
Abstract
Introduction
This 24-week randomized, double-blind, non-inferiority study compared the efficacy and safety of febuxostat, a xanthine oxidase inhibitor, with allopurinol using an up-titration method in hyperuricemic Chinese subjects with or without gout.
Methods
Eligible adults (serum uric acid [SUA] > 7.0 mg/dl with a history of gout, SUA ≥ 8.0 mg/dl with complications or SUA ≥ 9.0 mg/dl without complications) were randomized (1:1:1) to febuxostat 40 mg/day, 80 mg/day, or allopurinol 300 mg/day. Starting doses of febuxostat 20 mg/day and allopurinol 100 mg/day were up-titrated, up to 16 weeks, to the randomized doses and maintained to week 24. Primary endpoint was non-inferiority of febuxostat 40 mg/day versus allopurinol 300 mg/day based on the percentage of subjects with SUA ≤ 6.0 mg/dl at week 24. The same comparison was made between febuxostat 60 mg/day or 80 mg/day versus allopurinol 300 mg/day. Safety assessments included measurement of treatment-emergent adverse events (TEAEs).
Results
The per-protocol population comprised 472 subjects. Non-inferiority of febuxostat 40 mg/day versus allopurinol 300 mg/day was not demonstrated based on the protocol-defined margin of − 10% (44.7 vs. 50.0%; − 5.3% difference; 95% confidence interval [CI]: − 16.4%, 5.8%); however, superiority over allopurinol 300 mg/day was demonstrated for febuxostat 60 mg/day at week 16 (66.3 vs. 51.2%; a 15.0% difference; 95% CI: 4.2%, 25.9%) and febuxostat 80 mg/day at week 24 (70.0 vs. 50.0%; a 20.0% difference; 95% CI: 9.3%, 30.7%). The frequency of TEAEs was similar across groups, with gout flares occurring frequently.
Conclusions
Using a novel dose-titration method, although the primary endpoint of non-inferiority of febuxostat 40 mg/day versus allopurinol 300 mg/day was not reached, non-inferiority and superiority of febuxostat 60 mg/day and 80 mg/day versus allopurinol 300 mg/day was demonstrated at weeks 16 and 24, respectively. Febuxostat demonstrated an acceptable tolerability profile in the treatment of hyperuricemia in Chinese subjects with or without gout.
Trial Registration
JapicCTI-132106.
Funding
Astellas Pharma Global Development, Inc.
Similar content being viewed by others

Introduction
The global prevalence of hyperuricemia, defined as serum uric acid (SUA) levels of ≥ 6.8 mg/dl in the extracellular fluid, has increased over the past few decades; it continues to rise in developed countries such as the United States (21.2–21.6%) and in rapidly developing countries such as China (13.3%), where major shifts in lifestyle and diet have occurred over the past two decades [1,2,3,4]. Prolonged hyperuricemia can lead to the development of gout, manifesting as the deposition of monosodium urate crystals in and around the joints [5] and is associated with the development of metabolic syndrome components such as obesity, hypertension, diabetes, and dyslipidemia [6,7,8]. In addition, gout often causes severe pain that may lead to substantial morbidity [4], meaning that effective treatments are warranted.
Currently, gout management aims to reduce SUA levels to ≤ 6.0 mg/dl to prevent the deposition of monosodium urate crystals in and around the joints [9, 10]. Approved SUA-lowering treatments include xanthine oxidase inhibitors, such as allopurinol, which has been widely used for the treatment of gout since the 1960s [11,12,13], in combination with low-dose, daily colchicine for the prophylaxis of flares [14]. However, treatment with allopurinol is associated with a risk of hypersensitivity syndrome, including toxic epidermal necrolysis and Stevens–Johnson syndrome [15, 16]. Other studies have demonstrated that these hypersensitivity syndromes have a strong association with the HLA-B*58:01 allele, a genetic variant frequently observed in Asian populations [17].
Febuxostat is a more recently developed non-purine xanthine oxidase inhibitor approved for the treatment of subjects with hyperuricemia and/or gout [18]. In numerous randomized, controlled, phase 3 studies, febuxostat demonstrated superior efficacy for reducing SUA levels to ≤ 6.0 mg/dl compared with allopurinol 300 mg/day, with a comparable tolerability profile [19,20,21,22,23,24]. In Chinese subjects, the efficacy and safety of febuxostat compared with allopurinol has been studied [22,23,24]. One of these studies in Chinese subjects assessed febuxostat 40 mg/day and 80 mg/day versus allopurinol 300 mg/day over 52 weeks (prophylaxis for gout flares was allowed) [22]. The study achieved the primary endpoint of SUA < 6.0 mg/dl at the last three visits in a significantly greater number of subjects in the febuxostat 80 mg/day group (p < 0.001) compared with the allopurinol 300 mg/day group, but not in the febuxostat 40 mg/day group (p = 0.216) [22]. Another study in Chinese subjects assessed febuxostat 40 mg/day and 80 mg/day versus allopurinol 300 mg/day over 28 weeks (prophylaxis for gout flares was allowed during weeks 1–8) [24]. The study achieved the primary endpoint of SUA < 6.0 mg/dl at the last three monthly measurements in a significantly greater number of subjects in the febuxostat 80 mg/day group (p < 0.0001) compared with the allopurinol 300 mg/day group, and the febuxostat 40 mg/day group was non-inferior to the allopurinol 300 mg/day group [24]. However, in a study of the cardiovascular safety of febuxostat 40 mg/day and 80 mg/day versus allopurinol 300–600 mg, febuxostat was associated with a significant increased risk of death from any cause (p = 0.04) and cardiovascular death (p = 0.03) [25].
Our phase 3, randomized, double-blind, non-inferiority study aimed to contribute to the evidence base by investigating the efficacy and safety of febuxostat at daily doses of 40, 60, and 80 mg/day, compared with allopurinol 300 mg/day, over 24 weeks in hyperuricemic Chinese subjects with or without gout.
Methods
Study Design
This 24-week, phase 3, multicenter, randomized, double-blind, double-dummy, active-controlled, three-arm, parallel-group, non-inferiority study assessed the efficacy and safety of once-daily oral febuxostat (40 mg/day or 80 mg/day) compared with allopurinol 300 mg/day in reducing SUA levels in adults from China with hyperuricemia with or without gout (trial registration number JapicCTI-132106). The study included a screening period of at least 2 weeks, followed by a double-blind treatment phase of 24 weeks and a safety follow-up phase of 2 weeks (Fig. 1).

Study design and dose-titration scheme for febuxostat 40 mg/day, 80 mg/day, and allopurinol 300 mg/day groups. The study included a screening phase, a double-blind randomized treatment phase (including a dose-titration period and a dose maintenance period, totaling 24 weeks) and a safety follow-up phase (2 weeks). After the screening phase, subjects were randomized into a 24-week treatment phase in which febuxostat and allopurinol were gradually up-titrated to the randomized dose; this was followed-up with a 2-week safety phase. The last visit was an end-of-treatment visit or an early termination visit
At randomization (day − 1), subjects were assigned to one of three treatment groups in a 1:1:1 ratio: febuxostat 40 mg/day, febuxostat 80 mg/day, or allopurinol 300 mg/day. Subjects were randomized using an interactive web response system (IWRS) and were stratified by screening SUA level (< 9.0 mg/dl, ≥ 9.0 mg/dl to < 10.0 mg/dl and ≥ 10.0 mg/dl) and study center. Authorized personnel at each study center received a unique identity number and personal identification number for use with the IWRS to ensure secure and controlled access to the system to record which treatment group each subject was randomized to. The treatment that each subject received was not disclosed to investigators, site staff, subjects, the sponsor or their representatives until after the study database had been formally locked. The appearance of study medications and placebo were identical.
Rapid reductions in SUA levels during treatment initiation with SUA-reducing drugs are associated with acute gout flares. To avoid such flares, doses were gradually increased from initial to target doses of febuxostat and allopurinol. For both febuxostat dose groups, treatment was initiated at 20 mg/day on day 1 and was up-titrated to 40 mg/day on week 5. For the febuxostat 80 mg/day group, the dose was then further up-titrated to 60 mg/day on week 9 and to 80 mg/day on week 17. Allopurinol was initiated at 100 mg/day on day 1, increased to 200 mg/day on week 3, and to 300 mg/day on week 5 (Fig. 1). Treatment compliance was verified by tablet count at each study visit. Subjects who had taken < 80% or > 120% of the required number of tablets were considered non-compliant.
The study was conducted at 14 sites in China between April 2013 and January 2015. Ethical approval was obtained from the Independent Ethics Committees (IEC) from each study site (Supplementary Table 1) and all subjects provided written informed consent prior to randomization. The master IEC was Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China. The study was conducted in accordance with the Declaration of Helsinki on Ethical Principles, applicable provisions in Good Clinical Practice, and applicable drug and data protection laws and regulations in China.
Subjects
Eligible subjects were men or women aged between 18 and 85 years, with SUA levels of > 7.0 mg/dl with a history of gout, SUA levels of ≥ 8.0 mg/dl with complications (defined as a need for pharmacologic or other treatment for lithangiuria, hypertension, hyperlipidemia, or abnormal glucose tolerance) or SUA levels of ≥ 9.0 mg/dl without complications.
Subjects were excluded if they: reported an acute attack of gouty arthritis at the screening visit or the randomization visit (day − 1) or if they had recovered for less than 2 weeks from a previous gouty arthritis attack; had been routinely receiving non-steroidal anti-inflammatory drugs or corticosteroids (not including topical application) for a disease other than gouty arthritis; had a medical condition that would interfere with treatment, safety, or adherence to the protocol; were pregnant or lactating; had a history of drug-induced allergy or hypersensitivity; had renal dysfunction (serum creatinine ≥ 1.5 mg/dl or 133 μmol/l); had severe hypertension (systolic blood pressure ≥ 180 mmHg or diastolic blood pressure ≥ 110 mmHg) or blood pressure that was not well controlled with antihypertensive agents; or had received any investigational product within 90 days prior to the start of screening. Subjects were prohibited from taking any uric acid-reducing medication or any drugs for the prophylaxis of gout flares, such as colchicine, during the study. Subjects who took one or more prohibited medications during the 2 weeks prior to providing informed consent underwent a washout period of at least 2 weeks prior to randomization (Fig. 1).
Endpoints
The assessment of efficacy was based on centralized measures of SUA levels in blood samples taken at baseline and after 2, 4, 8, 12, 16, 20, and 24 weeks of treatment. The primary efficacy endpoint was non-inferiority of febuxostat 40 mg/day versus allopurinol 300 mg/day based on the percentage of subjects with SUA ≤ 6.0 mg/dl at week 24. Key secondary efficacy endpoints were non-inferiority of febuxostat 60 mg/day (at week 16) and 80 mg/day (at week 24) versus allopurinol 300 mg/day based on the percentage of subjects with SUA ≤ 6.0 mg/dl. Other secondary efficacy endpoints included the percentage of subjects with SUA ≤ 6.0 mg/dl at post-baseline visits, the percentage of subjects with SUA ≤ 7.0 mg/dl at week 24 and the mean percentage change in SUA level from baseline to each post-baseline visit.
Adverse events (AEs), serious AEs (SAEs), clinical laboratory tests, 12-lead electrocardiogram, vital signs and physical examination findings were assessed throughout the 24-week study. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 17.1. Gout attack during treatment was recorded where the lowest level term was gout, gout acute, gout attack, or gout flare.
Statistical Analysis
Based on the results of two previous up-titration febuxostat versus allopurinol clinical studies in Japan [26, 27], a sample of 474 subjects (158 per treatment group) was required to meet the non-inferiority criteria with 90% power to detect a 10% difference between the febuxostat 40 mg/day and allopurinol 300 mg/day group for the primary endpoint. The true response rates were assumed to be 76% for allopurinol and 81% for febuxostat 40 mg/day.
The safety analysis population included all subjects who were randomized and had received at least one dose of randomized study treatment. The full analysis set included all subjects who were randomized, had a SUA level > 7.0 mg/dl at randomization and had taken at least one dose of randomized study treatment. The per-protocol population included subjects in the full analysis set who were considered sufficiently compliant with the protocol.
Efficacy analyses were carried out on the per-protocol population and were repeated for the full analysis set to explore the robustness of the results. If SUA data were missing, data from the last available post-baseline assessment were used for the primary analysis. For the primary efficacy analysis, a non-inferiority margin of − 10% was used for the comparison of febuxostat 40 mg/day versus allopurinol 300 mg/day; this meant that non-inferiority would be demonstrated if the lower bound of the two-sided 95% exact binomial confidence intervals (CIs) for the difference in response rates between febuxostat 40 mg/day and allopurinol 300 mg/day was greater than − 10%. When analyzing the difference in response rates between allopurinol 300 mg/day and febuxostat 60 mg/day or febuxostat 80 mg/day, non-inferiority was assessed first. If non-inferiority was demonstrated, then superiority was assessed based on a margin of 0%; i.e., superiority would be demonstrated if the lower bound of the two-sided 95% exact binomial CIs for the difference in response rates was greater than 0%. Data for the percentage of subjects with SUA ≤ 6.0 mg/dl and the mean percentage change from baseline in SUA at week 24 were also summarized for subgroups based on baseline SUA levels.
Results
Study Population
A total of 763 subjects provided consent, of whom 599 were randomly assigned to treatment (Fig. 2). The safety analysis population included 590 subjects who received at least one dose of randomized study treatment. Five subjects received the wrong treatment and were excluded from the analysis populations. The full analysis set included 553 subjects. The most common reasons for exclusion from the full analysis set were SUA ≤ 7.0 mg/dl at day − 1 and withdrawal of consent (both n = 36). The per-protocol population included 472 subjects (150 in the febuxostat 40 mg/day group, 160 in the febuxostat 80 mg/day group, and 162 in the allopurinol 300 mg/day group).

Subject disposition. AE adverse event, FAS full analysis set, PPP per protocol population, SAF safety analysis set, SUA serum uric acid
Baseline demographics and disease characteristics were comparable across the three treatment groups (Table 1). All subjects were of Asian race and were mostly male (98.7%). Over half of the subjects (52.4%) had a history of alcohol abuse and most were overweight (61.1%) with a BMI of ≥ 25 kg/m2. The mean SUA at baseline ranged from 9.6 to 9.8 mg/dl. Mean treatment compliance was similar in all groups, ranging from 94.6 to 96.7% in the full analysis set.
Efficacy
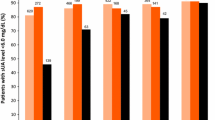
In the per-protocol population, the proportions of subjects achieving a week 24 SUA level of ≤ 6.0 mg/dl was 44.7% and 70.0% in the febuxostat 40 mg/day and 80 mg/day groups, respectively, versus 50.0% in the allopurinol 300 mg/day group (Fig. 3). The lower bound of the 95% CI for the difference in week 24 response rates between the febuxostat 40 mg/day and allopurinol 300 mg/day groups was outside the non-inferiority margin of − 10% (difference, − 5.3% [95% CI − 16.4%, 5.8%]); therefore, febuxostat 40 mg/day did not meet the primary endpoint of demonstrating non-inferiority to allopurinol 300 mg/day (Fig. 3a). At week 16, when the subjects in the febuxostat 80 mg/day group were receiving a 60 mg/day dose, the proportion of subjects achieving SUA ≤ 6.0 mg/dl was 66.3 vs. 51.2% in the allopurinol 300 mg/day group. The lower bound of the 95% CI for the difference in week 16 response rates between febuxostat 60 mg/day and allopurinol 300 mg/day was within both the non-inferiority margin of − 10% and the superiority margin of 0% (difference, 15.0% [95% CI 4.2%, 25.9%]); therefore, febuxostat 60 mg/day demonstrated both non-inferiority and superiority to allopurinol 300 mg/day (Fig. 3b). At week 24, the lower bound of the 95% CI for the difference in response rates between the febuxostat 80 mg/day and allopurinol 300 mg/day groups was within both the non-inferiority margin of − 10% and the superiority margin of 0% (difference, 20.0% [95% CI 9.3%, 30.7%]); therefore, febuxostat 80 mg/day demonstrated both non-inferiority and superiority to allopurinol 300 mg/day (Fig. 3c). Comparison of SUA ≤ 6.0 mg/dl response rates for the full analysis set demonstrated similar results (Fig. 3).

Percentage of subjects achieving SUA ≤ 6.0 mg/dl during treatment with a febuxostat 40 mg/day versus allopurinol 300 mg/day at week 24, b febuxostat 60 mg/day versus allopurinol 300 mg/day at week 16 and c febuxostat 80 mg/day versus allopurinol 300 mg/day at week 24 versus allopurinol 300 mg/day. CI confidence interval, SUA serum uric acid
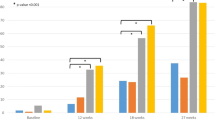
The mean percentage change from baseline SUA levels over time is shown in Fig. 4. At week 2, when all groups were at the starting dose (febuxostat 20 mg/day or allopurinol 100 mg/day), there was a larger mean percentage change from baseline SUA in both febuxostat treatment groups (− 27.65% and − 29.25% in the febuxostat 40 mg/day and febuxostat 80 mg/day groups, respectively) than in the allopurinol 300 mg/day group (− 17.99%). From week 12 onwards, after the dose of febuxostat had been increased to 60 mg/day in the febuxostat 80 mg/day group, the decrease in SUA levels was greater in this group than in the allopurinol 300 mg/day and febuxostat 40 mg/day groups. At week 24, the largest mean percentage decreases in SUA from baseline were observed in the febuxostat 80 mg/day group (45.2 vs. 32.6 and 33.7% in the febuxostat 40 mg/day and allopurinol 300 mg/day groups, respectively).

Change from baseline in mean SUA over time (per-protocol population). Subjects in both febuxostat groups received febuxostat 20 mg/day from randomization until week 4 and were then up-titrated to febuxostat 40 mg/day until week 8. Subjects in the febuxostat 40 mg/day group maintained this dose until week 24. From week 8, subjects in the febuxostat 80 mg/day group were up-titrated to febuxostat 60 mg/day until week 16 and then further up-titrated to febuxostat 80 mg/day until week 24. Subjects in the allopurinol 300 mg/day group received allopurinol 100 mg/day from randomization until week 2, received allopurinol 200 mg/day from week 2 until week 4 and received allopurinol 300 mg/day from week 5 until week 24. SD standard deviation, SUA serum uric acid
At week 24, mean SUA levels were lowest in the febuxostat 80 mg/day group (5.2 versus 6.4 and 6.4 mg/dl in the febuxostat 40 mg/day and allopurinol 300 mg/day groups, respectively). Similarly, the percentage of subjects with SUA ≤ 6.0 mg/dl at post-baseline visits increased gradually with dose titration in all three treatment groups. Response rates were comparable in the febuxostat 40 mg/day and allopurinol 300 mg/day groups, maintaining a range of 42.0–54.3% from week 12 onwards. In contrast, in the febuxostat 80 mg/day group the response rate increased to 70.6% by week 12 and remained above 60% at all subsequent time points. At week 24, the percentage of subjects with SUA levels ≤ 7.0 mg/dl was higher in the febuxostat 80 mg/day group (80.6% [129 responders]) compared with the febuxostat 40 mg/day group (67.3% [101 responders]) and the allopurinol 300 mg/day group (68.5% [111 responders]).
A subgroup analysis of response rate was performed according to baseline SUA levels (low < 9.0 mg/dl; moderate ≥ 9.0– < 10.0 mg/dl; high ≥ 10.0 mg/dl). Comparable response rates between subgroups were observed in the febuxostat 40 mg/day and allopurinol 300 mg/day groups (low 55.4 vs. 51.0%; moderate 55.3 vs. 69.6%; high 26.8 vs. 36.4%). Notably, response rates in the febuxostat 40 mg/day and allopurinol 300 mg/day groups were lowest among subjects with high baseline SUA levels; however, response rates in the febuxostat 80 mg/day group were comparable over baseline SUA levels (low 78.0%; moderate 60.9%; high 69.1%).
Safety
In the overall safety analysis population, 446 subjects (76%) experienced at least one treatment emergent adverse event (TEAE). The incidence of TEAEs of any grade and of serious TEAEs were similar across the three treatment groups (Table 2). One subject in the febuxostat 40 mg/day group died in a road traffic accident. Gout was the most frequent TEAE in all three groups and was experienced by 51.0% of subjects. The incidence of gout flare was highest during the first 5 weeks in each group and gradually decreased thereafter; for example, in the febuxostat 80 mg/day group, gout attacks were experienced by 28.6% of subjects during the first 2 weeks of treatment and by 16.6% of subjects during the last 4 weeks of treatment. The only other TEAEs occurring in > 5% of subjects in any one treatment group were increased alanine aminotransferase and upper respiratory tract infections. TEAEs led to the early withdrawal of study treatment on the investigator’s decision for four subjects (2.0%) in the febuxostat 80 mg/day group, nine subjects (4.7%) in the febuxostat 40 mg/day group and eight subjects (4.1%) in the allopurinol 300 mg/day group. These were considered related to treatment for three subjects in the febuxostat 80 mg/day group and eight subjects in the febuxostat 40 mg/day and allopurinol 300 mg/day groups.
Discussion
This randomized, double-blind, non-inferiority study did not meet the primary endpoint of non-inferiority of febuxostat 40 mg/day versus allopurinol 300 mg/day based on the percentage of subjects with SUA ≤ 6.0 mg/dl at week 24. This contrasts with previous clinical studies in North America [19,20,21] and China [22,23,24]; however, febuxostat at doses of 60 mg/day and 80 mg/day did demonstrate both non-inferiority and superiority to allopurinol 300 mg/day for the urate-lowering endpoint of SUA ≤ 6.0 mg/dl. In addition, febuxostat resulted in a prompt reduction in SUA (within 2 weeks) that was persistent across doses. This was the first study to examine a dose-titration method for febuxostat treatment in a Chinese population and, as the febuxostat dose was increased, SUA levels decreased. At week 24, mean SUA levels were reduced to 6.4, 5.2, and 6.4 mg/dl in the febuxostat 40 mg/day, 80 mg/day, and allopurinol 300 mg/day groups, respectively.
A previous Chinese study demonstrated a febuxostat response rate that was similar to the rate achieved in our study, but with lower response rates for allopurinol 300 mg/day (34.6% compared with 50.0% in this study) [22, 23]. In addition, a recent meta-analysis demonstrated that febuxostat was superior to other urate-lowering drugs with regard to safety and efficacy [28]. In the meta-analysis, the efficacy of treatments with increasing doses was determined using surface under the cumulative ranking curve (SUCRA) percentage, reporting response rates of 55.0, 61.6, 76.7, and 88.7% for febuxostat 40, 60, 80 mg/day and 120 mg/day, respectively [28]. The response rate at week 24 for febuxostat 40 mg/day was lower in our study (44.7%) than that observed in the meta-analysis (55.0%), which may have been owing to the exclusion of urate-lowering and prophylactic drugs in our study. Subjects in our current study were found to have more severe hyperuricemia at baseline (mean SUA range 9.6–9.8 mg/dl) than subjects in two Japanese studies (mean SUA range 8.3–8.9 mg/dl) in which response rates of 82.9–88.9% for febuxostat 40 mg/day, 83.3–100% for febuxostat 60 mg/day and 87.8% for febuxostat 80 mg/day were reported [26, 27]. This may have contributed to the absence of non-inferiority of febuxostat 40 mg/day versus allopurinol 300 mg/day, as responses were demonstrated to be lowest in subjects with high baseline SUA levels. In contrast, baseline SUA levels seemed to have little effect on the responses in the febuxostat 80 mg/day group. In addition, more subjects with hyperuricemia had gout in the current study compared with the Japanese studies (97.8 and 62.0%, respectively). It is also possible that the dose titration of febuxostat could have had an impact on final responses. In addition, the percentage of patients with asymptomatic hyperuricemia was low enough to not substantially contribute to the overall results, which was largely unexpected.
A limitation of this study may have been the underestimation of the non-inferiority margin. The 10% non-inferiority margin used in a previous study [21] was selected under the assumption of an 80% febuxostat response rate in the population. However, as the febuxostat 40 mg/day response rate in our study (44.7%) was lower than expected, and the standard error was almost 1.7 times larger than expected, a wider non-inferiority margin may have been more appropriate [24]. In addition, although allopurinol dosage should be titrated according to the SUA target [14], this study limited subjects to a maximum of allopurinol 300 mg/day. It has previously been demonstrated that only around half of patients reach their SUA target when receiving allopurinol 300 mg/day [29], yet allopurinol dosage continues to be poorly optimized globally [14, 30].
Overall, febuxostat at ascending doses of 20, 40, 60, and 80 mg/day was well tolerated in this population of hyperuricemic Chinese subjects with and without gout. Numbers of TEAEs were similar between groups, most of which were mild/moderate in intensity. A total of 22 subjects with TEAEs and seven subjects with SAEs discontinued study treatment. Discontinuation rates were similar in the febuxostat 40 mg/day and allopurinol 300 mg/day groups and lower in the febuxostat 80 mg/day group.
Paradoxical flares of gout are well documented during initial urate-lowering therapy and have important implications for gout management. Both the European League Against Rheumatism [31] and the American College of Rheumatology [32] guidelines advocate prophylactic treatment to reduce the occurrence of flares, recommending colchicine and non-steroidal anti-inflammatory drugs as first-line options. While prophylactic treatment can decrease the rate of gout flares triggered at the initiation of urate-lowering therapy, it does not completely prevent their occurrence [33]. Furthermore, as with all pharmacological treatments, potential TEAEs and drug–drug interactions may be a concern; for example, colchicine has a narrow therapeutic window, with inter-subject variability in tolerance and multiple drug–drug interactions [34], and non-steroidal anti-inflammatory drugs have cardiovascular, renal, and gastrointestinal side effects [35, 36]. In this study, prophylaxis for gout flares was prohibited, and instead, this study employed a gradual dose-titration method whereby febuxostat and allopurinol doses were increased incrementally to reach the target dose. This has been shown to be just as effective as colchicine prophylaxis for the prevention of gout flares [37]. The incidence of gout attacks in this study was higher than in previous studies that did not use a dose-titration method [21, 22]; however, in these prior studies, all subjects received prophylaxis for gout, and gout flares were only recorded when additional treatment was required for their management. In our study, the incidence of gout flare was highest in each group during the first 5 weeks and gradually decreased thereafter. Decreased incidence of gout flares over time during febuxostat treatment has been observed in clinical practice and in another Chinese study [22].
Conclusions
Our study did not meet the primary endpoint of non-inferiority of febuxostat 40 mg/day versus allopurinol 300 mg/day based on the percentage of subjects with SUA ≤ 6.0 mg/dl at week 24. However, febuxostat 60 mg/day and 80 mg/day not only demonstrated non-inferiority but also demonstrated superiority versus allopurinol 300 mg/day based on the percentage of subjects with SUA ≤ 6.0 mg/dl at week 16 and week 24, respectively. Treatment with febuxostat using a dose-titration method was effective at lowering SUA and was associated with an acceptable tolerability profile in the treatment of hyperuricemic Chinese subjects with or without gout.
References
Loeb JN. The influence of temperature on the solubility of monosodium urate. Arthritis Rheum. 1972;15(2):189–92.
Roddy E, Choi HK. Epidemiology of gout. Rheum Dis Clin North Am. 2014;40(2):155–75.
Liu R, Han C, Wu D, et al. Prevalence of hyperuricemia and gout in mainland China from 2000 to 2014: a systematic review and meta-analysis. Biomed Res Int. 2015;2015:762820.
Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011;63(10):3136–41.
Ruoff G, Edwards NL. Overview of serum uric acid treatment targets in gout: why less than 6 mg/dl? Postgrad Med. 2016;128(7):706–15.
Chen LY, Zhu WH, Chen ZW, et al. Relationship between hyperuricemia and metabolic syndrome. J Zhejiang Univ Sci B. 2007;8(8):593–8.
Neogi T. Clinical practice. Gout. N Engl J Med. 2011;364(5):443–52.
Johnson RJ, Kivlighn SD, Kim YG, Suga S, Fogo AB. Reappraisal of the pathogenesis and consequences of hyperuricemia in hypertension, cardiovascular disease, and renal disease. Am J Kidney Dis. 1999;33(2):225–34.
Schumacher HR Jr, Becker MA, Lloyd E, MacDonald PA, Lademacher C. Febuxostat in the treatment of gout: 5-year findings of the FOCUS efficacy and safety study. Rheumatology (Oxford). 2009;48(2):188–94.
Becker MA, Schumacher HR, MacDonald PA, Lloyd E, Lademacher C. Clinical efficacy and safety of successful long-term urate lowering with febuxostat or allopurinol in subjects with gout. J Rheumatol. 2009;36(6):1273–82.
Li EK. Gout: a review of its aetiology and treatment. Hong Kong Med J. 2004;10(4):261–70.
EMA. Allopurinol summary of product characteristics. https://www.medicines.org.uk/emc/product/5693/smpc. Accessed December 2018.
Underwood M. Diagnosis and management of gout. BMJ. 2006;332(7553):1315–9.
Pascart T, Liote F. Gout: state of the art after a decade of developments. Rheumatology (Oxford). 2019;58(1):27–44.
Ramasamy SN, Korb-Wells CS, Kannangara DR, et al. Allopurinol hypersensitivity: a systematic review of all published cases, 1950–2012. Drug Saf. 2013;36(10):953–80.
Halevy S, Ghislain PD, Mockenhaupt M, et al. Allopurinol is the most common cause of Stevens–Johnson syndrome and toxic epidermal necrolysis in Europe and Israel. J Am Acad Dermatol. 2008;58(1):25–32.
Ko TM, Tsai CY, Chen SY, et al. Use of HLA-B*58:01 genotyping to prevent allopurinol induced severe cutaneous adverse reactions in Taiwan: national prospective cohort study. BMJ. 2015;351:h4848.
EMA. Febuxostat summary of product characteristics. https://www.medicines.org.uk/emc/product/487/smpc. Accessed December 2018.
Becker MA, Schumacher HR Jr, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353(23):2450–61.
Schumacher HR Jr, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum. 2008;59(11):1540–8.
Becker MA, Schumacher HR, Espinoza LR, et al. The urate-lowering efficacy and safety of febuxostat in the treatment of the hyperuricemia of gout: the CONFIRMS trial. Arthritis Res Ther. 2010;12(2):R63.
Xu S, Liu X, Ming J, et al. A phase 3, multicenter, randomized, allopurinol-controlled study assessing the safety and efficacy of oral febuxostat in Chinese gout patients with hyperuricemia. Int J Rheum Dis. 2015;18(6):669–78.
Wang L, Zhao Y. Effect and safety of febuxostant versus allopurinol in reducing serum urate in subjects with hyperuricemia and gout: a multi-center, randomized, double-blind, parallel controlled trial. Chin J Clin (Electronic Edition). 2013;7:2798–803.
Huang X, Du H, Gu J, et al. An allopurinol-controlled, multicenter, randomized, double-blind, parallel between-group, comparative study of febuxostat in Chinese patients with gout and hyperuricemia. Int J Rheum Dis. 2014;17(6):679–86.
White WB, Saag KG, Becker MA, et al. Cardiovascular safety of febuxostat or allopurinol in patients with gout. N Engl J Med. 2018;378(13):1200–10.
Kamatani N, Fujimori S, Hada T, et al. Placebo-controlled double-blind dose-response study of the non-purine-selective xanthine oxidase inhibitor febuxostat (TMX-67) in patients with hyperuricemia (including gout patients) in Japan: late phase 2 clinical study. J Clin Rheumatol. 2011;17(4 Suppl 2):S35–43.
Kamatani N, Fujimori S, Hada T, et al. An allopurinol-controlled, multicenter, randomized, open-label, parallel between-group, comparative study of febuxostat (TMX-67), a non-purine-selective inhibitor of xanthine oxidase, in patients with hyperuricemia including those with gout in Japan: phase 2 exploratory clinical study. J Clin Rheumatol. 2011;17(4 Suppl 2):S44–9.
Li S, Yang H, Guo Y, et al. Comparative efficacy and safety of urate-lowering therapy for the treatment of hyperuricemia: a systematic review and network meta-analysis. Sci Rep. 2016;6:33082.
Khanna D, Fitzgerald JD, Khanna PP, et al. American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64(10):1431–46.
Singh JA, Akhras KS, Shiozawa A. Comparative effectiveness of urate lowering with febuxostat versus allopurinol in gout: analyses from large U.S. managed care cohort. Arthritis Res Ther. 2015;17(1):120.
Richette P, Doherty M, Pascual E, et al. 2016 updated EULAR evidence-based recommendations for the management of gout. Ann Rheum Dis. 2017;76(1):29–42.
Khanna D, Khanna PP, Fitzgerald JD, et al. American College of Rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken). 2012;64(10):1447–61.
Dalbeth N, So A. Hyperuricaemia and gout: state of the art and future perspectives. Ann Rheum Dis. 2010;69(10):1738–43.
Terkeltaub RA, Furst DE, Digiacinto JL, Kook KA, Davis MW. Novel evidence-based colchicine dose-reduction algorithm to predict and prevent colchicine toxicity in the presence of cytochrome P450 3A4/P-glycoprotein inhibitors. Arthritis Rheum. 2011;63(8):2226–37.
McGettigan P, Henry D. Cardiovascular risk with non-steroidal anti-inflammatory drugs: systematic review of population-based controlled observational studies. PLoS Med. 2011;8(9):e1001098.
Stamp LK, Chapman PT. Gout and its comorbidities: implications for therapy. Rheumatology (Oxford). 2013;52(1):34–44.
Yamanaka H, Tamaki S, Ide Y, et al. Stepwise dose increase of febuxostat is comparable with colchicine prophylaxis for the prevention of gout flares during the initial phase of urate-lowering therapy: results from FORTUNE-1, a prospective, multicentre randomised study. Ann Rheum Dis. 2018;77(2):270–6.
Acknowledgements
We thank the participants of the study.
Data Availability
Access to anonymized individual participant-level data collected during the trial, in addition to supporting clinical documentation, is planned for trials conducted with approved product indications and formulations, as well as compounds terminated during development. Conditions and exceptions are described under the Sponsor Specific Details for Astellas on www.clinicalstudydatarequest.com. Study-related supporting documentation is redacted and provided if available, such as the protocol and amendments, statistical analysis plan, and clinical study report. Access to participant-level data is offered to researchers after publication of the primary manuscript (if applicable) and is available as long as Astellas has legal authority to provide the data. Researchers must submit a proposal to conduct a scientifically relevant analysis of the study data. The research proposal is reviewed by an Independent Research Panel. If the proposal is approved, access to the study data is provided in a secure data sharing environment after receipt of a signed Data Sharing Agreement.
Funding
Astellas Pharma Global Development, Inc. funded this work and the journal’s Rapid Service Fee.
Medical Writing and Editorial Assistance
Medical writing and editorial assistance was provided by Aisling Koning and Matthew Reynolds of OPEN Health Medical Communications (London, UK), funded by Astellas Pharma Global Development, Inc.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Fengchun Zhang, Zhichun Liu, Lindi Jiang, Hao Zhang, Dongbao Zhao, Yang Li, Hejian Zou, Xiaoyue Wang, Xiangpei Li, Bingyin Shi, Jianhua Xu, Hongjie Yang, Shaoxian Hu and Shen Qu have nothing to disclose.
Compliance with Ethics Guidelines
Ethical approval was obtained from the Independent Ethics Committees (IEC) from each study site (Supplementary Table 1) and all subjects provided written informed consent prior to randomization. The master IEC was Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China. The study was conducted in accordance with the Declaration of Helsinki on Ethical Principles, applicable provisions in Good Clinical Practice, and applicable drug and data protection laws and regulations in China.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to: https://doi.org/10.6084/m9.figshare.9771893.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Zhang, F., Liu, Z., Jiang, L. et al. A Randomized, Double-Blind, Non-Inferiority Study of Febuxostat Versus Allopurinol in Hyperuricemic Chinese Subjects With or Without Gout. Rheumatol Ther 6, 543–557 (2019). https://doi.org/10.1007/s40744-019-00173-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-019-00173-8