
Abstract
Introduction
The biologics abatacept and adalimumab have different mechanisms of action (MoAs). We analyzed data from patients with rheumatoid arthritis treated in AMPLE (NCT00929864) to explore the pharmacodynamic effects of abatacept or adalimumab on anti-citrullinated protein antibodies (ACPAs) and gene expression.
Methods
AMPLE was a phase IIIb, 2-year, randomized, head-to-head trial of abatacept versus adalimumab. Post hoc analyses of baseline anti-cyclic citrullinated peptide-2 (anti-CCP2, an ACPA surrogate) positive (+) status and ACPA fine-specificity profiles over time, as well as transcriptional profiling (peripheral whole blood), were performed.
Results
Of 646 patients treated (abatacept, n = 318; adalimumab, n = 328), ACPA and gene expression data were available from 508 and 566 patients, respectively. In anti-CCP2+ patients (n = 388), baseline fine specificities for most ACPAs were highly correlated; over 2 years, levels decreased with abatacept but not adalimumab. By year 2, expression of genes associated with T cell co-stimulation and antibody production was lower for abatacept versus adalimumab; expression of genes associated with proinflammatory signaling was lower for adalimumab versus abatacept. Treatment modulated the expression of T- and B-cell gene signatures, with differences in CD8+ T cells, activated T cells, plasma cells, B cells, natural killer cells (all lower with abatacept versus adalimumab), and polymorphonuclear leukocytes (higher with abatacept versus adalimumab).
Conclusions
In AMPLE, despite similar clinical outcomes, data showed that pharmacodynamic/genetic changes after 2 years of abatacept or adalimumab were consistent with drug MoAs. Further assessment of the relationship between such changes and clinical outcomes, including prediction of response, is warranted.
Trial Registration
ClinicalTrials.gov identifier, NCT00929864.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Improved knowledge of the relevant pathological processes in rheumatoid arthritis (RA) has led to the development of targeted therapies with differing mechanisms of action (MoAs), such as adalimumab and abatacept. |
We aimed to provide new insights into the pharmacodynamic (PD) effects of treatment with abatacept versus adalimumab by profiling ACPAs and gene expression in patients from the AMPLE phase 3 clinical study. |
What was learned from this study? |
PD changes, as reflected by differing ACPA and gene expression profiles and immune cell signatures, observed after 2 years of abatacept or adalimumab treatment were consistent with the hypothesized MoAs of these agents; expression of genes related to activation of the immune system was lower with abatacept. |
These findings illustrate how gene expression studies can provide potentially valuable information in addition to that gained from conventional clinical assessments, as they investigate underlying processes that are beyond the clinical manifestations of disease. |
Further analysis of this data set from the AMPLE study is warranted and may provide valuable information regarding predictive biomarkers of response and disease progression. |
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease that primarily impacts joints and has a complex and still incompletely understood etiology, despite the remarkable innovations in available therapies [1]. In RA, constant renewal of the T cell–initiated immune response results in the production of autoantibodies, particularly anti-citrullinated protein antibodies (ACPAs), and the perpetuation of proinflammatory cytokines [1,2,3,4,5]. The understanding of the relevance of ACPAs in RA has evolved over time as their role in disease pathogenesis [6,7,8] and association with poor prognosis has been elucidated [1, 9]. This has led to their clinical utility being recognized; as such, ACPAs are included in the 2010 American College of Rheumatology (ACR)/European League Against Rheumatism diagnostic criteria [10]. In parallel, an understanding of the relevance of key cytokines and their impact on cellular pathways has led to better immunophenotyping approaches, and such information continues to guide targeted therapeutic development [11].
Biologic (b) disease-modifying antirheumatic drug (DMARD) therapies are included in the current treatment paradigm for patients with active RA. For those with an inadequate response to conventional synthetic (cs)DMARDs [9, 12], a treat-to-target approach is recommended, with the goal of suppressing inflammation and preventing joint damage that can lead to disability. Improved knowledge of the relevant pathological processes has led to the development of targeted therapies with differing mechanisms of action (MoAs), such as adalimumab and abatacept. Abatacept is an immunomodulator that disrupts the T cell activation that is characteristic of RA by blocking CD28-mediated activation [1, 13], while adalimumab is a monoclonal antibody that binds to tumor necrosis factor (TNF)-alpha and inhibits its functions [1]. Currently, there is no evidence-based approach to inform bDMARD treatment selection based on the particular characteristics of individual patients. A better understanding of how to match patients with RA to specific therapies is desirable.
The head-to-head abatacept versus adalimumab comparison in biologic-naive RA subjects with background methotrexate (AMPLE; NCT00929864) trial demonstrated non-inferiority for subcutaneous (SC) abatacept versus adalimumab at 1 year [14], with responses maintained over 2 years [15]. These group-level clinical effects were seen across multiple clinical outcomes, including radiographic assessments, for both drugs. Although the treatment groups in AMPLE showed similar responses, it is reasonable to ask if these therapies with different MoAs produced similar pharmacodynamic (PD) effects, or if, despite the similar clinical outcomes seen, there could still be other physiological differences that might have implications for treatment selection. For this reason, we initiated post hoc assessments of biomarkers from patients in the AMPLE trial. Group-level PD differences with potentially relevant clinical implications have been previously reported. Analysis of anti-cyclic citrullinated peptide-2 (anti-CCP2, an ACPA surrogate) positivity (+) at baseline was associated with better response in both the abatacept and adalimumab treatment groups [6]. Response was more pronounced in the subgroup of patients in the highest anti-CCP2+ concentration quartile receiving abatacept compared with adalimumab [6]. In addition, an association of anti-CCP status with abatacept but not TNF inhibitors (TNFis) has been observed in registry data [16, 17].
The clinical relevance of a reduction in levels of autoantibodies including ACPAs in response to treatment is unknown, but there is some evidence that, when tapering csDMARD or bDMARD therapy, the profile of autoantibody response against modified proteins, including citrullinated proteins, is associated with risk of relapse [18]. Furthermore, in an observational study of 100 patients with RA followed up for 2.5 years, differential effects of DMARD treatments on anti-CCP2 antibody levels have been shown: levels were significantly decreased by abatacept and rituximab treatment, and unchanged with methotrexate (MTX), tocilizumab, or TNFi treatment [19], which may reflect the different drug MoAs.
Based on our initial findings, we postulate that the underlying PD effects of abatacept and adalimumab have notable differences based on their MoAs. These differences hint at patient-level differential responses to therapy for some patients with RA, as well as the potential for biomarker-guided therapeutic decision making. The AMPLE trial is particularly well suited to explore the relationship between drug MoA, PD effects, and clinical outcomes due to its well-characterized, relatively large sample size and long observation period. In this post hoc analysis of AMPLE, we aimed to provide new insights into the PD effects of treatment with abatacept versus adalimumab by profiling ACPAs and whole-blood gene expression. Understanding the impact of specific treatments on ACPAs may improve our knowledge of the dynamic changes in autoantibody-mediated downstream effector pathways, whereas characterizing gene expression may help to reveal the broader impact of treatment on cellular functions. Specifically, the objectives of this analysis were to: (1) assess ACPA fine-specificity profiles and identify changes in ACPAs over time associated with abatacept or adalimumab treatment in patients who were anti-CCP2+ at baseline; (2) evaluate whole-blood transcriptional profiling data to determine the changes in mRNA gene expression over time; and (3) estimate changes in immune cell–type signatures associated with abatacept or adalimumab treatment.
Methods
Study Design, Patient Population, and Sample Collection
The design of the AMPLE trial has been reported previously [14]. Briefly, AMPLE was a phase IIIb, 2-year, multinational, prospective, randomized, investigator-blinded, head-to-head trial with a primary endpoint of treatment non-inferiority, assessed by the proportion of patients with 20% improvement in ACR criteria at 1 year. Biologic-naive patients with active RA and an inadequate response to MTX were randomized 1:1 to abatacept 125 mg SC weekly or adalimumab 40 mg SC bi-weekly, both with a stable dose of MTX.
Serum and peripheral blood mononuclear cell samples were collected, frozen, and stored for exploratory biomarker analysis. Collections were performed at baseline, day 85, year 1, and year 2.
Analysis Outcomes
The analyses reported here were performed post hoc. In the ACPA analysis, outcomes in patients who were anti-CCP2+ were the assessment of the correlation of different ACPA fine-specificity profiles at baseline and changes in ACPA reactivity over time. In the analysis of whole-blood gene expression, outcomes were the identification of specific genes showing PD effect in whole blood and the estimation of changes over time in levels of immune cells, including plasma cells, derived from gene expression surrogates.
ACPA Analysis
Serum samples collected at baseline, day 85, year 1, and year 2 were analyzed to determine ACPA fine-specificity profiles by Luminex Multiplex Assay (R&D Systems, MN, USA), as previously described [20]. A panel of 19 ACPAs (14 citrullinated peptides and five citrullinated proteins) was used for reactivity testing (peptide sequences are shown in Supplementary Material Table S1). Reactivity to two forms of CCP and rheumatoid factor testing were also included. Anti-CCP2+ status was determined at baseline using the commercial anti-CCP2 immunoglobulin G (IgG) enzyme-linked immunosorbent assay (Euro Diagnostica Immunoscan CCPlus, Malmö, Sweden; obtained from IBL America); patients with an anti-CCP2 IgG concentration of ≥ 25 AU/ml were considered to be positive. A linear regression mixed-effects model was applied to the profiles to evaluate treatment difference at year 2.
ACPA fine-specificity profiles were generated separately for the 30.2% of patients across both treatment groups who met the definition of major clinical response (MCR; defined as maintenance of 70% improvement in ACR criteria over a continuous 6-month period) during the study period.
Gene Expression Analysis
Peripheral whole blood was collected in PAXgene tubes (BD Biosciences, San José, CA, USA) at baseline, day 85, year 1, and year 2 for transcriptional profiling. RNA was isolated from all available samples (Qiagen Kit, Hilden, Germany) following the manufacturer’s instructions.
Transcriptional profiling was performed using Affymetrix U219 GeneChips. Blood samples were collected over a 3-year period (2009–2012). Samples were processed in two batches: an early cohort comprising baseline and day 85 and a late cohort comprising year 1 and year 2 samples. The strategy to remove batch effect was as follows: RNA profiling from a representative set of 54 patients (9.5% of the 568 patients at baseline) was repeated in the late cohort; a linear model was used to compute the batch-specific differences using the repeated samples. Poor probes and non-informative gene matches were sequentially eliminated via an alternative chip definition file (BrainArray Ensembl chip definition file based on Human Genome build GRCh37) [21] and I/NI filter, respectively.
Differential Gene Expression, Transcriptional Modules, and Molecular Pathway Analyses
Differential expression of genes over time and between treatment arms was assessed with a linear model using clinical covariates and a multiple testing correction. A mixed measures repeated model (MMRM) was employed. In the linear MMRM analysis, covariates were baseline Disease Activity Score in 28 joints using C-reactive protein, country, race, and sex; to capture individual-level variation in gene expression over time, ‘visit’ and ‘patient’ were used as random effects. A correction for multiple hypothesis testing was applied using the false discovery rate (FDR) method [22] (Supplementary Material). The model was as follows:
Differentially expressed genes were considered to be those with a fold-change of > 0.2 (up-regulated) and < − 0.2 (down-regulated), and adjusted p value < 0.05 from baseline to year 2 (see Supplementary Material for note on programming code). p values were calculated using the Imer4 package; an additional analysis was also implemented to test the robustness of random effect estimates using bootstrapping to estimate empirical p values. Genes that were differentially expressed between treatment arms over time with FDR < 0.1 are reported.
Genes that were differentially expressed between treatment arms over time with FDR < 0.05 were analyzed for molecular pathway enrichment using MetaCore process networks (Clarivate Analytics, Philadelphia, PA, USA). Single Sample Gene Set Enrichment Analysis [23] was used to calculate enrichment for transcriptional modules derived from the literature [24].
Analysis of Immune Cell Types by Gene Signature
Transcriptional profiling of naive and activated immune cell types was used to generate gene signatures specific to an immune cell type, as a proxy for circulating cell concentrations, using computational deconvolution methods. Gene signatures were derived from purified cell types and validated using a separate cohort from the Benaroya Research Institute at Virginia Mason (Seattle, WA, USA), comprising normal healthy volunteers, patients with systemic lupus erythematosus (SLE), and patients with multiple sclerosis (Supplementary Material Fig. S1) [25, 26]. Immune cells assessed were T and B lymphocytes, including CD4+, CD8+, and T cells stimulated with CD3/CD28 (CD4/CD8 mixed), polymorphonuclear (PMN) leukocytes, natural killer (NK) cells, and monocytes. The mean expression of cell type-specific signatures was used to compare treatment arms and a t test was applied to the profiles to evaluate treatment difference at year 2.
Compliance with Ethics Guidelines
All patients in AMPLE provided written informed consent. The trial was conducted in accordance with the Helsinki Declaration of 1964, and its later amendments, and the protocol was approved by the institutional review boards and independent ethics committees at the participating sites.
Results
Sample Availability and Baseline Characteristics of Patients
Overall, 646 patients were randomized and treated (abatacept, n = 318; adalimumab, n = 328). At baseline, serum for determination of anti-CCP2+ status and ACPA profiling was available from 251 patients treated with abatacept and 257 patients treated with adalimumab. Anti-CCP2 was not measured at later time points; ACPAs were also measured at day 85 (abatacept, n = 213; adalimumab, n = 215), year 1 (abatacept, n = 204; adalimumab, n = 199), and year 2 (abatacept, n = 139; adalimumab, n = 131). Affymetrix gene expression data were available for 566 patients at baseline, 493 patients at year 1, and 430 patients at year 2. Of 430 evaluable patients at year 2, 218 were treated with abatacept and 212 were treated with adalimumab.
Baseline demographic and clinical characteristics were generally similar for the ACPA analysis and gene expression analysis subsets, and the overall population (Table 1).
ACPA Profiles at Baseline and Over Time
We tested baseline serum samples for all available patients who were anti-CCP2+ (n = 388) for ACPA fine-specificity profiles against a panel of 14 citrullinated peptide and five citrullinated protein antigens (Fig. 1a). Most ACPAs on the panel were found to be highly correlated with other ACPAs at baseline. Inspection of the distributions showed that very few patients had high signal (Supplementary Material Fig. S2).

Fine-specificity ACPAs in patients who were anti-CCP2+ at baseline. a Correlation plot of ACPAs at baselinea; b Change in ACPA specificity profiles from baseline to year 2 in the overall population of patients treated with abatacept or adalimumab; c Change in ACPA specificity profiles from baseline to year 2 in patients with MCR treated with abatacept or adalimumab. aVertical and horizontal axes list the ACPAs tested in the analysis; at the intersection between each row/column, spots of varying shades are presented depending on how correlated the two ACPAs were. ACPA anti-citrullinated protein antibody, anti-CCP2 anti-cyclic citrullinated peptide-2, FDR false discovery rate, MCR major clinical response. c adapted from Connolly SC, et al. EULAR Congress 2014; June 11–14, 2014; Paris France; poster FRI0039 (with permission of the authors)
Reactivity profiles from baseline to year 2 differed by treatment (Fig. 1b; Supplementary Material Fig. S3). In general, levels decreased in both treatment groups in year 1 but in year 2, while the levels continued to decrease in the abatacept arm, they tended to increase in the adalimumab arm. Profiles that were significantly different between treatments were fibrinogen A 211–230, fibrinogen B 246–267, and apolipo E 277–296. In the subset of patients who achieved an MCR, the increase in median change from baseline in the adalimumab arm were more pronounced (Fig. 1c).
Identification of Differentially Expressed Genes, Transcriptional Modules, and Molecular Pathways
Results of gene expression analysis from baseline to year 2 showed notable differences between treatment groups (Table 2, Fig. 2), which appeared to be related to the respective MoAs. The expression of genes that encode proteins associated with T cell co-stimulation and antibody production was lower in the abatacept arm versus the adalimumab arm (Table 2). Genes transcribed in response to proinflammatory signals (CXCL1, IL1b, ORM1) were expressed at lower levels in the adalimumab arm compared with the abatacept arm (Table 2).

Genes differentially expressed between treatment arms at year 2. a Volcano plota; b Heat map of top 20 differentially expressed genes. aStatistical significance (adjusted p value) versus magnitude of change (effect size) is shown so that genes with large effect sizes that are also statistically significant can be quickly identified. FDR false discovery rate
Expression of modules of immune genes also differed between the two arms, with modules for ‘IL-2–activated NK cells’ and ‘B cells’ down-regulated with abatacept, and modules for ‘cell cycle metaphase’ down-regulated with adalimumab (Table 2).
The most differentially expressed genes at year 2 are shown (Fig. 2). There were 99 genes with FDR < 0.1, with a number of genes displaying lower expression levels with abatacept compared with adalimumab, and other genes showing higher expression levels with abatacept compared with adalimumab. Of the genes showing lower expression levels with abatacept compared with adalimumab treatment, CTLA4, CCR4, and SLAMF1 were the most statistically significant (Fig. 2a); the plasma cell gene IGHA1 had the largest magnitude of fold change (Fig. 2a and 2b). Among genes showing increased levels of expression with abatacept compared with adalimumab treatment, the PCSK5 gene was the most statistically significant (Fig. 2a); the PI3 gene had the largest magnitude of fold change (Fig. 2a and 2b).
Certain molecular pathways, including those involved in T-helper cell differentiation and T cell receptor signaling, were significantly lower in the abatacept treatment arm compared with the adalimumab arm (Table 3). Other pathways were significantly lower in their expression levels in the adalimumab treatment arm compared with the abatacept arm (Table 3).
Estimating Immune Cell Types by Gene Signatures
Results from transcriptional profiling of cell type signatures from baseline to year 2 are shown (Fig. 3). By year 2, both T- and B-cell gene signatures increased, but differences between treatment arms were observed, including in CD8 + T cells and activated T cells (anti CD3 and anti CD28), which were decreased with abatacept compared with adalimumab treatment. In addition, gene signatures of plasmablasts, B cells, and NK cells were decreased with abatacept treatment, and PMN leukocyte levels were increased compared with levels in patients treated with adalimumab.

Expression of immune cell type-specific gene signatures from baseline to year 2. *Profiles which were significantly different (p < 0.05; t test) between treatments at year 2. IL interleukin, IZ isoleucine zipper, MFI mean fluorescence intensity, NK natural killer, PMN polymorphonuclear, SC subcutaneous, SE standard error
Discussion
The AMPLE trial was the first head-to-head study in RA to compare two bDMARDs, abatacept and adalimumab, and the only trial to obtain controlled data over 2 years of treatment. To our knowledge, this post hoc analysis of data from the large randomized controlled AMPLE trial is the first of its kind to assess a range of PD effects over 2 years in response to therapies with different MoAs but with identical group-level responses, and to provide a detailed characterization of the impact of TNFis on ACPAs. We have shown broad and differential impact on the overall pattern of ACPA fine specificities, immune cell profiles, and gene expression during treatment, which provides insights into each drug’s respective effects on adaptive immunity and autoimmune processes. Distinguishing which of these effects represent abnormalities that drive early disease development and which reflect later phenotypic changes that amplify disease progression or are counter regulatory is an ongoing effort.
At baseline, individual ACPAs were highly correlated with each other in patients who were anti-CCP2+. ACPAs may be present in serum prior to clinical disease onset [20, 27]; therefore, the finding of high correlation between ACPAs suggests broad activation of ACPA-producing B cells prior to the onset of clinical RA symptoms. ACPA reactivity profiles are known to change over time; while the relevance of such changes to understanding response to specific therapies is of interest, it is not well established [28,29,30]. Here, changes in ACPA reactivity over time showed a prominent global effect of treatment in a large sample size, such that, over 2 years of treatment, ACPA reactivity profiles decreased with abatacept but less so with adalimumab. The findings are consistent with those previously observed for overall ACPA levels during abatacept treatment of anti-CCP2+ RA [31, 32]. The greater decrease in ACPA titers seen with abatacept is congruent with its MoA, whereby T cell-dependent B cell responses such as ACPA generation are inhibited. ACPA testing in a clinical setting has usually been relegated to a diagnostic role. These findings suggest that effective therapy can decrease ACPA levels but the relationship is complex. Both therapies initiated a decrease in overall titers that was sustained over 2 years in the abatacept group but that started to reverse in the adalimumab group after year 1. Paradoxically, this differential response was more pronounced, compared with the overall population, among the best responders (i.e., those who achieved MCR and who therefore represent a unique sample of patients with very deep and sustained responses to therapy). This suggests that an ongoing decrease in ACPA titers may not be associated with clinical efficacy, and also suggests that the impact of ACPA titers can be mitigated by specific therapy (i.e., TNF inhibition). It clearly shows that abatacept and adalimumab have a differential impact on a relevant pathogenic biomarker, which could suggest differential benefit in patients whose disease process is more driven by ACPAs.
RA is characterized by the dysregulation of multiple cytokines, chemokines, and cellular abnormalities. We wanted to describe the overall differential effects associated with abatacept and adalimumab therapy. Effects of treatment on gene expression profiles were consistent with the known MoA of each drug, but the specific genes affected were of interest. Genes known to be involved in T cell co-stimulation and antibody production were down-regulated with abatacept versus adalimumab, whereas proinflammatory signal genes were up-regulated. Some of the most differentially expressed genes have not been previously associated with RA, or are poorly studied (e.g., PI3/elafin, ALPL, PROK2, GBP5, and ZNF683) and warrant further investigation. Transcriptional modules of genes that are co-expressed have proven useful in identifying disease-specific gene expression patterns that inform disease pathogenesis [33]. Therefore, we extended our analysis beyond the level of individual genes to look at immune-specific transcriptional modules, in order to explore whether bDMARDs of differing MoAs would impact different transcriptional modules. We found that modules identified as being differentially expressed between treatment arms were consistent with the known MoAs. These gene modules were derived from a modular analysis framework of gene sets that co-cluster across diseases, which was previously used to characterize blood microarray transcriptional profiles from patients with SLE [24]. Use of such strategies for the analysis of large-scale transcriptomic data may provide insights into mechanisms of disease pathogenesis and provide a source of potential biomarkers.
Changes in levels of immune cell types over time were also in line with the known MoA of each drug. Levels of plasma cells, B cells, and NK cells were lower in the abatacept treatment arm compared with the adalimumab treatment arm, consistent with the mechanism of disruption of T cell activation and subsequent T cell help with abatacept. In a previous study of cell-specific gene signatures in patients with SLE, a higher proportion of activated (CD3/CD28-stimulated) T cells corresponded with higher interferon activity levels [25]; the significance of these findings is not fully understood. Distinct immune cell phenotypic clusters have been suggested to predict potential response to abatacept therapy [34]; although treatment response was not assessed in the current analysis, the clearest difference between abatacept and adalimumab was seen with the cluster enriched with antibody-producing plasma cells (and double-stranded [ds] DNA). A recent study of whole-blood cell signatures at baseline identified immune cell phenotypes that were associated with poor response to abatacept in patients with newly diagnosed type 1 diabetes, most notably a transient increase in activated B cells [35]. This finding suggests that baseline cell profiles may predict treatment resistance in type 1 diabetes, which may also be true for RA and other autoimmune disorders.
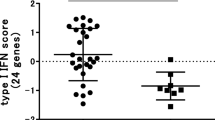
There is a scarcity of literature from other phase III trials investigating the effect of bDMARD treatment on gene expression in whole blood over time in patients with RA. A retrospective, observational study in patients with RA examined the gene expression signatures in patients treated with one of three bDMARDS, including abatacept, and found no overlap of gene sets between the three agents, suggesting that the molecular targets of each are distinct [36], which is in line with the results from the current analysis. Studies investigating gene expression biomarkers for MTX response have also been scarce, although a whole-blood transcript profiling study showed that changes in gene expression (specifically, type I interferon signaling pathway genes) soon after MTX initiation may provide an early classifier of those patients with RA who are unlikely to benefit from MTX over the longer term [37]. Although these data require validation, they support the importance of biomarker monitoring early in RA treatment.
The production of T cell-dependent autoantibodies such as ACPAs is a hallmark of RA. By disrupting T cell activation, and thus the provision of T cell help, abatacept inhibits downstream autoantibody production in RA [1, 13]. Previous analysis of data from the two RA trials that included both abatacept and TNFi treatment arms, AMPLE and ATTEST (abatacept or infliximab versus placebo, the Trial for Tolerability, Efficacy and Safety in Treating rheumatoid arthritis) showed that on-therapy autoantibody (antinuclear antibody [ANA] and anti-dsDNA) development occurred more frequently in the TNFi arm than in the abatacept treatment arm. In addition, in the ATTEST trial, most patients who had seroconverted from negative to positive (either ANA or anti-dsDNA) after 1 year of TNFi therapy, reverted to seronegative when switched to abatacept therapy [38]. These results are consistent with our current findings suggesting that abatacept can impact T cell-dependent autoantibody production.
The clinical relevance of the PD differences between abatacept and adalimumab observed in this study is not fully understood. These findings do illustrate how gene expression studies can provide insights in addition to those gained from conventional clinical assessments and should form the basis of future analyses for identification of relevant biomarkers with the potential to guide personalized treatment decision-making. RA is clearly a complex disease that can be subdivided into various subtypes (e.g., ACPA+ and ACPA–) which will each likely respond differently to different therapies [39]. The findings reported here and from related biomarker work led to a follow-on exploratory study of ACPA+/RF+ patients with early RA with the aim of further understanding the differential benefit of abatacept therapy, including the impact of HLA-DRB1 shared epitope status [40]. The results strongly suggested the differential benefit of the use of abatacept in this population, which is not likely limited to impacts on T cells [13, 41, 42]. A confirmatory study is underway to evaluate abatacept response in ACPA+ /RF+ patients with early RA (NCT04909801).
Limitations of this analysis should be acknowledged. Estimation of appropriate tests for significance in multiple measurements is an area of active investigation. The central issue is that, as repeated measures are correlated to each other in a mixed model framework where ‘patient’ is treated as a random effect, the null distribution is not known. Estimation of the null distribution can be performed using permutation tests or approximations (R-specific methods) [43]. Employment of the MMRM analysis for the multiple measures over time means that p values and resulting significant differences should be interpreted with caution and validated with orthogonal data. Although the software package Bioconductor LIMMA is often used for gene expression analyses, here MMRM analysis was deemed more appropriate due to the large sample size and multiple measurements. It should be noted that certain assumptions are made when conducting t tests (e.g., normally distributed data) and linear regression modeling (linear relationship in independent and dependent variables which may not always be biologically true; and the relationship is based on means, not on the complete distribution). Finally, while changes observed were statistically significant, they were numerically small, which could be attributed to the heterogeneity of a large trial and measurement of effects distal to the joints. Follow-up studies using serum proteins are ongoing and will be used to corroborate current findings.
Conclusions
PD changes, as reflected by differing ACPA and gene expression profiles and immune cell signatures, observed after 2 years of abatacept or adalimumab treatment were consistent with the hypothesized MoAs of these agents. The findings suggest that abatacept fundamentally down-regulates the adaptive anti-citrullinated protein immune response in RA, providing the potential to treat the adaptive immune response that drives the pathogenesis of RA. Further analysis of this data set from the AMPLE study is warranted and may provide valuable information regarding predictive biomarkers of response and disease progression.
References
Smolen JS, Aletaha D, Barton A, et al. Rheumatoid arthritis. Nat Rev Dis Primers. 2018;4:18001.
Edwards JC, Cambridge G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol. 2006;6:394–403.
Verpoort KN, Jol-van der Zijde CM, Papendrecht-van der Voort EA, et al. Isotype distribution of anti-cyclic citrullinated peptide antibodies in undifferentiated arthritis and rheumatoid arthritis reflects an ongoing immune response. Arthritis Rheum. 2006;54:3799–808.
Tan YC, Kongpachith S, Blum LK, et al. Barcode-enabled sequencing of plasmablast antibody repertoires in rheumatoid arthritis. Arthritis Rheumatol. 2014;66:2706–15.
Elliott SE, Kongpachith S, Lingampalli N, et al. Affinity maturation drives epitope spreading and generation of proinflammatory anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheumatol. 2018;70:1946–58.
Sokolove J, Schiff M, Fleischmann R, et al. Impact of baseline anti-cyclic citrullinated peptide-2 antibody concentration on efficacy outcomes following treatment with subcutaneous abatacept or adalimumab: 2-year results from the AMPLE trial. Ann Rheum Dis. 2016;75:709–14.
Lingampalli N, Sokolove J, Lahey LJ, et al. Combination of anti-citrullinated protein antibodies and rheumatoid factor is associated with increased systemic inflammatory mediators and more rapid progression from preclinical to clinical rheumatoid arthritis. Clin Immunol. 2018;195:119–26.
Robinson WH, Lindstrom TM, Cheung RK, Sokolove J. Mechanistic biomarkers for clinical decision making in rheumatic diseases. Nat Rev Rheumatol. 2013;9:267–76.
Smolen JS, Landewe RBM, Bijlsma JWJ, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. 2020;79:685–99.
Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62:2569–81.
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–19.
Singh JA, Saag KG, Bridges SL Jr, et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res. 2016;68:1–26.
Bonelli M, Scheinecker C. How does abatacept really work in rheumatoid arthritis? Curr Opin Rheumatol. 2018;30:295–300.
Weinblatt ME, Schiff M, Valente R, et al. Head-to-head comparison of subcutaneous abatacept versus adalimumab for rheumatoid arthritis: findings of a phase IIIb, multinational, prospective, randomized study. Arthritis Rheum. 2013;65:28–38.
Schiff M, Weinblatt ME, Valente R, et al. Head-to-head comparison of subcutaneous abatacept versus adalimumab for rheumatoid arthritis: two-year efficacy and safety findings from AMPLE trial. Ann Rheum Dis. 2014;73:86–94.
Harrold LR, Litman HJ, Connolly SE, et al. Effect of anticitrullinated protein antibody status on response to abatacept or antitumor necrosis factor-alpha therapy in patients with rheumatoid arthritis: a US national observational study. J Rheumatol. 2018;45:32–9.
Courvoisier DS, Chatzidionysiou K, Mongin D, et al. The impact of seropositivity on the effectiveness of biologic anti-rheumatic agents: results from a collaboration of 16 registries. Rheumatology (Oxford). 2020;60: keaa393.
Figueiredo CP, Bang H, Cobra JF, et al. Antimodified protein antibody response pattern influences the risk for disease relapse in patients with rheumatoid arthritis tapering disease modifying antirheumatic drugs. Ann Rheum Dis. 2017;76:399–407.
Wunderlich C, Oliviera I, Figueiredo CP, Rech J, Schett G. Effects of DMARDs on citrullinated peptide autoantibody levels in RA patients: a longitudinal analysis. Semin Arthritis Rheum. 2017;46:709–14.
Sokolove J, Bromberg R, Deane KD, et al. Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PLoS ONE. 2012;7: e35296.
17.0.0 Bi. Index of Brainarray Database. 2018. http://brainarray.mbni.med.umich.edu/Brainarray/Database/CustomCDF/17.0.0/ensg.asp. Accessed 30 Nov 2021.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol). 1995;57:289–300.
Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50.
Chaussabel D, Quinn C, Shen J, et al. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity. 2008;29:150–64.
Jabado O, Hu S, Carman J, et al. Deconvolution of immune cell proportions from whole blood RNA using next-generation sequencing [abstract]. Arthritis Rheumatol. 2016;68(suppl 10).
Gaujoux R, Seoighe C. CellMix: a comprehensive toolbox for gene expression deconvolution. Bioinformatics. 2013;29:2211–2.
van de Stadt LA, de Koning MH, van de Stadt RJ, et al. Development of the anti-citrullinated protein antibody repertoire prior to the onset of rheumatoid arthritis. Arthritis Rheum. 2011;63:3226–33.
Jonsson MK, Hensvold AH, Hansson M, et al. The role of anti-citrullinated protein antibody reactivities in an inception cohort of patients with rheumatoid arthritis receiving treat-to-target therapy. Arthritis Res Ther. 2018;20:146.
Kastbom A, Forslind K, Ernestam S, et al. Changes in the anticitrullinated peptide antibody response in relation to therapeutic outcome in early rheumatoid arthritis: results from the SWEFOT trial. Ann Rheum Dis. 2014. https://doi.org/10.1136/annrheumdis-2014-205698.
Bohler C, Radner H, Smolen JS, Aletaha D. Serological changes in the course of traditional and biological disease modifying therapy of rheumatoid arthritis. Ann Rheum Dis. 2013;72:241–4.
Jansen D, Emery P, Smolen JS, et al. Conversion to seronegative status after abatacept treatment in patients with early and poor prognostic rheumatoid arthritis is associated with better radiographic outcomes and sustained remission: post hoc analysis of the AGREE study. RMD Open. 2018;4: e000564.
Endo Y, Koga T, Kawashiri SY, et al. Anti-citrullinated protein antibody titre as a predictor of abatacept treatment persistence in patients with rheumatoid arthritis: a prospective cohort study in Japan. Scand J Rheumatol. 2020;49:13–7.
Morand EF, Furie R, Tanaka Y, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. 2020;382:211–21.
Bandyopadhyay S, Connolly SE, Jabado O, et al. Identification of biomarkers of response to abatacept in patients with SLE using deconvolution of whole blood transcriptomic data from a phase IIb clinical trial. Lupus Sci Med. 2017;4: e000206.
Linsley PS, Greenbaum CJ, Speake C, Long SA, Dufort MJ. B lymphocyte alterations accompany abatacept resistance in new-onset type 1 diabetes. JCI Insight. 2019;4: e126136.
Nakamura S, Suzuki K, Iijima H, et al. Identification of baseline gene expression signatures predicting therapeutic responses to three biologic agents in rheumatoid arthritis: a retrospective observational study. Arthritis Res Ther. 2016;18:159.
Plant D, Maciejewski M, Smith S, et al. Profiling of gene expression biomarkers as a classifier of methotrexate nonresponse in patients with rheumatoid arthritis. Arthritis Rheumatol. 2019;71:678–84.
Buch MH, Johnsen A, Schiff M. Can switching to abatacept therapy in patients with rheumatoid arthritis on background methotrexate reverse TNF-inhibitor-induced antinuclear autoantibody/double-stranded DNA autoantibody conversion? An analysis of the AMPLE and ATTEST trials. Clin Exp Rheumatol. 2019;37:127–32.
Scherer HU, Häupl T, Burmester GR. The etiology of rheumatoid arthritis. J Autoimmun. 2020;110:102400.
Rigby W, Buckner JH, Louis Bridges S, et al. HLA-DRB1 risk alleles for RA are associated with differential clinical responsiveness to abatacept and adalimumab: data from a head-to-head, randomized, single-blind study in autoantibody-positive early RA. Arthritis Res Ther. 2021;23:245.
Bonelli M, Ferner E, Goschl L, et al. Abatacept (CTLA-4IG) treatment reduces the migratory capacity of monocytes in patients with rheumatoid arthritis. Arthritis Rheum. 2013;65:599–607.
Axmann R, Herman S, Zaiss M, et al. CTLA-4 directly inhibits osteoclast formation. Ann Rheum Dis. 2008;67:1603–9.
Kuznetsova A, Brockhoff PB, Rune HBC. lmerTest package: tests in linear mixed effects models. J Stat Softw. 2017;82:1–26.
Acknowledgements
The authors thank the patients and families who made this study possible.
Funding
Sponsorship for this study and Rapid Service Fee were funded by Bristol Myers Squibb.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authors Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Study conception and design: OJ, MAM, MS, MEW, RF, WHR, AH, VP, SEC. Acquisition of data: MAM, MS, MEW, RF, WHR, AH, VP, SEC. Analysis of data: OJ, MAM, WHR, AH, VP, AG, JS, DG, SEC. Interpretation of data: OJ, MAM, MS, MEW, RF, WHR, AG, JS, DG, SEC
Medical Writing, Editorial, and Other Assistance
The authors would like to acknowledge the following contributors: Lauren Lahey (Stanford University School of Medicine) for Luminex assay data; Julie Carman, Steve Nadler (both of Bristol Myers Squibb), Cate Speake, and Peter Linsley (both of the Benaroya Research Institute) for immune cell deconvolution data; Somnath Bandyopadhyay and Scott Chasalow (both of Bristol Myers Squibb) for guidance in statistical analysis and interpretation of results; and Sandra Overfield (Bristol Myers Squibb) for assistance in initial trial design and operational aspects of the study. Professional medical writing and editorial assistance was provided by Katerina Kumpan, PhD, and Joanna Wright, DPhil, at Caudex, and was funded by Bristol Myers Squibb.
Disclosures
Michael A. Maldonado, Aiqing He, Jasmine Saini, David Galbraith, and Sean E. Connolly are employees of and shareholders in Bristol Myers Squibb. At the time this study was conducted, Omar Jabado, Vishal Patel, and Alex Greenfield were employees of and shareholders in Bristol Myers Squibb. Omar Jabado is now an employee of Genmab, Vishal Patel is now an employee of Merck & Co, and Alex Greenfield is now an employee of Valo Health. Michael Schiff has received speaker and consulting fees from Bristol Myers Squibb (less than $10,000 each). Michael E. Weinblatt has received research grants from Amgen, Bristol Myers Squibb, Crescendo Bioscience, Lilly, and Sanofi; has received consulting fees from Arena, Corrona, Gilead, Pfizer (more than $10,000 each) and AbbVie, Amgen, Bristol Myers Squibb, Canfite, Crescendo, GlaxoSmithKline, Horizon, Lilly, Lycera, Novartis, Roche, Samsung, Scipher, and Set Point (less than $10,000 each) and owns stock options in Canfite, Inmedix, Lycera, and Scipher. Roy Fleischmann has received consulting fees from AbbVie and Bristol Myers Squibb (less than $10,000 each); has received grant/research support from AbbVie and Bristol Myers Squibb; and is Editor-in-Chief of the journal, Rheumatology and Therapy. William H. Robinson has received consulting fees from Atreca, Bristol Myers Squibb, Celgene, Eli Lilly, and Sanofi (less than $10,000 each); and has received research grants from Bristol Myers Squibb, Gilead Sciences, and Sanofi.
Compliance with Ethics Guidelines
All patients in AMPLE provided written informed consent. The trial was conducted in accordance with the Helsinki Declaration of 1964, and its later amendments, and the protocol was approved by the institutional review boards and independent ethics committees at the participating sites.
Data Availability
Bristol Myers Squibb’s policy on data sharing may be found at https://www.bms.com/researchers-and-partners/independent-research/data-sharing-requestprocess.html.
Author information
Authors and Affiliations
Corresponding author
Additional information
O. Jabado’s, V. Patel’s and A. Greenfield’s affiliation was at the time the study was conducted.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Jabado, O., Maldonado, M.A., Schiff, M. et al. Differential Changes in ACPA Fine Specificity and Gene Expression in a Randomized Trial of Abatacept and Adalimumab in Rheumatoid Arthritis. Rheumatol Ther 9, 391–409 (2022). https://doi.org/10.1007/s40744-021-00404-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-021-00404-x