett UK, Rutkowski MR, Rauwerdink AM, Fields J, Escovar-Fadul X, Baird J, Cubillos-Ruiz JR, Jacobs AC, Gonzalez JL, Weaver J: Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J Exp Med. 2012, 209: 495-506. 10.1084/jem.20111413.
Faget J, Bendriss-Vermare N, Gobert M, Durand I, Olive D, Biota C, Bachelot T, Treilleux I, Goddard-Leon S, Lavergne E: ICOS-ligand expression on plasmacytoid dendritic cells supports breast cancer progression by promoting the accumulation of immunosuppressive CD4+ T cells. Cancer Res. 2012, 72: 6130-6141. 10.1158/0008-5472.CAN-12-2409.
Conrad C, Gregorio J, Wang YH, Ito T, Meller S, Hanabuchi S, Anderson S, Atkinson N, Ramirez PT, Liu YJ: Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3(+) T-regulatory cells. Cancer Res. 2012, 72: 5240-5249. 10.1158/0008-5472.CAN-12-2271.
Liu VC, Wong LY, Jang T, Shah AH, Park I, Yang X, Zhang Q, Lonning S, Teicher BA, Lee C: Tumor evasion of the immune system by converting CD4 + CD25- T cells into CD4 + CD25+ T regulatory cells: role of tumor-derived TGF-beta. J Immunol. 2007, 178: 2883-2892.
Curti A, Pandolfi S, Valzasina B, Aluigi M, Isidori A, Ferri E, Salvestrini V, Bonanno G, Rutella S, Durelli I: Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood. 2007, 109: 2871-2877.
Oleinika K, Nibbs RJ, Graham GJ, Fraser AR: Suppression, subversion and escape: the role of regulatory T cells in cancer progression. Clin Exp Immunol. 2013, 171: 36-45. 10.1111/j.1365-2249.2012.04657.x.
Steinman RM, Hawiger D, Nussenzweig MC: Tolerogenic dendritic cells. Annu Rev Immunol. 2003, 21: 685-711. 10.1146/annurev.immunol.21.120601.141040.
Ito T, Liu YJ, Kadowaki N: Functional diversity and plasticity of human dendritic cell subsets. Int J Hematol. 2005, 81: 188-196. 10.1532/IJH97.05012.
Scarlett UK, Cubillos-Ruiz JR, Nesbeth YC, Martinez DG, Engle X, Gewirtz AT, Ahonen CL, Conejo-Garcia JR: In situ stimulation of CD40 and Toll-like receptor 3 transforms ovarian cancer-infiltrating dendritic cells from immunosuppressive to immunostimulatory cells. Cancer Res. 2009, 69: 7329-7337. 10.1158/0008-5472.CAN-09-0835.
Zou W, Machelon V, Coulomb-L'Hermin A, Borvak J, Nome F, Isaeva T, Wei S, Krzysiek R, Durand-Gasselin I, Gordon A: Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nat Med. 2001, 7: 1339-1346. 10.1038/nm1201-1339.
Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, Shalabi A, Shin T, Pardoll DM, Tsuchiya H: B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001, 193: 839-846. 10.1084/jem.193.7.839.
Krempski J, Karyampudi L, Behrens MD, Erskine CL, Hartmann L, Dong H, Goode EL, Kalli KR, Knutson KL: Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J Immunol. 2011, 186: 6905-6913. 10.4049/jimmunol.1100274.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K: Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002, 8: 793-800.
Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ: Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3 + CD4+ regulatory T cells. Proc Natl Acad Sci U S A. 2008, 105: 9331-9336. 10.1073/pnas.0710441105.
Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, Messina JL, Chandler P, Koni PA, Mellor AL: Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. 2004, 114: 280-290.
Della Chiesa M, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, Moretta L, Moretta A, Vitale M: The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood. 2006, 108: 4118-4125. 10.1182/blood-2006-03-006700.
Wan T, Liu JH, Zheng LM, Cai MY, Ding T: [Prognostic significance of tumor-associated macrophage infiltration in advanced epithelial ovarian carcinoma]. Ai Zheng. 2009, 28: 323-327.
Bak SP, Alonso A, Turk MJ, Berwin B: Murine ovarian cancer vascular leukocytes require arginase-1 activity for T cell suppression. Mol Immunol. 2008, 46: 258-268. 10.1016/j.molimm.2008.08.266.
Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P: PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 2011, 71: 7463-7470. 10.1158/0008-5472.CAN-11-2449.
Ezernitchi AV, Vaknin I, Cohen-Daniel L, Levy O, Manaster E, Halabi A, Pikarsky E, Shapira L, Baniyash M: TCR zeta down-regulation under chronic inflammation is mediated by myeloid suppressor cells differentially distributed between various lymphatic organs. J Immunol. 2006, 177: 4763-4772.
Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, Segal DM: Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002, 168: 689-695.
Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI: Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007, 13: 828-835. 10.1038/nm1609.
Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH: Gr-1 + CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66: 1123-1131. 10.1158/0008-5472.CAN-05-1299.
Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, De Palma A, Mauri P, Monegal A, Rescigno M: Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. 2011, 208: 1949-1962. 10.1084/jem.20101956.
Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW: ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood. 2005, 106: 584-592. 10.1182/blood-2004-12-4942.
Sebti Y, Le Friec G, Pangault C, Gros F, Drenou B, Guilloux V, Bernard M, Lamy T, Fauchet R, Amiot L: Soluble HLA-G molecules are increased in lymphoproliferative disorders. Hum Immunol. 2003, 64: 1093-1101. 10.1016/j.humimm.2003.08.345.
Freedman RS, Deavers M, Liu J, Wang E: Peritoneal inflammation - A microenvironment for Epithelial Ovarian Cancer (EOC). J Transl Med. 2004, 2: 23-10.1186/1479-5876-2-23.
Nelson BH: The impact of T-cell immunity on ovarian cancer outcomes. Immunol Rev. 2008, 222: 101-116. 10.1111/j.1600-065X.2008.00614.x.
Lin A, Zhang X, Zhou WJ, Ruan YY, Xu DP, Wang Q, Yan WH: Human leukocyte antigen-G expression is associated with a poor prognosis in patients with esophageal squamous cell carcinoma. Int J Cancer. 2011, 129: 1382-1390. 10.1002/ijc.25807.
Rodriguez GC, Haisley C, Hurteau J, Moser TL, Whitaker R, Bast RC, Stack MS: Regulation of invasion of epithelial ovarian cancer by transforming growth factor-beta. Gynecol Oncol. 2001, 80: 245-253. 10.1006/gyno.2000.6042.
Ferrandina G, Lauriola L, Zannoni GF, Fagotti A, Fanfani F, Legge F, Maggiano N, Gessi M, Mancuso S, Ranelletti FO, Scambia G: Increased cyclooxygenase-2 (COX-2) expression is associated with chemotherapy resistance and outcome in ovarian cancer patients. Ann Oncol. 2002, 13: 1205-1211. 10.1093/annonc/mdf207.
Martinet L, Jean C, Dietrich G, Fournie JJ, Poupot R: PGE2 inhibits natural killer and gamma delta T cell cytotoxicity triggered by NKR and TCR through a cAMP-mediated PKA type I-dependent signaling. Biochem Pharmacol. 2010, 80: 838-845. 10.1016/j.bcp.2010.05.002.
Komarova S, Kawakami Y, Stoff-Khalili MA, Curiel DT, Pereboeva L: Mesenchymal progenitor cells as cellular vehicles for delivery of oncolytic adenoviruses. Mol Cancer Ther. 2006, 5: 755-766. 10.1158/1535-7163.MCT-05-0334.
Baratelli F, Lin Y, Zhu L, Yang SC, Heuze-Vourc'h N, Zeng G, Reckamp K, Dohadwala M, Sharma S, Dubinett SM: Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol. 2005, 175: 1483-1490.
Harden JL, Egilmez NK: Indoleamine 2,3-dioxygenase and dendritic cell tolerogenicity. Immunol Invest. 2012, 41: 738-764. 10.3109/08820139.2012.676122.
Qian F, Villella J, Wallace PK, Mhawech-Fauceglia P, Tario JD, Andrews C, Matsuzaki J, Valmori D, Ayyoub M, Frederick PJ: Efficacy of levo-1-methyl tryptophan and dextro-1-methyl tryptophan in reversing indoleamine-2,3-dioxygenase-mediated arrest of T-cell proliferation in human epithelial ovarian cancer. Cancer Res. 2009, 69: 5498-5504. 10.1158/0008-5472.CAN-08-2106.
Nonaka H, Saga Y, Fujiwara H, Akimoto H, Yamada A, Kagawa S, Takei Y, Machida S, Takikawa O, Suzuki M: Indoleamine 2,3-dioxygenase promotes peritoneal dissemination of ovarian cancer through inhibition of natural killercell function and angiogenesis promotion. Int J Oncol. 2011, 38: 113-120.
Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, Mellor AL: GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005, 22: 633-642. 10.1016/j.immuni.2005.03.013.
Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, Orabona C, Bianchi R, Belladonna ML, Volpi C: The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol. 2006, 176: 6752-6761.
Platten M, Wick W, Van den Eynde BJ: Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2011, 72: 5435-5440.
Winter WE, Maxwell GL, Tian C, Sundborg MJ, Rose GS, Rose PG, Rubin SC, Muggia F, McGuire WP: Tumor residual after surgical cytoreduction in prediction of clinical outcome in stage IV epithelial ovarian cancer: a Gynecologic Oncology Group Study. J Clin Oncol. 2008, 26: 83-89. 10.1200/JCO.2007.13.1953.
Covens AL: A critique of surgical cytoreduction in advanced ovarian cancer. Gynecol Oncol. 2000, 78: 269-274. 10.1006/gyno.2000.5926.
Hensler T, Hecker H, Heeg K, Heidecke CD, Bartels H, Barthlen W, Wagner H, Siewert JR, Holzmann B: Distinct mechanisms of immunosuppression as a consequence of major surgery. Infect Immun. 1997, 65: 2283-2291.
Baumgartner JM, McCarter MD: Suppressing the suppressor: Role of immunosuppressive regulatory T cells in cancer surgery. Surgery. 2009, 145: 345-350. 10.1016/j.surg.2008.12.013.
Zitvogel L, Kepp O, Kroemer G: Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol. 2011, 8: 151-160. 10.1038/nrclinonc.2010.223.
Ma Y, Aymeric L, Locher C, Mattarollo SR, Delahaye NF, Pereira P, Boucontet L, Apetoh L, Ghiringhelli F, Casares N: Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J Exp Med. 2011, 208: 491-503. 10.1084/jem.20100269.
Ramakrishnan R, Assudani D, Nagaraj S, Hunter T, Cho HI, Antonia S, Altiok S, Celis E, Gabrilovich DI: Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J Clin Invest. 2010, 120: 1111-1124. 10.1172/JCI40269.
Carson WE, Shapiro CL, Crespin TR, Thornton LM, Andersen BL: Cellular immunity in breast cancer patients completing taxane treatment. Clin Cancer Res. 2004, 10: 3401-3409. 10.1158/1078-0432.CCR-1016-03.
Zitvogel L, Apetoh L, Ghiringhelli F, Andre F, Tesniere A, Kroemer G: The anticancer immune response: indispensable for therapeutic success?. J Clin Invest. 2008, 118: 1991-2001. 10.1172/JCI35180.
Tesniere A, Apetoh L, Ghiringhelli F, Joza N, Panaretakis T, Kepp O, Schlemmer F, Zitvogel L, Kroemer G: Immunogenic cancer cell death: a key-lock paradigm. Curr Opin Immunol. 2008, 20: 504-511. 10.1016/j.coi.2008.05.007.
Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M: Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005, 202: 1691-1701. 10.1084/jem.20050915.
Kandalaft LE, Powell DJ, Singh N, Coukos G: Immunotherapy for ovarian cancer: what's next?. J Clin Oncol. 2011, 29: 925-933. 10.1200/JCO.2009.27.2369.
Preston CC, Goode EL, Hartmann LC, Kalli KR, Knutson KL: Immunity and immune suppression in human ovarian cancer. Immunotherapy. 2011, 3: 539-556. 10.2217/imt.11.20.
Leffers N, Daemen T, Helfrich W, Boezen HM, Cohlen BJ, Melief K, Nijman HW: Antigen-specific active immunotherapy for ovarian cancer. Cochrane Database Syst Rev. 2010, CD007287-10.1002/14651858.CD007287.pub2. Review. PMID: 20091627 [PubMed - indexed for MEDLINE], 1
Frederick PJ, Straughn JM, Alvarez RD, Buchsbaum DJ: Preclinical studies and clinical utilization of monoclonal antibodies in epithelial ovarian cancer. Gynecol Oncol. 2009, 113: 384-390. 10.1016/j.ygyno.2009.01.008.
Berek J, Taylor P, McGuire W, Smith LM, Schultes B, Nicodemus CF: Oregovomab maintenance monoimmunotherapy does not improve outcomes in advanced ovarian cancer. J Clin Oncol. 2009, 27: 418-425.
Reinartz S, Kohler S, Schlebusch H, Krista K, Giffels P, Renke K, Huober J, Mobus V, Kreienberg R, DuBois A: Vaccination of patients with advanced ovarian carcinoma with the anti-idiotype ACA125: immunological response and survival (phase Ib/II). Clin Cancer Res. 2004, 10: 1580-1587. 10.1158/1078-0432.CCR-03-0056.
Pfisterer J, du Bois A, Sehouli J, Loibl S, Reinartz S, Reuss A, Canzler U, Belau A, Jackisch C, Kimmig R: The anti-idiotypic antibody abagovomab in patients with recurrent ovarian cancer. A phase I trial of the AGO-OVAR. Ann Oncol. 2006, 17: 1568-1577. 10.1093/annonc/mdl357.
Makhija S, Amler LC, Glenn D, Ueland FR, Gold MA, Dizon DS, Paton V, Lin CY, Januario T, Ng K: Clinical activity of gemcitabine plus pertuzumab in platinum-resistant ovarian cancer, fallopian tube cancer, or primary peritoneal cancer. J Clin Oncol. 2010, 28: 1215-1223. 10.1200/JCO.2009.22.3354.
Konner JA, Bell-McGuinn KM, Sabbatini P, Hensley ML, Tew WP, Pandit-Taskar N, Vander Els N, Phillips MD, Schweizer C, Weil SC: Farletuzumab, a humanized monoclonal antibody against folate receptor alpha, in epithelial ovarian cancer: a phase I study. Clin Cancer Res. 2010, 16: 5288-5295. 10.1158/1078-0432.CCR-10-0700.
Heiss MM, Murawa P, Koralewski P, Kutarska E, Kolesnik OO, Ivanchenko VV, Dudnichenko AS, Aleknaviciene B, Razbadauskas A, Gore M: The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int J Cancer. 2010, 127: 2209-2221. 10.1002/ijc.25423.
Rosenblum MG, Verschraegen CF, Murray JL, Kudelka AP, Gano J, Cheung L, Kavanagh JJ: Phase I study of 90Y-labeled B72.3 intraperitoneal administration in patients with ovarian cancer: effect of dose and EDTA coadministration on pharmacokinetics and toxicity. Clin Cancer Res. 1999, 5: 953-961.
Diefenbach CS, Gnjatic S, Sabbatini P, Aghajanian C, Hensley ML, Spriggs DR, Iasonos A, Lee H, Dupont B, Pezzulli S: Safety and immunogenicity study of NY-ESO-1b peptide and montanide ISA-51 vaccination of patients with epithelial ovarian cancer in high-risk first remission. Clin Cancer Res. 2008, 14: 2740-2748. 10.1158/1078-0432.CCR-07-4619.
Odunsi K, Qian F, Matsuzaki J, Mhawech-Fauceglia P, Andrews C, Hoffman EW, Pan L, Ritter G, Villella J, Thomas B: Vaccination with an NY-ESO-1 peptide of HLA class I/II specificities induces integrated humoral and T cell responses in ovarian cancer. Proc Natl Acad Sci U S A. 2007, 104: 12837-12842. 10.1073/pnas.0703342104.
Leffers N, Lambeck AJ, Gooden MJ, Hoogeboom BN, Wolf R, Hamming IE, Hepkema BG, Willemse PH, Molmans BH, Hollema H: Immunization with a P53 synthetic long peptide vaccine induces P53-specific immune responses in ovarian cancer patients, a phase II trial. Int J Cancer. 2009, 125: 2104-2113. 10.1002/ijc.24597.
Disis ML, Gooley TA, Rinn K, Davis D, Piepkorn M, Cheever MA, Knutson KL, Schiffman K: Generation of T-cell immunity to the HER-2/neu protein after active immunization with HER-2/neu peptide-based vaccines. J Clin Oncol. 2002, 20: 2624-2632. 10.1200/JCO.2002.06.171.
Chianese-Bullock KA, Irvin WP, Petroni GR, Murphy C, Smolkin M, Olson WC, Coleman E, Boerner SA, Nail CJ, Neese PY: A multipeptide vaccine is safe and elicits T-cell responses in participants with advanced stage ovarian cancer. J Immunother. 2008, 31: 420-430. 10.1097/CJI.0b013e31816dad10.
Gulley JL, Arlen PM, Tsang KY, Yokokawa J, Palena C, Poole DJ, Remondo C, Cereda V, Jones JL, Pazdur MP: Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clin Cancer Res. 2008, 14: 3060-3069. 10.1158/1078-0432.CCR-08-0126.
Hernando JJ, Park TW, Fischer HP, Zivanovic O, Braun M, Polcher M, Grunn U, Leutner C, Potzsch B, Kuhn W: Vaccination with dendritic cells transfected with mRNA-encoded folate-receptor-alpha for relapsed metastatic ovarian cancer. Lancet Oncol. 2007, 8: 451-454. 10.1016/S1470-2045(07)70142-0.
Brossart P, Wirths S, Stuhler G, Reichardt VL, Kanz L, Brugger W: Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood. 2000, 96: 3102-3108.
Hernando JJ, Park TW, Kubler K, Offergeld R, Schlebusch H, Bauknecht T: Vaccination with autologous tumour antigen-pulsed dendritic cells in advanced gynaecological malignancies: clinical and immunological evaluation of a phase I trial. Cancer Immunol Immunother. 2002, 51: 45-52. 10.1007/s00262-001-0255-1.
Zhao X, Wei YQ, Peng ZL: Induction of T cell responses against autologous ovarian tumors with whole tumor cell lysate-pulsed dendritic cells. Immunol Invest. 2001, 30: 33-45. 10.1081/IMM-100103689.
Aoki Y, Takakuwa K, Kodama S, Tanaka K, Takahashi M, Tokunaga A, Takahashi T: Use of adoptive transfer of tumor-infiltrating lymphocytes alone or in combination with cisplatin-containing chemotherapy in patients with epithelial ovarian cancer. Cancer Res. 1991, 51: 1934-1939.
Freedman RS, Edwards CL, Kavanagh JJ, Kudelka AP, Katz RL, Carrasco CH, Atkinson EN, Scott W, Tomasovic B, Templin S: Intraperitoneal adoptive immunotherapy of ovarian carcinoma with tumor-infiltrating lymphocytes and low-dose recombinant interleukin-2: a pilot trial. J Immunother Emphasis Tumor Immunol. 1994, 16: 198-210. 10.1097/00002371-199410000-00004.
Fujita K, Ikarashi H, Takakuwa K, Kodama S, Tokunaga A, Takahashi T, Tanaka K: Prolonged disease-free period in patients with advanced epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Clin Cancer Res. 1995, 1: 501-507.
Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L: A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006, 12: 6106-6115. 10.1158/1078-0432.CCR-06-1183.
Dobrzanski MJ, Rewers-Felkins KA, Quinlin IS, Samad KA, Phillips CA, Robinson W, Dobrzanski DJ, Wright SE: Autologous MUC1-specific Th1 effector cell immunotherapy induces differential levels of systemic TReg cell subpopulations that result in increased ovarian cancer patient survival. Clin Immunol. 2009, 133: 333-352. 10.1016/j.clim.2009.08.007.
Curran KJ, Pegram HJ, Brentjens RJ: Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J Gene Med. 2012, 14: 405-415.
Sadelain M, Brentjens R, Riviere I: The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3: 388-398. 10.1158/2159-8290.CD-12-0548.
Berd D, Maguire HC, Mastrangelo MJ: Induction of cell-mediated immunity to autologous melanoma cells and regression of metastases after treatment with a melanoma cell vaccine preceded by cyclophosphamide. Cancer Res. 1986, 46: 2572-2577.
Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B: Metronomic cyclophosphamide regimen selectively depletes CD4 + CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007, 56: 641-648. 10.1007/s00262-006-0225-8.
Attia P, Maker AV, Haworth LR, Rogers-Freezer L, Rosenberg SA: Inability of a fusion protein of IL-2 and diphtheria toxin (Denileukin Diftitox, DAB389IL-2, ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J Immunother. 2005, 28: 582-592. 10.1097/01.cji.0000175468.19742.10.
Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ, Colligon TA, Trosko JA, Leinbach LI, Pletcher CH: CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med. 2012, 4: 134ra162-
Barnett B, Kryczek I, Cheng P, Zou W, Curiel TJ: Regulatory T cells in ovarian cancer: biology and therapeutic potential. Am J Reprod Immunol. 2005, 54: 369-377. 10.1111/j.1600-0897.2005.00330.x.
Sutmuller RP, van Duivenvoorde LM, van Elsas A, Schumacher TN, Wildenberg ME, Allison JP, Toes RE, Offringa R, Melief CJ: Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med. 2001, 194: 823-832. 10.1084/jem.194.6.823.
Wilke CM, Kryczek I, Wei S, Zhao E, Wu K, Wang G, Zou W: Th17 cells in cancer: help or hindrance?. Carcinogenesis. 2011, 32: 643-649. 10.1093/carcin/bgr019.
Greten TF, Zhao F, Gamrekelashvili J, Korangy F: Human Th17 cells in patients with cancer: Friends or foe?. Oncoimmunology. 2012, 1: 1438-1439. 10.4161/onci.21245.
Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C: T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009, 31: 787-798. 10.1016/j.immuni.2009.09.014.
Charles KA, Kulbe H, Soper R, Escorcio-Correia M, Lawrence T, Schultheis A, Chakravarty P, Thompson RG, Kollias G, Smyth JF: The tumor-promoting actions of TNF-alpha involve TNFR1 and IL-17 in ovarian cancer in mice and humans. J Clin Invest. 2009, 119: 3011-3023. 10.1172/JCI39065.
Wu X, Lee VC, Chevalier E, Hwang ST: Chemokine receptors as targets for cancer therapy. Curr Pharm Des. 2009, 15: 742-757. 10.2174/138161209787582165.
Pere H, Montier Y, Bayry J, Quintin-Colonna F, Merillon N, Dransart E, Badoual C, Gey A, Ravel P, Marcheteau E: A CCR4 antagonist combined with vaccines induces antigen-specific CD8+ T cells and tumor immunity against self antigens. Blood. 2011, 118: 4853-4862. 10.1182/blood-2011-01-329656.
Zollo M, Di Dato V, Spano D, De Martino D, Liguori L, Marino N, Vastolo V, Navas L, Garrone B, Mangano G: Targeting monocyte chemotactic protein-1 synthesis with bindarit induces tumor regression in prostate and breast cancer animal models. Clin Exp Metastasis. 2012, 29: 585-601. 10.1007/s10585-012-9473-5.
Sevko A, Umansky V: Myeloid-derived suppressor cells interact with tumors in terms of myelopoiesis, tumorigenesis and immunosuppression: thick as thieves. J Cancer. 2013, 4: 3-11. 10.7150/jca.5047.
Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, Lush RM, Antonia S, Gabrilovich DI: All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006, 66: 9299-9307. 10.1158/0008-5472.CAN-06-1690.
Shurin MR, Naiditch H, Gutkin DW, Umansky V, Shurin GV: ChemoImmunoModulation: immune regulation by the antineoplastic chemotherapeutic agents. Curr Med Chem. 2012, 19: 1792-1803. 10.2174/092986712800099785.
Melero I, Grimaldi AM, Perez-Gracia JL, Ascierto PA: Clinical development of immunostimulatory monoclonal antibodies and opportunities for combination. Clin Cancer Res. 2013, 19: 997-1008. 10.1158/1078-0432.CCR-12-2214.
Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP: Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009, 206: 1717-1725. 10.1084/jem.20082492.
Hodi FS, Butler M, Oble DA, Seiden MV, Haluska FG, Kruse A, Macrae S, Nelson M, Canning C, Lowy I: Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A. 2008, 105: 3005-3010. 10.1073/pnas.0712237105.
Abiko K, Mandai M, Hamanishi J, Yoshioka Y, Matsumura N, Baba T, Yamaguchi K, Murakami R, Yamamoto A, Kharma B: PD-L1 on Tumor Cells Is Induced in Ascites and Promotes Peritoneal Dissemination of Ovarian Cancer through CTL Dysfunction. Clin Cancer Res. 2013, 19: 1363-1374. 10.1158/1078-0432.CCR-12-2199.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M: Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012, 366 (26): 2443-2454. 10.1056/NEJMoa1200690. Epub 2012 Jun 2
Karim R, Jordanova ES, Piersma SJ, Kenter GG, Chen L, Boer JM, Melief CJ, van der Burg SH: Tumor-expressed B7-H1 and B7-DC in relation to PD-1+ T-cell infiltration and survival of patients with cervical carcinoma. Clin Cancer Res. 2009, 15: 6341-6347. 10.1158/1078-0432.CCR-09-1652.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K: Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012, 366: 2455-2465. 10.1056/NEJMoa1200694.
Hamid O, Carvajal RD: Anti-programmed death-1 and anti-programmed death-ligand 1 antibodies in cancer therapy. Expert Opin Biol Ther. 2013, 13 (6): 847-861. 10.1517/14712598.2013.770836. Epub 2013 Feb 19. PMID: 23421934 [PubMed - in process]
Benson DM, Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, Bakan C, Andre P, Efebera Y, Tiollier J: A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood. 2012, 120: 4324-4333. 10.1182/blood-2012-06-438028.
Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, Etienne A, Andre P, Romagne F, Benson D: A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood. 2012, 120: 4317-4323. 10.1182/blood-2012-06-437558.
Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, Leffet L, Hansbury MJ, Thomas B, Rupar M: Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010, 115: 3520-3530. 10.1182/blood-2009-09-246124.
Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S: Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007, 67: 4507-4513. 10.1158/0008-5472.CAN-06-4174.
Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, Hegmans JP: COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma, Celecoxib influences MDSC function. BMC Cancer. 2010, 10: 464-10.1186/1471-2407-10-464.
Arber N, Eagle CJ, Spicak J, Racz I, Dite P, Hajer J, Zavoral M, Lechuga MJ, Gerletti P, Tang J: Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006, 355: 885-895. 10.1056/NEJMoa061652.
Rogers TL, Holen I: Tumour macrophages as potential targets of bisphosphonates. J Transl Med. 2011, 9: 177-10.1186/1479-5876-9-177.
Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Hegmans JP, Aerts JG: Zoledronic acid impairs myeloid differentiation to tumour-associated macrophages in mesothelioma. Br J Cancer. 2010, 103: 629-641. 10.1038/sj.bjc.6605814.
Acknowledgements
The authors thank American Journal Experts for editing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Author’ contributions
VL, AT, FF, SH, VJ and FC reviewed the literature; VL, AT and FC wrote the paper; VL, JL, VC, MABR and FC proofread the final copy. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lavoué, V., Thédrez, A., Levêque, J. et al. Immunity of human epithelial ovarian carcinoma: the paradigm of immune suppression in cancer. J Transl Med 11, 147 (2013). https://doi.org/10.1186/1479-5876-11-147
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-5876-11-147