
Summary
Purpose
Biotechnological substances (BSs) are strongly relied upon to prevent rejection of transplanted organs, and to treat oncological, allergological, and other inflammatory diseases. Allergic reactions to partly foreign biologics can occur due to their potential immunogenicity. The severity of an immune response to a biological drug may range from no clinical significance to a severe, life-threatening anaphylactic reaction.
Methods
Detailed searches were performed on Pubmed, Web of Science, and Google Scholar to include all available publications. In addition, the Food and Drug Administration, the European Medicines Agency, and British Columbia Cancer Agency Drug Manual databases were screened for hypersensitivity reaction (HSR), infusion reaction, injection site reaction, urticaria, and anaphylaxis for individual BSs.
Results
Treatment with BSs can cause various types of HSR. These are mentioned in the literature with definitions such as allergic reactions, anaphylactoid reactions, anaphylaxis, HSR, infusion reactions, injection site reactions, cytokine release syndrome, and urticaria. Due to the overlap in signs and symptoms in the reported descriptions, it is not always possible to differentiate these reactions properly according to their pathomechanism. Similarly, many data reported as anaphylaxis actually describe severe anaphylactic reactions (grades III or IV).
Conclusion
There is an urgent need for a simpler symptom- or system-based classification and scoring system to create an awareness for HSRs to BSs. A better understanding of the pathophysiology of HSRs and increased clinical experience in the treatment of side effects will provide timely control of unexpected reactions. As a result, immunotherapy with BSs will become safer in the future.
Similar content being viewed by others


Introduction
Biotechnological substances (BSs), also known as biological agents, biologicals or biologics, are produced by living organisms or are synthesized from a product made by a living organism. Some BSs are directed against specific cytokines or cell surface signal receptors. In recent years, the BS industry has experienced rapid growth and yielded products for treatment of a broad spectrum of diseases and disorders. Biologics are strongly relied upon to prevent rejection after organ transplantation and to treat diseases related to allergology, pneumology, rheumatology, oncology, and dermatology. BSs have revolutionized the treatment of diseases such as rheumatoid arthritis, psoriatic arthritis, inflammatory bowel disease, and organ-specific cancers (lung, stomach, colon, cervix, breast, etc.). Other conditions commonly treated or managed with biologics include several types of asthma, plaque psoriasis, chronic urticaria, and vasculitis. Some new indications and less studied applications include interstitial lung disease, unstable angina pectoris during cardiac catheterization, paroxysmal nocturnal hemoglobinuria, neonatal bronchopulmonary dysplasia, and multiple sclerosis (Table 1).
Allergic reactions to partly foreign biologics can occur due to their potential immunogenicity. Adverse drug reactions (ADRs), however, can also result from an agent’s direct biological function. For instance, neutralization of an off-target cytokine’s activity can cause ADRs [1]. The severity of an immune response to a biological drug may range from no clinical significance to a severe, life-threatening anaphylactic reaction [2]. Thus it became evident that the nomenclature used to describe and document immunological adverse events was not always precise enough, or the cases had not been investigated in enough detail to clearly differentiate between truly allergic and other reactions. This review will evaluate reports of allergic and substance-specific infusion reactions (IR), injection-site reactions (ISR), hypersensitivity reactions (HSR), urticaria, and anaphylaxis caused by BSs.
Material and methods
BSs were investigated individually for HSR, IR, ISRs, and anaphylaxis from The Food and Drug Administration (FDA), European Medicines Agency (EMA), and British Columbia Cancer Agency (BCCA) Drug Manual database and created the main information pool in terms of these side effects. In addition, case reports, articles, and reviews on this subject were retrieved by searching three international databases (Pubmed, Web of Science, and Google Scholar). The search terms, such as specific BS, allergy, anaphylaxis, hypersensitivity, IR, ISR, and urticaria were determined for the search.
Overview of reported allergic reactions to biotechnological substances
BSs are engineered macromolecules that are quite similar to autologous proteins. Therefore, they vary, as do small-molecule pharmaceuticals, in the reactions they produce. In a comprehensive review of the Spanish Society of Rheumatology’s Biobadaser Database in 2018, 8253 side effects were reported in 4454 patients treated with BSs (mostly tumor necrosis factor [TNF]-α-blockers) [3]. Of these, 7133 (86.4%) were classified as non-serious, 1089 (13.2%) as serious, and 31 (0.38%) as fatal.
The symptoms mentioned in the various classifications of HSRs and anaphylaxis have many common features. Due to the overlap in these common signs and symptoms, it is not possible to differentiate between IRs, anaphylaxis, or other HSRs in the reported descriptions and publications. In this review, we will elaborate IR, ISR, HSR, urticaria, as well as anaphylaxis. Drugs are described individually under subheadings naming the various medical disciplines in which they are used.
Pulmonology
The most common indications for the use of biologics in lung diseases are allergic and severe uncontrolled asthma. Biologics are used more rarely in interstitial lung diseases (Table 2). Their use in lung cancer will be mentioned in the section Oncology. Benralizumab, lebrikizumab, mepolizumab, reslizumab, and omalizumab are used in asthmatic patients. The primary indication for nintedanib and pirfenidone is idiopathic pulmonary fibrosis.
Benralizumab
Benralizumab is the newest anti-asthmatic and anti-inflammatory drug in the monoclonal antibody (mAb) family developed for the treatment of severe eosinophilic and allergic asthma. It exerts its effects by binding to the α‑subunit of the interleukin (IL)-5 receptor on eosinophils and basophils.
In a phase IIb study conducted between 2011 and 2012, the ISR rate was 16% and anaphylaxis was 0% [4]. FDA labels show a 3% incidence of HSRs (anaphylaxis, urticaria, rash, and angioedema) in the treatment group receiving benralizumab and placebo [5]. The frequency of ISR (erythema, local pain, papule, or pruritus) was 2.2% in patients treated with benralizumab [5]. These reactions usually occurred within the first hours after s.c. administration of the drug. The most reported ADR was upper respiratory tract infection and mild to moderate nasopharyngitis [6]. In a recent study in which 13 patients were treated, HSR and anaphylaxis were not reported; only slight fever with chills was observed in two patients [7].
In the meta-analysis of eight randomized controlled trials, the overall risk of ADRs and severe side effects was lower and the risk of headache and pyrexia higher in patients treated with benralizumab compared to placebo [8]. In addition, an increased incidence of ISRs, hypersensitivity, and death was not observed compared to placebo. The ISRs were reported in 2.6% of patients treated with 30 mg benralizumab. Post-marketing side effect recording is ongoing.
Lebrikizumab
Lebrikizumab is a novel humanized mAb that specifically inhibits the activity of IL-13. Clinical studies for the treatment of asthma that is uncontrolled by inhaled corticosteroids, chronic obstructive pulmonary disease (COPD), atopic dermatitis, and idiopathic pulmonary fibrosis (IPF) are ongoing.
In one placebo-controlled study using higher doses, ISRs (pain, erythema) were found to be more prevalent, 20.5% and 20.3% in groups receiving 125 and 250 mg doses of lebrikizumab, 11.1% in a group receiving 37.5 mg, and 6% in the group given placebo [9]. In the phase III study (LAVOLTA) of lebrikizumab for asthma, ISRs occurred in 6% of participants receiving 37.5 mg s.c. injections, 10% of participants receiving 125 mg s.c. injections, and 8% of participants receiving a placebo once every 4 weeks [10]. In the same study, anaphylaxis was reported to be below 1%.
Korenblat et al. reported anaphylaxis in one patient (1%) treated with lebrikizumab, and this reaction was attributed to a known peanut allergy [11]. In another study, lebrikizumab was well tolerated, and death and anaphylactic reactions were not reported [12]. ISRs rarely occurred (1.3% in the lebrikizumab and 1.9% in the placebo group).
Mepolizumab
Mepolizumab is a comparatively new anti-inflammatory, anti-asthmatic BS consisting of a mAb that binds to IL‑5. Therefore, it was also approved by the FDA in December 2017 for use in the treatment of Churg-Strauss syndrome (eosinophilic granulomatosis with polyangiitis).
In a multicenter study covering the years 2009–2011 (DREAM study), the most commonly reported drug-related adverse events were non-allergic, infusion-related reactions [13]. Infusion-related reactions occurred in 5% of participants receiving 75 mg of mepolizumab, 8% of participants receiving 250 mg, 12% of participants receiving 750 mg, and 6% of participants receiving placebo. HSRs thought to be associated with the study drug occurred in 0% of participants receiving 75 mg mepolizumab, <1% of participants receiving 250 mg, 1% of participants receiving 750 mg, and 2% of participants receiving placebo. No anaphylactic reaction was reported.
Anaphylaxis has not been reported on FDA labels [14]. However, in one case, type-IV delayed HSR was observed in the 9th month of treatment, 3 days after subcutaneous (SC) administration of 100 mg mepolizumab. Local ISRs (8% after 100 mg application, 3% after 75 mg application, 3% after placebo), and only few allergic reactions (≤2%) have been reported.
Lugogo et al. [15] conducted a study on SC administration of mepolizumab in severe eosinophilic asthma patients. In this study, HSRs (type IV) occurred in ≤1% of participants, local ISRs occurred in 3%, and <1% of participants experienced injection-related reactions (non-allergic). There was no report of mepolizumab-related anaphylaxis.
Finally, in a recent study of 347 severe eosinophilic asthma patients, local ISRs (12%), allergic/HSRs (2%), and non-allergic systemic reactions (<1%) were reported [16]. Anaphylaxis was not observed.
Reslizumab
Reslizumab is a biologically active humanized mAb used for adjunctive therapy of severe asthma with an eosinophilic phenotype. This drug binds and inhibits the cytokine IL‑5, which is responsible for growth, differentiation, recruitment, activation, and survival of eosinophils.
In a study conducted by Castro et al. [17] in 2015, ISRs (hematoma, rash, and local pain) were reported to be less than 2%, and an anaphylactic reaction was reported in two patients. Anti-drug antibodies (ADA) for reslizumab were found to be negative in these patients, and therefore no further information could be obtained.
In FDA labels, treatment-related anaphylaxis was reported in three patients in the reslizumab 3 mg/kg treatment group [18]. The same label states that this drug is produced in the murine cell line, which introduces the galactose-α‑1,3‑galactose (α-gal) carbohydrate sequence as a post-translational modification into the carbohydrate side chains of the mAb. This explains why this drug is immunogenic for humans, but it is unclear how α‑gal acts and which role it plays in these reactions. ADA were positive in approximately 6% of patients treated with reslizumab, but there was no correlation with HSR and allergic reactions.
Murphy and colleagues [19] investigated the long-term effects of reslizumab and reported two HSRs (<1%), two drug eruptions (<1%), and very rare local infusion-related AEs (e.g., pain at injection site) (<1%). They did not report anaphylaxis during the follow-up period.
In a recent study, one case of toxicoderma (the 4th dose of the drug 12 h after administration) was reported [20]. The patient developed a symmetrical rash on the trunk and proximal limbs, and the symptoms improved after administration of systemic corticosteroids and antihistamines.
Omalizumab
Omalizumab is a humanized mAb commonly used as an adjunctive and second-line treatment for severe allergic asthma and chronic spontaneous urticaria. The effect is due to selective binding to immunoglobulin E (IgE).
The task force report published by the American Allergy Academy in 2007 states that the rate of anaphylaxis associated with omalizumab treatment is 0.09% [21]. Of the reactions observed to date, 61% occurred within 2 h after one of the first three doses, and 14% occurred within 30 min after the fourth or a later dose. Omalizumab is produced in the ovarian cells of a Chinese hamster and does not express the α‑1,3‑galactosyl transferase, and therefore does not contain α‑gal. As a consequence, pre-existing IgE-antibodies as have been described in patients with anaphylaxis to cetuximab were not detectable in humans [21].
In a large-scale real life study conducted between 2007 and 2016, ISRs, immediate local reactions, urticaria, and anaphylaxis were reported to be 3.4%, 1.1%, 1.0%, and 0%, respectively [22].
In 2011, a sequel of the task force report of the American Allergy Academy written in 2007 was published on the basis of post-marketing safety data provided by Genentech/Novartis [23]. In this additional report, a total of 77 of the 127 patients (60.6%) that developed anaphylaxis against omalizumab reported that these reactions occurred within the recommended hospital waiting period (especially within the first 2 h after the first three injections).
In FDA labels, anaphylaxis with symptoms such as angioedema, bronchospasm, urticaria, hypotension, and syncope has been reported in 0.1% of patients [24]. This can be seen after the first dose, as well as 1 year later. ISRs of any severity (pain, burning, induration, warmth, redness, bruising, itching, granuloma formation, etc.) were observed in 45% of patients compared with placebo (43%). More severe reactions have been reported in 12% of patients receiving omalizumab and 9% in placebo. Most of these reactions occur 1 h after injection and last for less than 8 days.
Nintedanib
Nintedanib is an antiproliferative and antitumoral oral BS of the multikinase receptor inhibitor group usually used in the treatment of idiopathic pulmonary fibrosis and non-small cell lung cancer.
The Australian Public Assessment report and the FDA label did not observe an increase in the incidence of severe immunological and anaphylactic reactions related to the use of nintedanib [25, 26]. Both reports documented hepatotoxicity, bleeding and thromboembolic events, gastrointestinal toxicity (diarrhea, nausea, and vomiting) and perforation, myocardial infarction, and hypertension as side effects. In post-marketing studies, it is reported that pruritus and rash may be seen, but the rate is not clear (referenced in [26]). Nintedanib capsules contain the following excipients; (i) capsule content: triglycerides, hard fat, lecithin (soya); (ii) capsule shell: gelatin, glycerol, titanium dioxide, iron oxide red and yellow; (iii) printing ink: shellac glaze, iron oxide black, and propylene glycol [26]. In addition, the summary of product characterics indicate that those patients with soy and peanut allergy should be treated with caution, but more detailed information as to the reason for legume allergy to be considered as a risk is lacking.
Insufficient data were found in our literature review to assess the prevalence of allergic reactions, HSR, anaphylaxis, and urticaria due to the use of this BS. This is probably due to the fact that other side effects were considered as having higher priority.
Pirfenidone
Pirfenidone is an oral BS with antifibrotic and anti-inflammatory properties. Its only indication is the treatment of mild to moderate idiopathic pulmonary fibrosis. It exerts its effect by inhibiting transforming growth factor (TGF)-β1.
Skin rash was reported in 32% of patients treated with pirfenidone and in 12% of patients treated with placebo [27]. In addition, phototoxic burn-like skin rashes on sun-exposed body areas and erythematous (edematous or non-edematous) lesions were reported in 12% of patients and in 2% with placebo.
In newly published FDA labels, photosensitivity and rash were reported at a rate of 9%, but HSR and anaphylaxis were not mentioned in this report [28].
Dermatology
Indications for which BSs are developed in dermatology include moderate to severe psoriasis, chronic urticaria, and atopic dermatitis (Table 3). Currently prescribed BSs include alefacept, efalizumab, ixekizumab, secukinumab, ustekinumab, dupilumab, quilizumab, ligelizumab, and omalizumab. TNF‑α inhibitors such as etanercept, infliximab, and adalimumab have also been approved by the FDA for treatment of moderate to severe psoriasis and psoriatic arthritis [29]. Off-label indications for TNF‑α inhibitors include autoimmune bullous disease, pemphigus vulgaris, and pyoderma gangrenosum [30]. Rarer indications include connective tissue disorders such as scleroderma, dermatomyositis, systemic lupus erythematosus, Sweet’s syndrome, sarcoidosis, granuloma annulare, toxic epidermal necrolysis, pityriasis rubra pilaris, and Behcet’s disease [29]. BSs used in the treatment of psoriatic arthritis will be mentioned in the section Rheumatology.
Alefacept
Alefacept is a fully human recombinant lymphocyte function-associated antigen-3 (LFA-3) immunoglobulin G1 fusion protein with a dual action mechanism that targets T cells, and can be administered intramuscularly or intravenously on a weekly basis. Its primary function is to interact with CD2 in the membrane of CD4 + and CD8 + T cells, inhibiting activation and thus regulating CD2/LFA‑3 interaction. A secondary mechanism of action is the induction of apoptosis in memory-effect T lymphocytes.
According to FDA labels, four out of 1869 patients (0.2%) reported angioedema in clinical trials: two of these patients were hospitalized and treated [31]. However, urticaria was seen in six patients (<1%) during the 24-week period. In one patient, therapy needed to be terminated due to allergic reactions. ISRs were reported in 16% of patients receiving alefacept by intramuscular administration, compared with 8% of patients treated with placebo. Repeated doses did not change the rate of incidence. ISRs were usually mild and were reported to manifest as pain (7%), inflammation (4%), bleeding (4%), edema (2%), local granuloma (1%), and nonspecific reaction or skin hypersensitivity (<1.0%). It has been reported that approximately 3% of patients developed low titer antibodies to the fusion protein, but a long-term effect was not known. FDA approval was withdrawn in September 2012 after a decision was made by the manufacturer to stop production of the drug, and the drug was never approved for the European market. There is no information about the frequency of anaphylaxis in the literature.
Efalizumab
Efalizumab is a humanized mAb that binds to the CD11a subunit of lymphocyte function-associated antigen‑1 (LFA-1) on the surface of lymphocytes. It is used to treat adult patients with moderate to severe psoriasis.
In a randomized controlled study involving 556 patients conducted by Gordon et al., 2% of patients developed antibodies to efalizumab, but no anaphylaxis was observed [32].
In controlled clinical trials involving 1213 patients, 8% experienced at least one HSR (e.g., asthma, dyspnea, angioedema, urticaria, maculopapular rash) and 1% experienced urticaria. Other reported side effects include angioedema, laryngospasm, erythema multiforme, serum sickness-like reaction and allergic drug eruption. A low concentration of antibodies to protein components of the drug have been reported in 6.3% of patients (referenced in [33]).
In a retrospective cohort study conducted by Brunasso et al., ISRs (local erythema, itching, burning, pain, edema, or urticaria) were observed in 4% of participants [34].
Ixekizumab
Ixekizumab is an immunosuppressive and anti-inflammatory mAb used to treat moderate to severe plaque psoriasis and can be administered subcutaneously. Its mechanism of action involves binding of the cytokine IL-17A, an important factor in keratinocyte activation and proliferation.
According to FDA labels, ISRs were reported in 17% of patients. The most common type of ISRs were erythema and pain [35]. Less frequently, serious HSRs such as angioedema and urticaria (≤0.1% each) have also occurred. In the post-marketing period, rare events of anaphylaxis requiring hospitalization have been reported.
In a long-term clinical investigation by Strober and colleagues [36] following 4209 patients, no anaphylactic reactions were reported. In this study, ISR, local erythema, and drug hypersensitivity were reported in 6.8%, 2.1%, and 0.1% of patient-years, respectively.
Secukinumab
Secukinumab is a mAb that is administered subcutaneously to treat plaque psoriasis, ankylosing spondylitis, and psoriatic arthritis. Its anti-inflammatory and immunomodulatory properties result from its binding to and inactivation of IL-17A.
In the assessment report of the EMA in 2015, no cases of anaphylaxis or angioedema were reported [37]. Urticaria was rarely (<1.0%) observed. Injection-site pain was reported in 5.6% of cases, and general HSR occurred in 6.5–11.2%. The most common HSRs in any secukinumab group were reported to be dermatitis (1.2%), eczema (1.4%), and rash (1.8%). Less than 1% of patients developed antibodies during sekukinumab treatment for up to 52 weeks.
In one study, erythema and itching at the injection site were reported in 3.0% of participants and anaphylaxis in 2.0% [38]. According to the FDA label, urticaria was reported in 0.6–1.2% of patients depending on the dose, and less than 1% of patients developed ADA [39]. Although this report states that anaphylactic and allergic reactions are rarely seen, the rate was not given. In a recent study, HSR was reported to be 2.4% per 100 patient-years and ISR in 0.8–1.3%; in addition, ADAs were reported in <1% of patients receiving treatment for 52 weeks [40].
Ustekinumab
Ustekinumab is a mAb that acts as an interleukin inhibitor and was approved for administration at 3‑month intervals as a second-line treatment for patients with moderate to severe plaque psoriasis. It has immunosuppressant and anti-inflammatory effects and neutralizes IL-12 and IL-23.
In the EMA evaluation report for 2017, ISRs were reported to occur in 3.0%. Intravenous infusion was not associated with anaphylaxis, IR, or serum sickness-like reactions [41].
FDA labels show a 1–2% incidence of ISRs (bruising, hemorrhage, induration, irritation, pain, pruritus, and swelling) [42]. In addition, one patient (0.1%) experienced signs and symptoms (flushing, tightness of the throat, shortness of breath) consistent with anaphylaxis after initial SC injection, and one patient (0.08%) experienced signs and symptoms consistent with or related to an HSR (urticaria, flushing, chest discomfort, and increased body temperature) after initial intravenous injection. In addition, approximately 6–12.4% of patients developed ADAs, which were generally of low titer (referenced in [42]).
A recent study examined the side effects of ustekinumab in psoriasis, psoriatic arthritis, and Crohn’s disease [43]. No serum sickness-like reactions or serious anaphylactic events were reported. Two patients (<1.0%) had temporary signs of treatment-associated hypersensitivity. The patients developed flushing, shortness of breath, and throat tightness after the first SC administration. In addition, fever, flushing, chest discomfort, and urticaria were observed after the initial intravenous administration.
Long-term safety and side effect studies are ongoing (C0168Z03 [PSOLAR] and CNTO1275PSO4005 [Nordic Database Initiative]).
Dupilumab
Dupilumab is a relatively new BS in the mAb group, with anti-inflammatory and selective immunosuppressive properties. It is used as second-line therapy to treat moderate to severe atopic dermatitis. It exerts its effect by binding the alpha subunit of the IL‑4 receptor, eliminating the biological effects of IL‑4 and IL-13. Since May 2019, it has been additionally approved for severe uncontrolled asthma with type‑2 inflammation.
In the FDA drug report, hypersensitivity reactions such as serum sickness, serum sickness-like reaction, and generalized urticaria occurred with a frequency of <1%, and ISRs were observed in 10% of cases [44]. ADA were detected in 2–8% of treated patients, and two subjects developed serum sickness or serum sickness-like reactions. In these patients, a high antibody titer for dupilumab was reported during treatment. There is no information about anaphylaxis.
In a meta-analysis conducted by Ou et al., ISRs were reported in 13.2% of cases [45]. There is no mention of anaphylaxis, HSR, or urticaria, but it is stated that the high incidence of ISRs needs to be explained.
The EMA 2019 report shows an HSR in 3.0–4.3% of adult patients and ISRs in 16.0–20.1% (referenced in [46]). Severe ISRs were reported in 0.3–1.4%. Most ISRs were mild to moderate. The other most commonly reported adverse events were urticaria (0.5–1.3%) and rash (0.5–0.6%), depending on the dose, but with a similar incidence to placebo. Anaphylactic reaction or angioedema was reported in three patients (0.2%), hypersensitivity in two patients (0.1%), drug hypersensitivity in two patients (0.1%), and anaphylactic shock in one patient (<0.1%).
Ligelizumab
Ligelizumab is a newly developed humanized mAb against human IgE and belongs to the IgG1/κ isotype subclass. This BS has 50-fold greater affinity to human IgE compared with omalizumab. A recent phase II clinical trial demonstrated that a higher percentage of patients had complete control of symptoms of chronic spontaneous urticaria with ligelizumab therapy of 72 mg or 240 mg than with omalizumab or placebo [47]. In the 72-mg treatment group in this study, mild injection site erythema was observed in 2.0% and mild to moderate ISRs in 4.0% of patients, while in the 240-mg-treated group, these were reported in 6.0% and 7.0%, respectively. In addition, serious side effects were reported in 2% of patients in both treatment groups, but no mortality or anaphylaxis occurred. Phase III studies are ongoing and expected to be finalized by 2021.
Quilizumab
Quilizumab is a newly developed humanized mAb against the M1-prime segment of membrane-expressed IgE. It leads to depletion of IgE-producing and memory B cells, thus blocking IgE production. Its primary indications are chronic spontaneous urticaria and uncontrolled allergic asthma. In one study assessing efficacy and safety, 6.9% of patients reported ISRs (most often local pain) [48]. Further studies were discontinued due to the lack of efficacy in adults.
Oncology
The use of BSs in oncology is quite extensive. The literature shows that these drugs are used for the treatment of bladder, breast, cervical, head and neck, gastrointestinal (stomach, bowel, colorectal), lung, and kidney cancers, as well as leukemia and lymphoma. Details of the reactions are given in Table 4.
Aflibercept
Aflibercept is a human recombinant fusion protein targeting vascular endothelial growth factor 1 (VEGF-1) receptors used in colorectal carcinoma with metastasis, as well as retinal diseases such as macular degeneration.
In the EMA 2019 assessment report and FDA 2019 label, it was stated that HSRs were observed at a rate of 0.3% (placebo 0.5%) [49, 50]. Severe HSRs such as anaphylaxis, angioedema, bronchospasm, and dyspnea have been described as uncommon (≥1/1000 to <1/100) [49]. However, more detailed information has not been provided. In addition, ADA has been reported to develop in 1.4–3.1% of patients, and the clinical significance of their neutralization has not been assessed due to limited data [50]. Maculopapular rash may develop after intravitrael administration of this drug in retinal diseases [51].
Alemtuzumab
Alemtuzumab is a monoclonal IgG1kappa antibody and a cytolytic agent used intravenously to treat leukemia and multiple sclerosis. This drug binds the glycoprotein CD52 on the surface of lymphocytes and other immune cells and leads to cell death.
In the 2013 EMA assessment report, allergic side effects were reported with relatively high frequencies (rash in 48%, urticaria in 17%, and pruritus in 16.5% of patients) [52]. Serious and severe urticaria was seen in 0.4% of cases. Severe infusion-associated reactions, including pyrexia, urticaria, atrial fibrillation, nausea, chest discomfort, hypotension, and grade IV anaphylaxis were reported in 2.2–2.8% of cases.
The British Columbia Cancer Agency (BCCA) Drug Manual reported anaphylactic reactions and angioedema in <1%, ISRs in 1.0% (severe in 0%) of intravenous users and ISRs in 90% (severe in 2%) of SC users. Reactions occurred between 24 h after the first dose and the administration of the fourth dose. In addition, SC injection site pain was reported in 61.0% (severe in 2.0%). Pruritus and urticaria were reported in 21–30% (1–5% of these reactions were severe; 0% of the reactions was associated with SC use) (referenced in [53]).
In the last published FDA label, IRs and cytokine release syndrome were reported in 92% of treated patients, but epinephrine or atropine was administered in 0.6% of these patients [54]. In some patients, infusion reactions occurred 24 h after infusion. Symptoms include fatigue, dyspepsia, pruritus, flushing, urticaria, dyspnea, pain, nausea, dizziness, and pulmonary infiltrates. Severe reactions occurred in approximately 3% of patients. Two patients experienced anaphylaxis (including anaphylactic shock). Symptoms include headache, rash, chest pain, angioedema, bradycardia, tachycardia (including atrial fibrillation), bronchospasm, hypotension, hypertension, transient neurological symptoms, and pyrexia. The most common side effects were rash (53%), urticaria (16%), pruritus (14%), and flushing (10%). Anti-alemtuzumab antibodies were detected in 62%, 67%, 29%, and 75% of the patients at 1, 3, 12, and 24 months of treatment, respectively.
In a recent study that evaluated efficacy and safety, 95.5% of patients had IRs such as fever, shivers, rash, and headache [55].
Atezolizumab
Atezolizumab is an IgG‑1 class humanized antibody that acts by binding to programmed death ligand 1 (PD-L1) used to treat bladder, breast, and lung cancer. PD-L1 is the immune checkpoint protein, which is expressed by tumor cells and inhibits the anti-tumor function of the T cell. Inhibition of the ligand’s interaction with its receptor by BS enhances the anti-tumoral activity of T cells and improves the immune system of the patients.
According to the FDA 2016 label, severe IRs were observed in 1.3–1.7% of patients [56]. These reactions include the following symptoms: back or neck pain, chills or shaking, dizziness, feeling like passing out, fever, flushing, itching or rash, dyspnea or wheezing, and swelling of face or lips. However, immune-related adverse reactions (such as colitis, hepatitis, hypophysitis, meningoencephalitis, pancreatitis, and pneumonitis) have been reported that affect various organs.
In the EMA 2019 assessment report, HSR such as anaphylaxis and IRs is reported to be seen in up to 10% of patients [57]. In addition, itching of the skin and rash are reported to be seen in more than 10% of patients.
According to the recent BCCA Drug Manual, it was reported that HSR including anaphylaxis can develop in ≤1% (severe <1%), IRs in 1% (severe ≤1%), and immune-mediated rash in 8–18% (severe ≤1%) of patients [58].
Bevacizumab
Bevacizumab is a mAb used to treat various cancers including kidney, stomach, colon, breast, and lung cancer. The antibody inhibits binding of VEGF to its receptors, reducing angiogenesis (blood vessel formation) and tumor growth.
According to the BCCA Drug Manual, IRs/HSRs with symptoms (dyspnea, redness, rash, hypo- or hypertension, oxygen desaturation, chest pain, tremor, and nausea/vomiting) were reported in up to 5% of patients (referenced in [59]). Serious hypersensitivity reactions involving anaphylactic and anaphylactoid responses have occurred, but no rates have been reported. In addition, rash was reported in 13%, rhinorrhoea in 4%, and flushing in 1% of patients.
In the EMA assessment report, IRs occurred in 0.3–6.1% of cases, and the most commonly reported IR was hypertension (2.8–3.1%) (referenced in [60]). In this report, HSRs and anaphylaxis were described as treatment-emergent adverse events (TEAE) and were reported to be any grade of TEAEs in 36.9%, grade 3 or higher TEAEs in 5.9%, and serious TEAEs in 2.5% of cases. Further information on reactions is not available.
IR and symptoms (chest pain, chills, grade 3 hypersensitivity, diaphoresis, headache, hypertensive crises associated with hypertension, hypoxia, neurological signs and symptoms, and wheezing) have been reported in clinical trials and post-marketing studies, as indicated on the FDA label [61]. Any grade of IRs are reported to be rare (<3%), and serious IRs have occurred in 0.2% of the patients. Symptoms include chest pain, diaphoresis, hypertensive crises, hypoxia, rigors, and wheezing. Although urticaria and anaphylaxis are not mentioned in this label, exfoliative rash was seen in 23% with the administration of this BS in combination with the chemotherapy protocol.
Blinatumomab
Blinatumomab is a bispecific mAb used for cancer immunotherapy and specific forms of acute lymphoblastic leukemia (ALL). It exerts its effect in part by simultaneously binding CD19 on B cells and CD3 on T cells.
This BS can lead to cytokine release syndrome (CRS) in 11–12% of patients, with 1% of these cases being severe, to hypersensitivity in 2% of patients, and infusion reactions in 29% of patients, according to the BCCA’s 2017 report (referenced in [62]). Symptoms of this syndrome include asthenia, nausea, pyrexia, headache, hypotension, and increased liver enzymes. It was reported that CRS peaked within the first 2 days of treatment and was not easily differentiated from IR. Although anaphylaxis and urticaria were not mentioned in this report, the incidence of rash was reported to be 12–21% (severe 2–3%).
According to the EMA assessment report, CRS occurred in 0.5–11.4% of patients, rash occurred in 4.3%, and IRs occurred in 67.2% (grade ≥3 in 14.3%). In total, severe IRs were very rarely seen (4.3%) [63]. Fatal IRs were not reported. Additionally, maculopapular rash and flushing have been reported in 4.3% of patients.
According to the recent FDA label, this BS caused CRS in 7–15% of patients (3% of these instances were severe), skin rash in 16% of patients (<1% of these instances were severe) and IRs in 77% of patients (5% of these instances were severe) [64]. Symptoms of CRS included asthenia, headache, nausea, fever, hypotension, increased liver enzymes, increased total bilirubin, and diffuse intravascular coagulation. IRs usually occurred during the first 48 h of infusion and lasted less than 2 days. Clinical findings included hypotension, hypertension, rash, generalized pruritus, pyrexia, periorbital edema, and cytokine release syndrome. Events defined as rash included maculopapular rash, erythema, contact dermatitis, and eczema.
Cetuximab
Cetuximab is an intravenously administered epidermal growth factor receptor (EGFR) inhibitor used to treat advanced colorectal cancer, head and neck squamous cell carcinoma, pancreatic cancer, and less frequently, non-small cell lung cancer. This antibody can reduce cancer cell survival, neovascularization, cell migration, and metastasis. Cetuximab was the first anti-EGFR mAb and BS approved for cancer treatment; therefore, its immunological and allergic side effects are well studied.
In a review of acute infusion reactions induced by mAbs, acute IRs with cetuximab were in the range of 1.2–15% (referenced in [65]). It is noted in that particular review that the pathogenic mechanism of mAb-related IRs has not yet been fully defined, with the exception of several suggested mechanisms by W. J. Pichler [66].
HSRs have been reported in 7.4% of participants in a safety study with 419 patients, with 2.6% of reported HSRs being of grade 3 severity, and 1.4% being of grade 4 anaphylactic reactions [67]. The majority of reactions occurred after the first dose, and all grade 4 reactions occurred within minutes following the first infusion.
A 2015 review reported HSR to be in the 1.1–5.0% range, and infusion reactions in the 15.0–21.0% range in patients treated with cetuximab (referenced in [68]).
According to the FDA label, mild IRs (fever, chills, shortness of breath, bronchospasm, angioedema, urticaria, hypertension, and hypotension) occurred in 8.4% of patients, and severe IRs occurred in 2.2% of patients, with 90% of events occurring after the first infusion [69]. In addition, 82% of patients developed acneiform rash during treatment, 9.7% of which were severe. Anaphylactic reactions during treatment have been reported to be increased in association with tick bites by Amblyomma americanum, a history of red meat allergy, all associated with IgE-antibodies against α‑gal [69,70,71].
In 2010, reactions during the first infusion of cetuximab and delayed type anaphylactic reactions to red meat were investigated in detail [70]. In both diseases, patients were shown to have increased IgE antibodies specific for α‑gal, a mammalian disaccharide, not expressed in humans but on the tissue of mammalians. In most patients with an HSR and anaphylaxis, IgE antibodies to cetuximab were also present in serum before treatment, which prompted the investigations into the sensitization route [71]. It was shown that α‑gal is also the main cause of delayed anaphylaxis against red meat and that this was associated with the occurrence of the American tick Amblyomma americanum [72]. Presently, tick bites are thought to be the predominant sensitization route for anti-α-GAL IgE induction.
α‑GAL, however, has another implication for drug-induced hypersensitivity reactions: It is present in mammalian gelatine and, therefore, may be part of vaccines, gelatine-containing infusion solutions, ovula, capsules and pills, suppositories, snake venom antisera, and animal-derived heart valves (referenced in [73]).
However, in a study involving 92 patients, 14 (15.2%) reported HSR, eight of these (57.1%) reported severe HSR [74]. Anti-cetuximab IgE levels were detected in seven of the eight patients with severe HSR.
Durvalumab
Durvalumab is a PD-L1 binding and blocking mAb used in non-small cell lung carcinoma and metastatic urothelial carcinoma.
In the FDA 2019 labels, IRs were reported to occur in 2.2% (grade 3 in 0.3%), and immune-mediated rash in 11–26.0% (grade 3–4 in 0.6–1.0%) of patients [75]. ADA developed in 2.9% of patients, but its clinical significance has not been established. HSRs and anaphylaxis are not mentioned in these labels.
In the EMA 2020 assessment report, any grade of IR has been reported to occur in 1.9% (grade 3 in 0.3%), and skin rash in 21.7% (grade 3 in 0.6%) of patients [76]. Symptoms of IR include: chills or shaking, dizziness, dyspnea or wheezing, fever, flushing, and itching or rash. HSRs and anaphylaxis are not mentioned in these labels.
According to the BCCA’s 2020 drug report, IRs can be observed in 1.0–2.0% (severe in <1%) of patients, and skin rash in 14–26.0% (severe in <1%) of patients [77].
Gemtuzumab ozogamicin
Gemtuzumab (-ozogamicin) is an antibody against glycoprotein CD33, which is administered intravenously to treat acute myeloid leukemia (AML). This antibody combined with a cell-toxic substance induces apoptosis of the targeted cancer cells.
In a prospective observational study involving 225 patients, as described in the FDA report, IRs were observed in 8.0% of patients, and fatal reactions in 0.4% [78]. In addition, local reactions to the application occurred in 22.0%. Pruritus was described in 6.0% and rash in 18.0% of patients. Infusion-related reactions usually developed within 24 h after infusion. Signs and symptoms were similar to anaphylactic reactions. These included chills, hypoxia, fever, tachycardia, bronchospasm, and respiratory failure.
According to the 2018 EMA assessment report, IRs, including anaphylaxis, were reported in 7.6% of patients, and severe reactions in 3.6% of patients [79]. Symptoms included fever, chills, injection-site urticaria, and less frequently, hypotension, tachycardia, and respiratory symptoms. All symptoms occurred during the first 24 h after drug administration. In addition, rash was reported in 19.9% (severe in 5.8%), local erythema in 9.3% (severe in 2.1%), and pruritus in 5.4% (severe in 0.3%) of patients.
The literature contains one case report of HSR mortality, which was associated with platelet transfusion after gemtuzumab treatment [80].
Ibritumomab tiuxetan
Ibritumomab (-tiuxetan) is used in non-Hodgkin’s lymphoma patients or for cancers that are refractory to rituximab. It acts by binding to the antigen CD20, which is present on the surface of malignant and normal B‑lymphocytes.
In the first FDA report, allergic reactions were reported in 2% of patients, urticaria in 4%, and severe life-threatening reactions such as angioedema, lung edema, tachycardia, subdural hematoma, and pulmonary embolus in <1% of patients [81].
In the EMA evaluation report, allergic (hypersensitivity) reactions and infusion reactions were described [82]. Common symptoms (up to one in 10 patients) include skin reactions, difficulty in breathing, swelling, itching, redness, chills, and dizziness (a potential marker for hypotension). Severe hypersensitivity reactions involving anaphylaxis occurred in <1% of patients.
According to the BCCA’s drug report, ISRs (dermatitis, desquamation, and ulcer following extravasation) occurred in 1.0% of patients, angioedema in 5.0% (severe angioedema in 1.0%), pruritus in 9.0%, rash in 8.0%, and mucocutaneous reactions in <1% of patients (referenced in [83]). That particular monograph states that rituximab is an essential component of ibritumomab-based treatment regimens. Infusion reactions may occur frequently after pretreatment with rituximab, or during or after ibritumomab administration. Asthenia, cough, dizziness, nausea, pruritus, pyrexia, rash, rigors, tachycardia, and vomiting have been reported among the symptoms of IRs.
In a recent case report, patients were shown to develop human anti-murine antibodies (HAMA) after exposure to murine antibodies [84]. The low incidence of hypersensitivity reactions in patients with high HAMA titers is particularly promising for ibritumomab.
Imatinib
Imatinib is an active ingredient in capsules or tablets containing a group of kinase inhibitors used to treat chronic myeloid leukemia. Other and rarer indications include acute lymphoblastic leukemia, skin tumors, some gastrointestinal tumors, hypereosinophilic syndrome, atypical myelodysplastic or myeloproliferative disorders, as well as systemic mastocytosis.
The first FDA report does not include information on hypersensitivity and anaphylactic reactions. This report includes only skin rash in 32–39% of patients (severe skin rash in 3–4%), and pruritus in 6–10% (severe in 0.4–1.0%) [85].
BCCA drug manuals describe cutaneous reactions in <1%, rash in 32–39% (severe rash in 3–4%), and pruritus in 6–10% (with severe pruritus in 0–1%) of patients. This manual does not include information on hypersensitivity and anaphylactic reactions [86].
Ipilimumab
Ipilimumab is a fully-human mAb used primarily for the treatment of metastatic melanoma, metastatic colorectal carcinoma, and renal cell carcinoma. This BS shows its effect by binding and inhibiting human cytotoxic T lymphocyte antigen 4 (CTLA-4). As a result, T cell infiltration increases in tumor cells and tissues, thereby leading to their destruction. In addition, studies are ongoing for its use in the treatment of lung cancer, bladder cancer, and metastatic prostate cancer.
In the FDA 2019 labels, IRs were reported to occur in 4.2–5.1% of patients and urticaria in 2.0% of patients [87]. Symptoms of IR include: chills or shaking, dizziness, dyspnea, feeling like passing out, fever, flushing, and itching or rash.
In the EMA 2020 assessment report, HSRs are reported as uncommon (≥1/1000–<1/100), urticaria as common (≥1/100 to <1/10), and anaphylactic reaction as very rare (<1/10,000) [88]. IRs were observed at a rate of 2.2–4.0% depending on the dose of the drug, but grade 3–5 IRs were not reported.
According to the BCCA’s 2020 drug report, IRs can be observed in 2.0–6.0% (severe in <1%) of patients, pruritus in 24–26.0% of patients, and skin rash in 19–26.0% (severe in <1%) of patients [89]. Additionally, a case that developed a delayed type of HSR during melanoma treatment has been reported [90].
Necitumumab
Necitumumab is a human mAb used in chemotherapy combinations in the treatment of non-small cell lung cancer, and shows its effect by binding to the EGFR. EGFR plays an important role in the uncontrolled growth and proliferation of tumor cells and tissue as a result of apoptosis inhibition.
In the FDA 2015 labels, IRs of any severity have been reported to occur in 1.5% (grade 3 in 0.4%) of patients [91]. These reactions have been frequently observed after the first two doses.
The same data are also reported in the EMA 2016 assessment report [92]. In addition, it is stated that skin reactions can be seen in 77.9% (grade 3 in 6.3%) of patients. Symptoms of IR include: chills, dyspnea, or fever. Urticaria and anaphylaxis are not mentioned in that report. In addition, sudden death or cardiorespiratory arrest was reported in 2.8% of patients, which may also be a symptom of anaphylaxis.
Nivolumab
Nivolumab is a human mAb that is used in the treatment of advanced renal cell carcinoma, hepatocellular carcinoma, Hodgkin’s lymphoma, melanoma, non-small cell lung cancer, squamous cell cancer of the head and neck, as well as urothelial cancer and acts by binding to the programmed cell death protein (PD)-1-receptor.
In the FDA 2019 labels, IRs were reported to occur in 6.4% of patients [93]. The IR causing permanent discontinuation or withholding of the BS was reported in 0.5–1.4% of patients within 48 h after infusion. Anaphylaxis and hypersensitivity have not been reported, but it has been reported that the incidence of symptoms such as abdominal pain, arthralgia, asthenia, back pain, constipation, cough, decreased appetite, diarrhea, dyspnea, fatigue, headache, musculoskeletal pain, nausea, pruritus, pyrexia, rash, upper respiratory tract infection, and vomiting is more than 20%. Some of these symptoms can be seen in anaphylaxis. In treatment with this BS, immune-mediated organopathies such as colitis, encephalitis, hepatitis, pneumonitis, endocrinopathies, and nephritis can also be observed. The clinical significance of ADAs for these side effects is still unknown.
In the EMA 2020 assessment report, IRs and HSRs are stated as common (≥1/100–<1/10), rash and prutitus as very common (≥1/10), and anaphylactic reaction as rare (≥1/10,000–<1/1000) [94].
According to the BCCA’s 2020 drug report, IRs can be observed in 2.0–4.0% of patients, pruritus in 7–17.0% of patients, skin rash in 11–21.0% (severe in 1.0%), and urticaria in 1.0% of patients [95].
Allergic symptoms (eyelid angioedema, flushing, and hives on the neck and face) were reported in one patient during treatment with nivolumab [96]. However, a case with cytokine release syndrome has been reported [97]. In a recent case report, a patient who had recurrent infusion reactions despite premedication against nivolumab was successfully treated with pembrolizumab, another PD-L1/PD-L2-inhibitor [98].
Panitumumab
Panitumumab is a mAb EGFR inhibitor delivered intravenously to treat metastatic colorectal cancer. It acts by binding and inhibiting EGFR. Panitumab appears to be a safe option for treatment and potential side effects in patients with anti-α-gal IgE antibodies [99]. This is due to the fact that α‑galactosylated glycans are absent in purely human mAbs and that the system in which it is expressed does not seem to induce posttranslational modifications like allergenic glycosylation. The chimeric mAb cetuximab is not suitable in this sensitive patient group due to its high α‑gal content.
In the 2015 FDA report, IRs were reported in 4% of patients (severe reactions in 1%) [100]. Severe IRs included anaphylactic reactions, bronchospasm, and hypotension. Angioedema and fatal reactions have also been documented in post-marketing reports.
In the BCCA drug monograph, HSR was reported in 1% of patients (occurring within 24 h), while IRs were reported in 3–4% (severe IRs were seen in 1%, occurring within 24 h) (referenced in [101]). Angioedema was seen very rarely. Some cases were fatal, with possible late onset more than 24 h post infusion. The symptoms of most IRs were mild and included chills, fever, or dyspnea. In addition, pruritus was reported in 34–69% (severe in 1–4%) and rash in 20–78% (severe in 1–3%).
At the annual congress of the European Academy of Allergy and Clinical Immunology (EAACI) in 2019, a case of a successful desensitization was presented in a patient that had developed anaphylaxis against panitumumab [102].
Pembrolizumab
Pembrolizumab is a monoclonal antibody that is used to treat non-small cell lung cancer, lymphoma, melanoma, and urothelial carcinoma and which binds to the PD-1-receptor and inhibits its interaction with its ligands.
In the EMA 2019 assessment report, IRs are stated as common (≥1/100–<1/10), rash and pruritus as very common (≥1/10), and gastrointestinal symptoms (such as abdominal pain, constipation, diarrhea, nausea, vomiting) as very common (≥1/10) [103]. According to the BCCA’s 2019 drug report, IRs can be observed in <1.0% of patients, pruritus in 12–30.0% of patients, and skin rash in 1.0–29.0% (severe in 1.0%) of patients [104].
In the FDA 2020 labels, severe or life-threatening anaphylaxis, HSRs or IRs were reported in 0.2% of patients [105]. Symptoms of IR include: chills, dizziness, fever, flushing, hypotension, hypoxemia, pruritus, rash, rigors, and wheezing. However, the incidence of ADAs against pembrolizumab was 1.8%, and it was reported that ADA had no clinical relevance [106].
Pertuzumab
Pertuzumab is a humanized mAb that acts by blocking the human epidermal growth factor receptor 2 protein (HER2), and is used in combination with trastuzumab and docetaxel in the treatment of breast cancer. HER2 plays an important role in the growth and differentiation of tumor cells, and its blocking results in apoptosis.
According to the BCCA’s 2014 drug report, HSRs can be observed in 11.0% (severe in 2–5%) of patients, IRs in 13–19.0%, rash in 10–20% (severe in 2%), and pruritus in 10% of patients [107]. Symptoms of IR include: asthenia, chills, fatigue, fever, hypersensitivity, and vomiting.
In the EMA 2020 assessment report, IRs and rash are stated as very common (≥1/10), HSRs as common (≥1/100 to <1/10), anaphylaxis as uncommon (≥1/1000–<1/100), and CRS as rare (≥1/10,000–<1/1000) [108]. The most common IRs have been reported as asthenia, chills, fatigue, headache, hypersensitivity, pyrexia, and vomiting.
In the FDA 2019 labels, IRs have been reported to occur in 13–21.0% (severe less than 1.0%), and hypersensitivity/anaphylaxis in 5.0–11.0% of patients [109]. It is stated that CRS may also develop.
Ramucirumab
Ramucirumab is a human mAb used in the treatment of gastric, breast, lung, and colorectal cancers, showing its effect by selectively inhibiting VEGF receptor‑2. VEGF plays an important role in angiogenesis, cell division, and migration in vascular endothelium.
According to the BCCA’s 2017 drug report, IRs can be observed in 1.0–16.0%, and rash in 4.0% of patients [110]. Symptoms of IR include: back pain, chest pain, chills, dyspnea, flushing, paresthesias, rigors, and wheezing. Some cases have been observed with more severe symptoms such as bronchospasm, hypotension, and supraventricular tachycardia.
In the FDA 2019 labels, IRs have been reported to occur in 1.0–9.0% (severe in <1.0%) of patients [111]. Symptoms of IR include: back pain/spasms, chills, chest pain and/or tightness, dyspnea, flushing, hypoxia, paresthesia, rigors/tremors, and wheezing.
In the EMA 2019 assessment report, IRs can be observed in up to 10.0% of patients [112]. Symptoms of IR include: back or chest pain, chest tightness, chills, dyspnea, feeling of tingling or numbness in hands or feet, flushing, increased muscle tension, and wheezing. Some cases have been observed with more severe symptoms such as breathing distress, feeling faint, and tachycardia. Anaphylaxis and hypersensitivity are not mentioned in that report.
Trastuzumab
Trastuzumab is an anti-HER2-antibody combined with or conjugated to a cytotoxic drug that is used to treat local or advanced inoperable breast and gastric cancer.
In the BCCA drug assessment report (referenced in [113]), IRs occurred in 21–40% of cases, with severe reactions in 1%. Allergic reactions were reported in 3% of cases. IRs occurred in 40% of patients with their first infusion. Mild to moderate symptoms included headache, dizziness, rash, cough, nausea, vomiting, pain, chills, and asthenia. Severe reactions and symptoms were most often respiratory distress, bronchospasm, wheezing, low oxygen saturation, and hypo- or hypertension. Galvão et al. (referenced in [68]) reported an HSR rate of 0.6–5.0% and an infusion-related reaction rate of 40%.
According to the EMA assessment report 2018, HSR was reported in 0.9–3.5%, IRs in 8.5–37.1%, and anaphylaxis in 0–0.9% of patients [114].
According to the FDA’s 2019 report, IRs occur in 1.4–1.6% of patients and include symptoms such as fever, chills, redness, shortness of breath, bronchospasm, wheezing, tachycardia, and hypotension. In the same report, pruritus was seen in 6.0% and drug hypersensitivity was found to occur in 2.2–2.7% of patients. In preliminary studies, anaphylaxis was seen in one patient [115].
Rheumatology
BSs are often used to treat rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, inflammatory bowel diseases (ulcerative colitis, Crohn’s disease), Still’s disease, and vasculitis in rheumatology. Details on the reactions are given in Table 5.
Adalimumab
Adalimumab is a recombinant human IgG1 that binds to TNF‑α with high affinity. It acts by inhibiting the binding of TNF‑α to p55 and p75 surface receptors on target cells.
In a study on 201 patients conducted by Puxeddu et al. [116], hypersensitivity reactions occurred in seven patients (3.5%), local reactions occurred in three patients (1.5%), and urticaria and angioedema occurred in three patients (1.5%). Anaphylaxis was not observed in this study, but has been reported very rarely in the post-marketing period. Another study conducted by Tarkiainen et al. [117] reported HSR frequencies of 18.1%, ISRs frequencies of 17.0%, and allergic symptoms in 1.1% of the patients.
FDA 2018 labeling showed that local ISRs are more common in patients treated with the TNF‑α inhibitor than in those treated with placebo [118]. The incidence of ISRs including erythema, itching, hemorrhage, and pain or swelling is 8.0–20.0% in patients treated with adalimumab vs 1% in patients treated with placebo. Anaphylaxis and angioneurotic edema have rarely been reported following the application of this BS, and in the first 48 weeks of treatment, approximately 6% of patients had non-severe local allergic hypersensitivity reactions and allergic rashes.
Anakinra
Anakinra is a genetically engineered and recombinant human IL‑1 receptor antagonist. Its main effect is by neutralizing the biological activity of IL-1α and IL-1β, a proinflammatory cytokine. As a result, it contributes to the reduction of synovial inflammation by reducing many cellular responses.
However, anakinra has the following therapeutic indications: rheumatoid arthritis (RA), cryopyrin-associated periodic syndrome (CAPS), and Still’s disease.
In the EMA summary of product characteristics (SmPC) report, ISRs are generally reported to occur in the first 2 weeks of treatment (for RA and Still patients) and resolve within 4–6 weeks [119]. In that particular report, allergic reactions such as anaphylactic reactions, angioedema, urticaria and pruritus are expected to range from 1/100 to 1/1000.
According to the FDA’s 2018 label, the most frequently reported adverse reactions were ISRs (71.0%), which were mild to moderate in the majority of patients [120]. Only 3.2% of these reactions were classified as severe, with the majority including ecchymosis, erythema, inflammation, and local pain. Reactions usually occurred during the first month. This label states that anaphylaxis, angioedema, urticaria, and rash may occur, but no specific rate or detailed information is provided.
In the literature, it has been reported that anakinra treatment may cause anaphylaxis, cutaneous drug reactions, erythematous plaques, pruritic rash, and severe HSRs [121]. In a pediatric case, a patient with anakinra-induced anaphylaxis was successfully treated with canakinumab, an alternative anti-IL‑1 agent [122]. However, a desensitization protocol is also available [123].
Belimumab
Belimumab is a recombinant human IgG1 mAb that binds and inhibits the biological activity of the soluble B lymphocyte stimulator (BLyS), a member of the TNF ligand superfamily. It is used in the treatment of systemic lupus erythematosus and is applied subcutaneously or intravenously.
In the EMA 2018 assessment report, any event of ISRs (local pain, erythema, hematoma, induration, and pruritus) were reported in 6.1% of patients, HSRs in < 10.0% of patients, and post-injection anaphylactic reactions were reported in 0.1–1.0% of patients (serious reactions, 0.9%) [124]. HSR and anaphylactic reaction are mentioned to occur mostly during the first two infusions.
In the newly published FDA report, intravenous administration was evaluated [125]. HSRs were reported in 13.0% (in the placebo group 11%), anaphylaxis in 0.6% (placebo 0.4%) of patients. Symptoms included angioedema, dyspnea, hypotension, pruritus, rash, or urticaria. IRs were observed in 17% of patients and severe IRs in 0.5%, with placebo in 15% and 0.4%, respectively. It is stated that it is not easy to differentiate between HSRs and IRs due to the overlap in signs and symptoms. However, it has been reported that there is insufficient evidence as to how the premedication of some patients affects the frequency or severity of reactions.
Canakinumab
Canakinumab is a human mAb against IL‑1 beta and indicated in periodic fever syndromes, CAPS, tumor necrosis factor receptor-associated periodic syndrome (TRAPS), familial Mediterranean fever (FMF), Still’s disease, and gouty arthritis.
According to the FDA’s 2016 label, the most frequent adverse drug reactions were infections predominantly of the upper respiratory tract, and mild to moderate ISRs equal or greater than 10.0% [126]. No anaphylactoid or anaphylactic reactions were observed in more than 2600 patients whose clinical development was tested for this biologic, but the risk of severe hypersensitivity reactions was mentioned.
No cases of canakinumab-related anaphylaxis have been reported in the current literature [127]. In addition, patients with anaphylaxis to anakinra have been shown to tolerate canakinumab very well [122].
Etanercept
Etanercept is a subcutaneously administered selective immunosuppressive and anti-inflammatory BS belonging to the group of TNF‑α inhibitors used to treat rheumatoid arthritis and other rheumatic diseases. It exerts its effect by inhibiting the activity of TNF‑α.
Puxeddu et al. reported a 5.3% rate of HSRs, a 0.8% rate of anaphylaxis, a 2.0% rate of angioedema or urticaria, and a 1.6% rate of local reactions in a study involving 245 patients treated with etanercept [116]. In another study, the rate of HSRs was 11.3%, that of ISRs was 7.5%, and reactions at sites other than the injection site occurred at a rate of 4.2% [117].
The FDA drug label published in 2018 reports that allergic reactions associated with this treatment occur in <2% of patients, and mild to moderate ISRs (erythema, itching, pain, swelling, bleeding, bruising) occur in 15–37% of patients during the first 3 months of treatment [128]. ISRs were usually seen in the first month, and with decreased frequency thereafter.
Golimumab
Golimumab is a selective immunosuppressive and anti-inflammatory TNF-α-inhibitor applied subcutaneously to treat rheumatoid and psoriatic arthritis, as well as ankylosing spondylitis. It is a mAb that inhibits the activity of soluble and membrane-bound forms of the proinflammatory cytokine TNF‑α by binding TNF‑α and inhibiting its receptor binding.
In a 3-year safety study, the frequency of ISRs was 4.7–11.6%, depending on the drug dose. No anaphylactic reactions or serum-like reactions have been reported to date during the follow-up period of up to 160 weeks [129].
The FDA 2018 drug label reports an ISR frequency of 3.4–6.0% [130]. The reactions were often mild and included erythema, urticaria, induration, and pain. No anaphylactic reactions were observed in phase II and III studies with the drug.
Infliximab
Infliximab is an intravenously administered selective immunosuppressive and anti-inflammatory TNF‑α inhibitor used to treat rheumatoid and psoriatic arthritis, Crohn’s disease, and ankylosing spondylitis. This BS is a human–murine chimeric monoclonal IgG1κ antibody that binds TNF‑α and inhibits its effects.
In a review by Maggi et al. summarizing nine studies, infusion reactions occurred in 1–27% of patients depending on the disease being treated (referenced in [65]). In a study on 225 patients, Puxeddu and colleagues reported hypersensitivity reactions in 13.8%, anaphylaxis in 9.3%, urticaria or angioedema in 4.4%, and local reactions in 0% of patients [116]. Another study reported a 34.1% rate of infusion-related HSRs, and a 1.9% rate of anaphylaxis [117].
The FDA 2019 drug label reports IRs in 18% of patients during or within 1 h after the infusion [131]. A total of 3% of all patients given infliximab infusions experienced nonspecific symptoms (fever or tremor), while 1% experienced cardiopulmonary reactions (chest pain, hypotension, hypertension, or dyspnea). Symptoms related to pruritus, urticaria, and cardiopulmonary reactions were detected in 1% of patients. Serious infusion reactions such as anaphylaxis, convulsions, erythematous rash, and hypotension have occurred in <1% of patients. Delayed type HSR was detected in approximately 1% of patients. This usually occurred within 2 weeks of re-infusion, and was a combination of fever and/or rash symptoms with arthralgia and/or myalgia. These findings have been classified and reported as serum disease.
In a study conducted by Vultaggio et al., it was reported that IgE or IgM antibodies against infliximab could be detected and may play a role in IgE- and non-IgE-mediated anaphylaxis [132]. Skin testing and infliximab-specific antibody detection have been shown to be useful in predicting and preventing severe drug-related reactions [133].
Certolizumab
Certolizumab is a subcutaneously administered selective immunosuppressive and anti-inflammatory TNF-α-inhibitor used to treat rheumatoid and psoriatic arthritis, spondyloarthritis, and Crohn’s disease. It is a mAb that binds to and blocks the activity of the proinflammatory cytokine TNF‑α.
In the EMA 2015 assessment report, mild to moderate ISRs were reported in 6.4% of patients, injection-related acute systemic HSR (pre-syncope) was reported in 0.2%, and delayed HSR was seen in 1.1% of patients [134].
Recently published FDA labels have reported that HSRs may rarely occur and include allergic dermatitis, rash, angioedema, dizziness, pyrexia, flush, ISRs, malaise, serum sickness, hypotension, dyspnea, and vasovagal syncope [135]. It is noted that some of these reactions were observed after the first administration. ISRs were reported in 1.7–3.2% of patients.
Although the literature contains no clear information on rates of hypersensitivity and anaphylaxis, it is stated that no single-drug related anaphylactic shock was observed in the one study conducted in 2013 [136].
Rituximab
Rituximab is an intravenously delivered mAb that selectively binds to the CD20 antigen of B lymphocytes. Indications for its use include non-Hodgkin’s lymphoma, rheumatoid arthritis, and chronic lymphocytic leukemia.
Maggi et al. have reported acute infusion reaction frequencies of 10–77% during the first infusion (referenced in [65]). The FDA drug label for subcutaneous use [137] reports a 16–26% rate of local ISRs (pain, swelling, induction, hemorrhage, rash, pruritus, and erythema). Incidence rates reportedly decrease subsequent to the initial injection. The label also states that severe cytokine release syndrome and its symptoms (fever, tremor, urticaria, angioedema, bronchospasm, and severe dyspnea) may be seen within 1–2 h of infusion. However, the ratio is not specified in this label.
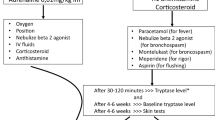
The recent FDA drug label for intravenous use [138] reports a ≥25.0% rate of IRs that typically (77%) occurred at the first infusion with the time onset between 30–120 min. This rate decreased after each ongoing infusion. Severe IRs may include symptoms such as acute respiratory distress syndrome, anaphylactic/anaphylactoid events, angioedema, bronchospasm, cardiogenic shock, hypoxia, hypotension, myocardial infarction, pulmonary infiltrates, urticaria, ventricular fibrillation, and death.
According to the BCCA drug assessment report (referenced in [139]) HSRs occurred in 1–10.0% of patients, IRs in 14–77.0% (severe, 0–7.0%), ISRs in 20.0%, and urticaria in 7.0% (severe, 1.0%) of patients. ISRs include local erythema, hemorrhage, induration, pain, pruritus, rash, and swelling at the injection site.
In a study conducted in 2009, the presence of serum anti-rituximab antibodies was reported in some patients in association with a less favourable treatment outcome [140]. Vultaggio and colleagues have shown that rituximab plays a role in specific T‑helper 2 cell and IgE responses in acute infusion reactions in addition to cytokine release mechanisms [141].
Transplantation
A less frequent use of BSs is to minimize the rejection of transplanted organs (mostly kidneys, hearts, and livers). Basiliximab, muromonab, and daclizumab have been developed for this purpose. Today, only basiliximab is approved for this application. The other two drugs have been withdrawn from the market for a variety of reasons. Details about the reactions are given in Table 6.
Basiliximab
Basiliximab is an immunosuppressive intravenously administered chimeric (75% human, 25% murine) mAb. It is used in combination with other immunosuppressive drugs to prevent acute graft rejection following renal transplantation. It acts by binding the alpha chain of the IL‑2 receptor on the surface of T lymphocytes.
The FDA drug label reports a >10.0% rate of side effects including nausea, vomiting, fever, dyspnea, and hypertension, and 3.0–10.0% of side effects include angina pectoris, cardiac failure, tachycardia, dizziness, bronchospasm, pulmonary edema, rhinitis, rash, and pruritus [142]. More detailed information is not provided. In a study conducted in 2010, infusion-related ISRs (pain, swelling, erythema) were found in 4.3% of patients [143]. In post-marketing surveys, HSRs have been reported in patients undergoing basiliximab therapy, including one case of IgE-mediated anaphylaxis [144].
Data on this BS are relatively limited, but according to the reports it seems to be mainly well tolerated by the patients [145]. Allergic reactions and anaphylaxis do not seem to be easily identified, since the patients experience intense immunosuppressive effects due to the concomitant medication. It does not induce CRS like muromonab-CD3, and its side effects are reported to be similar to placebo.
Belatacept
Belatacept is a human CTLA-4/human IgG1 fusion protein that selectively inhibits T cell activity by binding to CD80 and CD86 of the antigen presenting cells.
According to the FDA 2017 labels [146], no patient had developed anaphylaxis or HSRs at the 3‑year follow-up. However, milder IRs (5%) were reported, similar to placebo, within 1 h of infusion. In post-marketing experiences, a case of anaphylaxis has been reported.
In the EMA 2019 assessment report [147], acute infusion events (flushing, headache, hypotension, hypertension) were reported in 4.4–5.5% of patients. Symptoms of diarrhea and pyrexia (≥2%), as well as hypertension, peripheral edema, nausea, and vomiting (≥20%), which may be “possible peri-infusional events,” were reported in 3‑year follow-up of the patients. There was no association found between these reactions and belatacept ADAs. Anaphylactic events have been observed in the post-marketing period, but no incidence was given.
Muromonab
Muromonab is an immunosuppressive mAb given to reduce acute rejection in patients with kidney, liver, or heart transplants. It exerts its effect by targeting the CD3 receptor, a membrane protein expressed on the surface of T cells. Developments and progress of medical treatment in transplant medicine led to its withdrawal in 2010.
The literature contains information on cytokine release syndrome and anaphylaxis. In a book on this subject, symptoms of anaphylaxis such as pyrexia (90%; 40.0 °C or higher in 19% of cases), chills (59%), dyspnea (21%), nausea and vomiting (19%), chest pain (14%), diarrhea (14%), tremor (13%), bronchospasm (13%), headache (11%), tachycardia (10%), stiffness (8%), and hypertension (8%) were reported [148]. Pilot studies of this drug have been performed orally in patients with moderate to severe ulcerative colitis [149]. In this study, no severe side effects or CRS have been reported.
Daclizumab
Daclizumab is a subcutaneously administered, immunosuppressive mAb used to treat relapsing-remitting multiple sclerosis and to prevent rejection of transplanted kidneys. The antibody reduces the number of circulating activated T cells by binding to the alpha subunit of the IL‑2 receptor on T lymphocytes and inhibiting interaction with IL‑2. In 2018, the drug was withdrawn from the market owing to reports of severe hepatotoxic side effects and encephalopathies.
Other
Other indications for the use of biologicals include multiple sclerosis, paroxysmal nocturnal hemoglobinuria, instable angina pectoris during insertion of cardiac catheters, congenital bronchopulmonary dysplasia, and congenital heart disease.
Abciximab
Abciximab is an intravenously administered chimeric mAb fragment that inhibits binding of platelet adhesion proteins (such as fibrinogen, fibronectin, von-Willebrand-Factor) to glycoprotein IIb/IIIa receptors on the surface of platelets. It is frequently used to prevent ischemic complications associated with invasive heart procedures, as well as to prolong cardiac infarct prophylaxis in patients with unstable angina pectoris due to its antithrombotic effect.
The registry study of the drug reported hypersensitivity rates of 0.3–0.6%, an urticaria rate of 0.1%, and no cases of anaphylaxis or angioedema [150]. In the same study, human anti-chimeric antibodies were found to be associated with thrombocytopenia. The literature contains a very small number of case reports. One developed an anaphylaxis 2–4 h after treatment [151]. The FDA label reported bronchospasm (0.3%), injection site pain (0.1–3.6%), and pruritus (0.5%) [152]. No incidence of anaphylaxis has been reported in any of the phase III studies (clinical trials).
Eculizumab
Eculizumab is the humanized mAb used primarily for the treatment of paroxysmal nocturnal hemoglobinuria and atypical haemolytic uremic syndrome. It can also be used in the treatment of generalized myasthenia gravis and neuromyelitis optica spectrum disorder with antibody positivity. This drug acts by binding to the C5 component of the complement system, inhibiting its activation, and consequently contributes to reducing erythrocyte destruction and hemolysis.
FDA labels reported that IRs, HRs, and anaphylaxis may develop during treatment, but IRs requiring discontinuation of the drug have not been reported [153]. In the EMA 2019 assessment report, HSRs, IRs, urticaria, and anaphylaxis are stated as uncommon (≥1/1000–<1/100) [154].
Lanadelumab
Lanadelumab is a human mAb (class IgG1 kappa) that specifically inhibits the activity of plasma kallikrein, used to reduce attacks and representing a prophylaxis measure in patients with hereditary angioedema [155].
In a study published in 2018, 52.4% of patients reported ISRs (34.1% in the placebo group) with symptoms such as bruising, dizziness, erythema, and local pain [156]. Only one patient (1.2%) developed mild-to-moderate HSRs, which included transient symptoms of oral tingling and pruritus, and resolved spontaneously within 1 day after onset without need for medication.
The EMA 2018 evaluation report [157] provides more detailed data on the study conducted by Banerji et al. [156]. In this report, ISRs are given as follows; pain (41.7%), erythema (9.5%), bruising (6.0%), discomfort (3.6%), hemorrhage (3.6%), pruritus (3.6%), swelling (3.6%), hematoma (2.4%), induration (2.4%), paresthesia (2.4%), warmth (2.4%), edema (1.2%), and rash (1.2%). In addition, no events of anaphylactoid reaction or anaphylaxis were reported (referenced in [157]).
Natalizumab
Natalizumab is an intravenously administered, immunosuppressive recombinant humanized IgG4 antibody that is produced in mouse cells. It acts by binding integrin a4 and is used to treat relapsing-remitting multiple sclerosis.
Maggi and colleagues (referenced in [65]) reported IRs in 1–4% of the patients in their review.
According to the EMA 2016 evaluation report [158], 23.1% of patients experienced infusion-related reactions, HSRs occurred in up to 4% of patients, and anaphylactic/anaphylactoid reactions in less than 1%. HSRs usually occurred during infusion or within 1 h after completion. Hypersensitivity symptoms included rash and urticaria, hypotension, hypertension, chest pain, chest complaints, dyspnea, and angioedema.
The FDA’s 2019 label reported infusion-related reactions in 11–24% of patients (placebo 7–18.0%), acute urticaria in 1–2%, acute HSRs in 1.5%, pruritus in 4%, and serious IRs and anaphylaxis in <1% [159]. Symptoms frequently associated with these reactions were fever, chills, rash, urticaria, pruritus, dizziness, nausea, flush, hypotension, dyspnea, and chest pain. The reactions occurred mostly within 2 h after infusion.
Palivizumab
Palivizumab is an intramuscularly administered humanized mAb (95% human and 5% murine) that is used in pediatric patients to prevent respiratory syncytial virus (RSV) infections. The antibody binds to coat proteins on the viral surface, thereby inhibiting its entry into host cells. It is used in patients with bronchopulmonary dysplasia, cystic fibrosis, and neonatal heart disease that are considered to be at high risk of severe RSV-associated disease.
Serious acute HSRs were rare among the 400,000 patients treated to date (>2 million doses) [160]. Anaphylactic reactions to repeated dosing or during the follow-up period (<1 case per 100,000 patients) have also been very rare [160]. HSRs include dyspnea, respiratory insufficiency, cyanosis, urticaria, pruritus, angioedema, hypotonia, and unresponsiveness. No fatal reactions have been observed in any patient.
In a study conducted by Chen et al. [161] on 13,025 infants (that received 57,392 injections), HSR was observed at a rate of 0.05%. In another study, injections were generally well tolerated, and drug-associated side effects were observed at a rate of 3.4% [162].
Discussion
BSs are increasingly used today and play an important role in the treatment of allergic, pulmonary, dermatological, oncological, rheumatological, as well as various other diseases. These drugs can be used as first-line or second-line treatment, as well as for prevention purposes. Observed HSRs may be mild, moderate, but may also be life-threatening by severely affecting vital parameters. All organ systems can be affected during reactions, so clinical symptoms are quite diverse. Reactions may occur due to allergic, non-allergic, or immunological side effects. The risk of allergic reactions decreases proportionally with the increase in human homology of the BS (Fig. 1; [163]). Vultaggio and co-authors explain the reactions to BSs with two possible main mechanisms: (i) loss of immune tolerance due to side effect of the BSs; and (ii) triggering of immune responses and reactions to non-self epitopes in BSs [164].

Risk assessment of biotechnological substances
This comprehensive review reveals a hypersensitivity prevalence against various BSs and an inconsistency and lack of precision regarding the nomenclature used for immune reactions to BSs (hypersensitivity, anaphylaxis, anaphylactoid, and infusion reactions) in the literature. Theoretically defined hypersensitivity and the hypersensitivity that is observed and classified in clinical practice seem to be different. Similarly, many data reported as anaphylaxis actually describe severe anaphylactic reactions (grades III or IV). It became evident that these definitions describe many common symptoms. Due to the overlap in signs and symptoms in the reported descriptions, it is not always possible to differentiate these reactions properly according to their pathomechanism. The common points of all these reactions are indicated in Table 7 [165,166,167].
In addition, reactions such as pruritus, flush, and urticaria are also included in the definitions of IR, HSR, anaphylaxis, and CRS. A summary view of the reactions according to severity is shown in Fig. 2. Fig. 3 shows a synopsis of the incidences of HSRs (Fig. 3a), acute IRs (Fig. 3b), and anaphylaxis to BSs (Fig. 3c) as extracted by the authors from the comprehensive review of the databases. There is an urgent need to define and classify these reactions more precisely in the future. Furthermore, the development of a scoring system, including symptom- or system-based approaches, can make it easier for us to identify the reactions of our patients and treat them adequately. This scoring can be evaluated according to which organ system (cardiac, gastrointestinal, hematologic, muscular, neurological, renal, respiratory, skin, vascular) is affected and the severity of symptoms.

Synopsis of all reactions to biotechnological substances

a A descriptive graph on the incidence of hypersensitivity reactions to biotechnological substances according to the reviewed databases (for references see the description of the respective BS in the text). b A descriptive graph on the incidence of acute infusion reactions to biotechnological substances according to the reviewed databases (for references see the description of the respective BS in the text). c A descriptive graph on the incidence of anaphylaxis to biotechnological substances according to the reviewed databases (for references see the description of the respective BS in the text)
One of the important points that may have been overlooked is the understanding of whether some symptoms (such as nasopharyngitis, conjunctivitis, rhinitis, thrombocytopenia, anemia, or gastrointestinal symptoms, etc.) have an immunological origin. The role of ADAs in these symptoms is not yet known. In addition, the risk of patients at each injection/infusion should be defined according to real life. In general, the literature gives percentages by calculation and only follow-up reactions in the treatment process. The long-term evaluations seem to be rather small in number.
CRS, which is a form of severe IR, can also be observed during alemtuzumab, anti-CD28 monoclonal antibody TGN1412, anti-thymocyte globulin, blinatumomab, brentuximab, dacetuzumab, muromonab-CD3, nivolumab, rituximab, and obinutuzumab treatments (referenced in [167]). A large majority of patients may have sepsis/septic shock fragments, as well as symptoms that may also be confused with tumor lysis syndrome. IL‑6 plays an important role in the development of CRS; therefore siltuximab or tocilizumab, which is a chimeric mAb against IL‑6 and receptors, is used for the treatment of this condition [167]. In cases where CRS developed during treatment, the relationship between the drug itself and ADA is so far unknown.
Conclusion
BSs are important immunotherapeutic tools used in the treatment of many diseases today. Various side effects and reactions can be observed during treatment, but there are deficiencies in their identification and classification. During this comprehensive research, it became evident that a more precise nomenclature should be applied. In order to achieve a harmonization in this regard, a simpler symptom- or system-based classification and scoring system is needed. A better understanding of the pathophysiology of hypersensitivity reactions and increased clinical experience in the treatment of side effects will provide timely control of unexpected reactions. As a result, immunotherapy with BSs will become safer in the future.
Abbreviations
- α‑Gal:
-
Galactose-α‑1,3‑galactose
- ADA:
-
Anti-drug antibodies
- ADR:
-
Adverse drug reaction
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- ARCN:
-
Airway Research Center North
- BCCA:
-
British Columbia Cancer Agency
- BLyS:
-
B lymphocyte stimulant
- BMBF:
-
Federal Ministry for Science and Education
- BS:
-
Biotechnological substance
- CAPS:
-
Cryopyrin-associated periodic syndromes
- COPD:
-
Chronic obstructive pulmonary disease
- CRS:
-
Cytokine release syndrome
- CTLA‑4:
-
Cytotoxic T lymphocyte-associated antigen 4
- EAACI:
-
European Academy of Allergy and Clinical Immunology
- EGFR:
-
Epidermal growth factor receptors
- EMA:
-
European Medicines Agency
- Fab:
-
Antigen-binding fragment
- FDA:
-
Food and Drug Administration
- FMF:
-
Familial Mediterranean fever
- HAMA:
-
Human anti-murine antibodies
- HER2:
-
Human epidermal growth factor receptor 2 protein
- HSR:
-
Hypersensitivity reaction
- IgE:
-
Immunoglobulin E
- IL:
-
Interleukin
- IPF:
-
Idiopathic pulmonary fibrosis
- IR:
-
Infusion reaction
- ISR:
-
Injection-site reaction
- LFA‑1:
-
Lymphocyte function-associated antigen‑1
- mAb:
-
Monoclonal antibody
- PD-L1:
-
Programmed death ligand 1
- RA:
-
Rheumatoid arthritis
- SC:
-
Subcutaneous
- SmPC:
-
Summary of product characteristic
- TEAE:
-
Treatment-emergent adverse event
- TGF:
-
Transforming growth factor
- TNF:
-
Tumor necrosis factor
- TRAPS:
-
Tumor necrosis factor receptor-associated periodic syndrome
- VEGF:
-
Vascular endothelial growth factor
References
Scherer K, Spoerl D, Bircher AJ. Adverse drug reactions to biologics. J Dtsch Dermatol Ges. 2010;8(6):411–26.
Doessegger L, Banholzer ML. Clinical development methodology for infusion-related reactions with monoclonal antibodies. Clin Transl Immunology. 2015;4(7):e39.
Sociedad Española de Reumatología. Informe Anual Biobadaser 2018. Registro Español de acontecimientos adversos de terapia biológica en enfermedades reumáticas. 2018. https://biobadaser.ser.es/docs/2BIOBADASER%20Informe%20Web%202018.pdf. Accessed 5 Aug 2019.
Castro M, Wenzel SE, Bleecker ER, Pizzichini E, Kuna P, Busse WW. Benralizumab, an anti-interleukin 5 receptor α monoclonal antibody, versus placebo for uncontrolled eosinophilic asthma: a phase 2b randomised dose-ranging study. Lancet Respir Med. 2014;2:879–90.
US Food and Drug Administration. FDA labels for benralizumab. 2017. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761070Orig1s000SumR.pdf. Accessed 1 May 2018.
Park HS, Lee SH, Lee SY, Kim MK, Lee BJ, Werkström V, et al. Efficacy and safety of benralizumab for Korean patients with severe, uncontrolled eosinophilic asthma. Allergy Asthma Immunol Res. 2019;11:508–18.
Pelaia C, Busceti MT, Vatrella A, Rago GF, Crimi C, Terracciano R, et al. Real-life rapidity of benralizumab effects in patients with severe allergic eosinophilic asthma: assessment of blood eosinophils, symptom control, lung function and oral corticosteroid intake after the first drug dose. Pulm Pharmacol Ther. 2019;58:101830. https://doi.org/10.1016/j.pupt.2019.101830.
Liu W, Ma X, Zhou W. Adverse events of benralizumab in moderate to severe eosinophilic asthma: a meta-analysis. Medicine (Baltimore). 2019;98:e15868. https://doi.org/10.1097/MD.0000000000015868.
Hanania NA, Noonan M, Corren J, Korenblat P, Zheng Y, Fischer SK, et al. Lebrikizumab in moderate-to-severe asthma: pooled data from two randomised placebo-controlled studies. Thorax. 2015;70:748–56.
Hanania NA, Korenblat P, Chapman KR, Bateman ED, Kopecky P, Paggiaro P, et al. Efficacy and safety of lebrikizumab in patients with uncontrolled asthma (LAVOLTA I and LAVOLTA II): replicate, phase 3, randomised, double-blind, placebo-controlled trials. Lancet Respir Med. 2016;4:781–96.
Korenblat P, Kerwin E, Leshchenko I, Yen K, Holweg CTJ, Anzures-Cabrera J, et al. Efficacy and safety of lebrikizumab in adult patients with mild-to-moderate asthma not receiving inhaled corticosteroids. Respir Med. 2018;134:143–9.
Simpson EL, Flohr C, Eichenfield LF, Bieber T, Sofen H, Taïeb A, et al. Efficacy and safety of lebrikizumab (an anti-IL-13 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical corticosteroids: a randomized, placebo-controlled phase II trial (TREBLE). J Am Acad Dermatol. 2018;78:863–71.
Pavord ID, Korn S, Howarth P, Bleecker ER, Buhl R, Keene ON, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet. 2012;380:651–9.
US Food and Drug Administration. FDA labels for mepolizumab (Nucala®). 2015. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/125526Orig1s000SumR.pdf. Accessed 9 Aug 2019.
Lugogo N, Domingo C, Chanez P, Leigh R, Gilson MJ, Price RG, et al. Long-term efficacy and safety of mepolizumab in patients with severe eosinophilic asthma: a multi-center, open-label, phase IIIb study. Clin Ther. 2016;38(9):2058–2070.e1. https://doi.org/10.1016/j.clinthera.2016.07.010.
Khatri S, Moore W, Gibson PG, Leigh R, Bourdin A, Maspero J, et al. Assessment of the long-term safety of mepolizumab and durability of clinical response in patients with severe eosinophilic asthma. J Allergy Clin Immunol. 2019;143:1742–51.
Castro M, Zangrilli J, Wechsler ME, Bateman ED, Brusselle GG, Bardin P, et al. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir Med. 2015;3:355–66.
US Food and Drug Administration. FDA labels for reslizumab (Cinqair®). 2016. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761033Orig1s000SumR.pdf. Accessed 9 Aug 2019.
Murphy K, Jacobs J, Bjermer L, Fahrenholz JM, Shalit Y, Garin M, et al. Long-term safety and efficacy of reslizumab in patients with eosinophilic asthma. J Allergy Clin Immunol Pract. 2017;5:1572–81.
Pérez de Llano LA, Cosío BG, Domingo C, Urrutia I, Bobolea I, Valero A, et al. Efficacy and safety of reslizumab in patients with severe asthma with inadequate response to omalizumab: a multicenter, open-label pilot study. J Allergy Clin Immunol Pract. 2019;7:2277–83.
Cox L, Platts-Mills TA, Finegold I, Schwartz LB, Simons FE, Wallace DV. American Academy of Allergy, Asthma & Immunology/American College of Allergy, Asthma and Immunology Joint Task Force report on omalizumab-associated anaphylaxis. J Allergy Clin Immunol. 2007;120:1373–7.
Di Bona D, Fiorino I, Taurino M, Frisenda F, Minenna E, Pasculli C, et al. Long-term “real-life” safety of omalizumab in patients with severe uncontrolled asthma: a nine-year study. Respir Med. 2017;130:55–60.
Cox L, Lieberman P, Wallace D, Simons FE, Finegold I, Platts-Mills T, et al. American Academy of Allergy, Asthma & Immunology/American College of Allergy, Asthma & Immunology Omalizumab-Associated Anaphylaxis Joint Task Force follow-up report. J Allergy Clin Immunol. 2011;128:210–2.
US Food and Drug Administration. FDA labels for omalizumab (Xolair®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/103976s5234lbl.pdf. Accessed 12 Aug 2019.
Therapeutic Goods Administration. Australian public assessment report for nintedanib esilate. Proprietary product name: Ofev and Vargatef. 2016. https://www.tga.gov.au/sites/default/files/auspar-nintedanib-esilate-160208.pdf. Accessed 13 Aug 2019.
US Food and Drug Administration. FDA labels for nintedanib (OFEV®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/205832s012lbl.pdf. Accessed 13 Aug 2019.
Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–9.
US Food and Drug Administration. FDA labels for pirfenidone (Esbriet®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/022535s012,208780s002lbl.pdf. Accessed 13 Aug 2019.
Sehgal VN, Pandhi D, Khurana A. Biologics in dermatology: an integrated review. Indian J Dermatol. 2014;59:425–41.
Graves JE, Nunley K, Heffernan MP. Off-label uses of biologics in dermatology: rituximab, omalizumab, infliximab, etanercept, adalimumab, efalizumab, and alefacept (part 2 of 2). J Am Acad Dermatol. 2007;56:55–79.
US Food and Drug Administration. FDA labels for alefacept (AMEVIVE®). 2012. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/125036s0144lbl.pdf. Accessed 13 Aug 2019.
Gordon KB, Papp KA, Hamilton TK, Walicke PA, Dummer W, Li N, et al. Efalizumab Study Group. Efalizumab for patients with moderate to severe plaque psoriasis: a randomized controlled trial. JAMA. 2003;290:3073–80.
US Food and Drug Administration. FDA labels for efalizumab (RAPTIVA®). 2009. https://www.fda.gov/downloads/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/UCM143346.pdf. Accessed 13 Aug 2019.
Brunasso AM, Puntoni M, Salvini C, Delfino C, Curcic P, Gulia A, et al. Tolerability and safety of biological therapies for psoriasis in daily clinical practice: a study of 103 Italian patients. Acta Derm Venereol. 2011;91:44–9.
US Food and Drug Administration. FDA labels for ixekizumab (TALTZ®). 2017. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125521s004lbl.pdf. Accessed 14 Aug 2019.
Strober B, Leonardi C, Papp KA, Mrowietz U, Ohtsuki M, Bissonnette R, et al. Short- and long-term safety outcomes with ixekizumab from 7 clinical trials in psoriasis: etanercept comparisons and integrated data. J Am Acad Dermatol. 2017;76:432–40.
European Medicines Agency. Assessment report of secukinumab (COSENTYX®). 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/003729/WC500199574.pdf. Accessed 14 Aug 2019.
Schwensen JF, Clemmensen A, Sand C, Gniadecki R, Skov L, Zachariae C, et al. Effectiveness and safety of secukinumab in 69 patients with moderate to severe plaque psoriasis: a retrospective multicenter study. Dermatol Ther. 2017;30:e12550.
US Food and Drug Administration. FDA labels for secukinumab (COSENTYX®). 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125504s013lbl.pdf. Accessed 15 Aug 2019.
Deodhar A, Mease PJ, McInnes IB, Baraliakos X, Reich K, Blauvelt A, et al. Long-term safety of secukinumab in patients with moderate-to-severe plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis: integrated pooled clinical trial and post-marketing surveillance data. Arthritis Res Ther. 2019;21:111.
European Medicines Agency. Assessment report of ustekinumab (STELARA®). 2017. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/000958/WC500217159.pdf. Accessed 15 Aug 2019.
US Food and Drug Administration. FDA labels for ustekinumab (STELARA®). 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/761044lbl.pdf. Accessed 15 Aug 2019.
Ghosh S, Gensler LS, Yang Z, Gasink C, Chakravarty SD, Farahi K, et al. Correction to: Ustekinumab safety in psoriasis, psoriatic arthritis, and Crohn’s disease: an integrated analysis of phase II/III clinical development programs. Drug Saf. 2019;42:809.
US Food and Drug Administration. FDA labels for dupilumab (DUPIXENT®). 2017. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761055lbl.pdf. Accessed 16 Aug 2019.
Ou Z, Chen C, Chen A, Yang Y, Zhou W. Adverse events of dupilumab in adults with moderate-to-severe atopic dermatitis: a meta-analysis. Int Immunopharmacol. 2018;54:303–10.
European Medicines Agency. Assessment report of dupilumab (DUPIXENT®). 2019. https://www.ema.europa.eu/en/documents/variation-report/dupixent-h-c-4390-x-0004-g-epar-assessment-report-extension_en.pdf. Accessed 04 May 2020.
Maurer M, Giménez-Arnau AM, Sussman G, Metz M, Baker DR, Bauer A, et al. Ligelizumab for chronic spontaneous urticaria. N Engl J Med. 2019;381(14):1321–32.
Harris JM, Maciuca R, Bradley MS, Cabanski CR, Scheerens H, Lim J, et al. A randomized trial of the efficacy and safety of quilizumab in adults with inadequately controlled allergic asthma. Respir Res. 2016;17:29.
European Medicines Agency. Assessment report of aflibercept (ZALTRAP®). 2019. https://www.ema.europa.eu/en/documents/product-information/zaltrap-epar-product-information_en.pdf. Accessed 17 Feb 2020.
US Food and Drug Administration. FDA labels for aflibercept (ZALTRAP®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125418s045lbl.pdf. Accessed 17 Feb 2020.
Nagai N, Ibuki M, Shinoda H, Kameyama K, Tsubota K, Ozawa Y. Maculopapular rash after intravitreal injection of an antivascular endothelial growth factor, aflibercept, for treating age-related macular degeneration: a case report. Medicine (Baltimore). 2017;96(21):e6965.
European Medicines Agency. Assessment report of alemtuzumab (LEMTRADA®). 2013. https://www.ema.europa.eu/documents/assessment-report/lemtrada-epar-public-assessment-report_en.pdf. Accessed 17 Aug 2019.
BC Cancer Agency. Cancer drug manual, drug name: alemtuzumab. 2015. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Alemtuzumab_Monograph_1May2015.pdf. Accessed 17 Aug 2019.
US Food and Drug Administration. FDA labels for alemtuzumab (LEMTRADA®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/103948s5172lbl.pdf. Accessed 17 Aug 2019.
Frau J, Coghe G, Lorefice L, Fenu G, Musu L, Cocco E. Efficacy and safety of alemtuzumab in a real-life cohort of patients with multiple sclerosis. J Neurol. 2019;266:1405–11.
US Food and Drug Administration. FDA labels for atezolizumab (TECENTRIQ®). 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/761041s000lbl.pdf. Accessed 17 Feb 2020.
European Medicines Agency. Assessment report of atezolizumab (TECENTRIQ®). 2019. https://www.ema.europa.eu/en/documents/product-information/tecentriq-epar-product-information_en.pdf. Accessed 17 Feb 2020.
BC Cancer Agency. Cancer drug manual, drug name: atezolizumab. 2019. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Atezolizumab_Monograph.pdf. Accessed 17 Feb 2020.
BC Cancer Agency. Cancer drug manual, drug name: bevacizumab. 2016. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Bevacizumab_monograph.pdf. Accessed 04 May 2020.
European Medicines Agency. Assessment report bevacizumab (ZIRABEV®). 2018. https://www.ema.europa.eu/en/documents/assessment-report/zirabev-epar-public-assessment-report_en.pdf. Accessed 17 Aug 2019.
US Food and Drug Administration. FDA labels for bevacizumab (AVASTIN®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125085s331lbl.pdf. Accessed 17 Aug 2019.
BC Cancer Agency. Cancer drug manual, drug name: blinatumomab. 2017. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Blinatumomab_monograph.pdf. Accessed 17 Aug 2019.
European Medicines Agency. Assessment report of blinatumomab (BLINCYTO®). 2018. https://www.ema.europa.eu/documents/variation-report/blincyto-h-c-3731-ii-0018-epar-assessment-report-variation_en.pdf. Accessed 17 Aug 2019.
US Food and Drug Administration. FDA labels for blinatumomab (BLINCYTO®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125557s015s016lbl.pdf. Accessed 17 Aug 2019.
Maggi E, Vultaggio A, Matucci A. Acute infusion reactions induced by monoclonal antibody therapy. Expert Rev Clin Immunol. 2011;7:55–63.
Pichler WJ. Adverse side-effects to biological agents. Allergy. 2006;61:912–20.
Needle MN. Safety experience with IMC-C225, an anti-epidermal growth factor receptor antibody. Semin Oncol. 2002;29:55–60.
Galvão VR, Castells MC. Hypersensitivity to biological agents-updated diagnosis, management, and treatment. J Allergy Clin Immunol Pract. 2015;3:175–85.
US Food and Drug Administration. FDA labels for cetuximab (ERBITUX®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125084s273lbl.pdf. Accessed 18 Aug 2019.
Commins SP, Platts-Mills TA. Allergenicity of carbohydrates and their role in anaphylactic events. Curr Allergy Asthma Rep. 2010;10:29–33.
Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha‑1,3‑galactose. N Engl J Med. 2008;358:1109–17.
Commins SP, James HR, Kelly LA, Pochan SL, Workman LJ, Perzanowski MS, et al. The relevance of tick bites to the production of IgE antibodies to the mammalian oligosaccharide galactose-α-1,3-galactose. J Allergy Clin Immunol. 2011;127(5):1286–93.
Wedi B. Alpha-Gal in therapeutics: more relevant than thought? Hautarzt. 2017;68:246–7.
Mariotte D, Dupont B, Gervais R, Galais MP, Laroche D, Tranchant A, et al. Anti-cetuximab IgE ELISA for identification of patients at a high risk of cetuximab-induced anaphylaxis. MAbs. 2011;3:396–401.
US Food and Drug Administration. FDA labels for durvalumab (IMFINZI®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761069s013lbl.pdf. Accessed 04 May 2020.
European Medicines Agency. Assessment report of durvalumab (IMFINZI®). 2020. https://www.ema.europa.eu/en/documents/product-information/imfizi-epar-product-information_en.pdf. Accessed 18 Feb 2020.
BC Cancer Agency. Cancer drug manual, drug name: durvalumab. 2020. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Durvalumab_monograph.pdf. Accessed 18 Feb 2020.
US Food and Drug Administration. FDA labels for gemtuzumab (MYLOTARG®). 2006. https://www.accessdata.fda.gov/drugsatfda_docs/label/2006/021174s020lbl.pdf. Accessed 19 Aug 2019.
European Medicines Agency. Assessment report of gemtuzumab (MYLOTARG®). 2018. https://www.ema.europa.eu/en/documents/assessment-report/mylotarg-epar-public-assessment-report_en.pdf. Accessed 19 Aug 2019.
Hanbali A, Wollner I, Neme K, Ulreich C. Fatal hypersensitivity reaction to gemtuzumab ozogamicin associated with platelet transfusion. Am J Health Syst Pharm. 2007;64:1401–2.
US Food and Drug Administration. FDA labels for ibritumomab (ZEVALIN®). 2002. https://www.accessdata.fda.gov/drugsatfda_docs/label/2002/ibriide021902lb.pdf. Accessed 19 Aug 2019.
European Medicines Agency. Assessment report of ibritumomab (ZEVALIN®). 2017. https://www.ema.europa.eu/en/documents/product-information/zevalin-epar-product-information_en.pdf. Accessed 19 Aug 2019.
BC Cancer Agency. Cancer drug manual, drug name: ibritumomab. 2016. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Ibritumomab_interim%20monograph.pdf. Accessed 19 Aug 2019.
Mukherjee S, Ayanambakkam A, Ibrahimi S, Schmidt S, Charkrabarty JH, Khawandanah M. Ibritumomab tiuxetan (Zevalin) and elevated serum human anti-murine antibody (HAMA). Hematol Oncol Stem Cell Ther. 2018;11:187–8.
US Food and Drug Administration. FDA labels for imatinib (GLEEVEC®). 2001. https://www.accessdata.fda.gov/drugsatfda_docs/label/2001/21335lbl.pdf. Accessed 19 Aug 2019.
BC Cancer Agency. Cancer drug manual, drug name: imatinib. 2017. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Imatinib_Monograph_1Mar2017.pdf. Accessed 19 Aug 2019.
US Food and Drug Administration. FDA labels for ipilimumab (YERVOY®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125377s104lbl.pdf. Accessed 18 Feb 2020.
European Medicines Agency. Assessment report of ipilimumab (YERVOY®). 2020. https://www.ema.europa.eu/en/documents/product-information/yervoy-epar-product-information_en.pdf. Accessed 18 Feb 2020.
BC Cancer Agency. Cancer drug manual, drug name: ipilimumab. 2020. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Ipilimumab_monograph.pdf. Accessed 18 Feb 2020.
Ludlow SP, Kay N. Delayed dermatologic hypersensitivity reaction secondary to ipilimumab. J Immunother. 2015;38(4):165–6.
US Food and Drug Administration. FDA labels for necitumumab (PORTRAZZA®). 2015. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/125547s000lbl.pdf. Accessed 18 Feb 2020.
European Medicines Agency. Assessment report of necitumumab (PORTRAZZA®). 2016. https://www.ema.europa.eu/en/documents/product-information/portrazza-epar-product-information_en.pdf. Accessed 18 Feb 2020.
US Food and Drug Administration. FDA labels for nivolumab (OPDIVO®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125554s075lbl.pdf. Accessed 18 Feb 2020.
European Medicines Agency. Assessment report of nivolumab (OPDIVO®). 2020. https://www.ema.europa.eu/en/documents/product-information/opdivo-epar-product-information_en.pdf. Accessed 18 Feb 2020.
BC Cancer Agency. Cancer drug manual, drug name: nivolumab. 2020. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Nivolumab_monograph.pdf. Accessed 18 Feb 2020.
Sáenz de Santa María García M, Noguerado-Mellado B, Rojas-Pérez-Ezquerra P, Prieto-García A, Bartolomé-Zavala B, Tornero P. First case of allergy to nivolumab. J Allergy Clin Immunol Pract. 2017;5(4):1140–1.
Oda H, Ishihara M, Miyahara Y, Nakamura J, Kozuka Y, Iwasa M et al. First case of cytokine release syndrome after nivolumab for gastric cancer. Case Rep Oncol. 2019;12(1):147–56.
Choi B, McBride A, Scott AJ. Treatment with pembrolizumab after hypersensitivity reaction to nivolumab in a patient with hepatocellular carcinoma. Am J Health Syst Pharm. 2019;76(21):1749–52.
Caponetto P, Biedermann T, Yazdi AS, Fischer J. Panitumumab: a safe option for oncologic patients sensitized to galactose-α‑1,3‑galactose. J Allergy Clin Immunol Pract. 2015;3:982–3.
US Food and Drug Administration. FDA labels for panitumumab (VECTIBIX®). 2015. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/125147s200lbl.pdf. Accessed 19 Aug 2019.
BC Cancer Agency. Cancer drug manual, drug name: panitumumab. 2020. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Panitumumab%20monograph.pdf. Accessed 04 May 2020.
Mersin SS, Bulut I, Yakut T, Örçen C, Tepetam FM. Successful desensitization with panitumumab; a case report. Allergy. 2019;74:741–2.
European Medicines Agency. Assessment report of pembrolizumab (KEYTRUDA®). 2019. https://www.ema.europa.eu/en/documents/product-information/keytruda-epar-product-information_en.pdf. Accessed 18 Feb 2020.
BC Cancer Agency. Cancer drug manual, drug name: pembrolizumab. 2019. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Pembrolizumab_monograph.pdf. Accessed 18 Feb 2020.
US Food and Drug Administration. FDA labels for pembrolizumab (KEYTRUDA®). 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125514s067lbl.pdf. Accessed 18 Feb 2020.
van Vugt MJH, Stone JA, De Greef RHJMM, Snyder ES, Lipka L, Turner DC, et al. Immunogenicity of pembrolizumab in patients with advanced tumors. J Immunother Cancer. 2019;7(1):212.
BC Cancer Agency. Cancer drug manual, drug name: pertuzumab. 2014. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Pertuzumab_monograph_1Jan2014.pdf. Accessed 19 Feb 2020.
European Medicines Agency. Assessment report of pertuzumab (PERJETA®). 2019. https://www.ema.europa.eu/en/documents/product-information/perjeta-epar-product-information_en.pdf. Accessed 19 Feb 2020.
US Food and Drug Administration. FDA labels for pertuzumab (PERJETA®). 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125409s124lbl.pdf. Accessed 19 Feb 2020.
BC Cancer Agency. Cancer drug manual, drug name: ramucirumab. 2017. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Ramucirumab_monograph_1May2017.pdf. Accessed 19 Feb 2020.
European Medicines Agency. Assessment report of ramucirumab (CYRAMZA®). 2019. https://www.ema.europa.eu/en/documents/product-information/cyramza-epar-product-information_en.pdf. Accessed 19 Feb 2020.
US Food and Drug Administration. FDA labels for ramucirumab (CYRAMZA®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125477s033lbl.pdf. Accessed 19 Feb 2020.
BC Cancer Agency. Cancer drug manual, drug name: trastuzumab. 2020. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Trastuzumab_monograph.pdf. Accessed 04 May 2020.
European Medicines Agency. Assessment report of trastuzumab (TRAZIMERA®). 2018. https://www.ema.europa.eu/en/documents/assessment-report/trazimera-epar-public-assessment-report_en.pdf. Accessed 06 May 2020.
US Food and Drug Administration. FDA labels for trastuzumab (KADCYLA®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125427s105lbl.pdf. Accessed 19 Aug 2019.
Puxeddu I, Giori L, Rocchi V, Bazzichi L, Bombardieri S, Tavoni A, et al. Hypersensitivity reactions during treatment with infliximab, etanercept, and adalimumab. Ann Allergy Asthma Immunol. 2012;108:123–4.
Tarkiainen M, Tynjälä P, Vähäsalo P, Lahdenne P. Occurrence of adverse events in patients with JIA receiving biologic agents: long-term follow-up in a real-life setting. Rheumatology (Oxford). 2015;54:1170–6.
US Food and Drug Administration. FDA labels for TNF inhibitors: adalimumab (HUMIRA®). 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125057s410lbl.pdf. Accessed 19 Aug 2019.
European Medicines Agency. Summary of product characteristic. Anakinra (KINERET®). 2014. https://ec.europa.eu/health/documents/community-register/2014/20140627128993/anx_128993_en.pdf. Accessed 28 Oct 2019.
US Food and Drug Administration. FDA labels for anakinra (KINERET®). 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/103950s5182lbl.pdf. Accessed 04 May 2020.
Desai D, Goldbach-Mansky R, Milner JD, Rabin RL, Hull K, Pucino F. Anaphylactic reaction to anakinra in a rheumatoid arthritis patient intolerant to multiple nonbiologic and biologic disease-modifying antirheumatic drugs. Ann Pharmacother. 2009;43(5):967–72.
Aguiar CL, Pan N, Adams A, Barinstein L, Lehman TJ. Anaphylaxis to anakinra in a pediatric patient with systemic juvenile idiopathic arthritis successfully treated with canakinumab: a case-based review. Clin Rheumatol. 2015;34(10):1821–4.
Şoyyiğit S, Kendirlinan R, Aydın O, Çelik GE. Successful desensitization with anakinra in a case with immediate hypersensitivity reaction. Ann Allergy Asthma Immunol. 2014;113(3):325–6.
European Medicines Agency. Assessment report of belimumab (BENLYSTA®). 2018. https://www.ema.europa.eu/en/documents/product-information/benlysta-epar-product-information_en.pdf. Accessed 21 Aug 2019.
US Food and Drug Administration. FDA labels for belimumab (BENLYSTA®). 2019. http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125370s064,761043s007lbl.pdf. Accessed 21 Aug 2019.
US Food and Drug Administration. FDA labels for canakinumab (ILARIS®). 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125319s088lbl.pdf. Accessed 29 Oct 2019.
Bettiol A, Lopalco G, Emmi G, Cantarini L, Urban ML, Vitale A et al. Unveiling the efficacy, safety, and tolerability of anti-interleukin‑1 treatment in monogenic and multifactorial autoinflammatory diseases. Int J Mol Sci. 2019;20(8):1898.
US Food and Drug Administration. FDA labels for etanercept (ENBREL®). 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/103795s5569lbl.pdf. Accessed 20 Aug 2019.
Kay J, Fleischmann R, Keystone E, Hsia EC, Hsu B, Mack M, et al. Golimumab 3‑year safety update: an analysis of pooled data from the long-term extensions of randomised, double-blind, placebo-controlled trials conducted in patients with rheumatoid arthritis, psoriatic arthritis or ankylosing spondylitis. Ann Rheum Dis. 2015;74:538–46.
US Food and Drug Administration. FDA labels for TNF inhibitors: golimumab (SIMPONI®). 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125289s139lbl.pdf. Accessed 20 Aug 2019.
US Food and Drug Administration. FDA labels for TNF inhibitors: infliximab (RENFLEXIS®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761054s004lbl.pdf. Accessed 20 Aug 2019.
Vultaggio A, Matucci A, Nencini F, Pratesi S, Parronchi P, Rossi O, et al. Anti-infliximab IgE and non-IgE antibodies and induction of infusion-related severe anaphylactic reactions. Allergy. 2010;65:657–61.
Vultaggio A, Matucci A, Nencini F, Pratesi S, Maggi E. Skin testing and infliximab-specific antibodies detection as a combined strategy for preventing infusion reaction. Intern Emerg Med. 2012;7:77–9.
European Medicines Agency. Assessment report of certolizumab (CIMZIA®). 2015. https://www.ema.europa.eu/en/documents/variation-report/cimzia-h-c-1037-ii-0045-epar-assessment-report-variation_en.pdf. Accessed 20 Aug 2019.
US Food and Drug Administration. FDA labels for certolizumab (CIMZIA®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125160s289lbl.pdf. Accessed 20 Aug 2019.
Bykerk VP, Cush J, Winthrop K, Calabrese L, Lortholary O, de Longueville M, et al. Update on the safety profile of certolizumab pegol in rheumatoid arthritis: an integrated analysis from clinical trials. Ann Rheum Dis. 2015;74:96–103.
US Food and Drug Administration. FDA labels for rituximab (RITUXAN HYCELA®). 2017. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761064s000lbl.pdf. Accessed 20 Aug 2019.
US Food and Drug Administration. FDA labels for rituximab (RITUXAN®). 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/103705s5453lbl.pdf. Accessed 20 Aug 2019.
BC Cancer Agency. Cancer drug manual, drug name: rituximab. 2018. http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Rituximab_monograph.pdf. Accessed 20 Aug 2019.
Schmidt E, Hennig K, Mengede C, Zillikens D, Kromminga A. Immunogenicity of rituximab in patients with severe pemphigus. Clin Immunol. 2009;132:334–41.
Vultaggio A, Matucci A, Nencini F, Pratesi S, Petroni G, Cammelli D, et al. Drug-specific Th2 cells and IgE antibodies in a patient with anaphylaxis to rituximab. Int Arch Allergy Immunol. 2012;159:321–6.
US Food and Drug Administration. FDA labels for basiliximab (SIMULECT®). 2001. https://www.accessdata.fda.gov/drugsatfda_docs/label/2001/basnova032301LB.pdf. Accessed 21 Aug 2019.
Erickson AL, Roberts K, Malek SK, Chandraker AK, Tullius SG, Gabardi S. Analysis of infusion-site reactions in renal transplant recipients receiving peripherally administered rabbit antithymocyte globulin as compared with basiliximab. Transpl Int. 2010;23:636–40.
Baudouin V, Crusiaux A, Haddad E, Schandene L, Goldman M, Loirat C, et al. Anaphylactic shock caused by immunoglobulin E sensitization after retreatment with the chimeric anti-interleukin‑2 receptor monoclonal antibody basiliximab. Transplantation. 2003;76:459–63.
BC Transplant. Clinical guidelines for transplant medications. Section: Basiliximab. 2018. http://www.transplant.bc.ca/Documents/Health%20Professionals/Clinical%20guidelines/Clinical%20Guidelines%20for%20Transplant%20Medications%20-%20June%202018.pdf. Accessed 20 Aug 2019, p 8–12.
US Food and Drug Administration. FDA labels for belatacept (NULOJIX®). 2017. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125288s074lbl.pdf. Accessed 21 Aug 2019.
European Medicines Agency. Assessment report of belatacept (NULOJIX®). 2019. https://www.ema.europa.eu/en/documents/product-information/nulojix-epar-product-information_en.pdf. Accessed 29 Nov 2019.
Becker H, Reichert JM. Muromonab-CD3 (orthoclone OKT3). In: Dübel S, Reichert JM, editors. Handbook of therapeutic antibodies. 2014. pp. 1663–4. https://doi.org/10.1002/9783527682423.ch60.
Boden EK, Canavan JB, Moran CJ, McCann K, Dunn WA, Farraye FA, et al. Immunologic alterations associated with oral delivery of anti-CD3 (OKT3) monoclonal antibodies in patients with moderate-to-severe ulcerative colitis. Crohns Colitis 360. 2019;1(2):otz9. https://doi.org/10.1093/crocol/otz009.
Dery JP, Braden GA, Lincoff AM, Kereiakes DJ, Browne K, Little T, et al. ReoPro readministration registry investigators. Final results of the ReoPro readministration registry. Am J Cardiol. 2004;93:979–84.
Iakovou Y, Manginas A, Melissari E, Cokkinos DV. Acute profound thrombocytopenia associated with anaphylactic reaction after abciximab therapy during percutaneous coronary angioplasty. Cardiology. 2001;95:215–6.
US Food and Drug Administration. FDA labels for abciximab (REOPRO®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/103575s5318lbl.pdf. Accessed 21 Aug 2019.
US Food and Drug Administration. FDA labels for eculizumab (SOLIRIS®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125166s431lbl.pdf. Accessed 20 Feb 2020.
European Medicines Agency. Assessment report of eculizumab (SOLIRIS®). 2019. https://www.ema.europa.eu/en/documents/product-information/soliris-epar-product-information_en.pdf. Accessed 20 Feb 2020.
Wedi B. Lanadelumab to treat hereditary angioedema. Drugs Today. 2019;55(7):439–48.
Banerji A, Riedl MA, Bernstein JA, Cicardi M, Longhurst HJ, Zuraw BL, et al. HELP investigators. Effect of lanadelumab compared with placebo on prevention of hereditary angioedema attacks: a randomized clinical trial. JAMA. 2018;320(20):2108–21.
European Medicines Agency. Summary of product characteristic. Lanadelumab (TAKHZYRO®). 2018. https://www.ema.europa.eu/en/documents/assessment-report/takhzyro-epar-public-assessment-report_en.pdf. Accessed 29 Oct 2019.
European Medicines Agency. Assessment report of natalizumab (TYSABRI®). 2016. http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/Tysabri_20/European_Commission_final_decision/WC500202392.pdf. Accessed 21 Aug 2019.
US Food and Drug Administration. FDA labels for natalizumab (TYSABRI®). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125104s966lbl.pdf. Accessed 21 Aug 2019.
US Food and Drug Administration. FDA labels for palivizumab (SYNAGIS®). 2009. https://www.accessdata.fda.gov/drugsatfda_docs/bla/2003/103770orig1s5033.pdf. Accessed 22 Aug 2019.
Chen JJ, Chan P, Paes B, Mitchell I, Li A, Lanctôt KL, et al. Serious adverse events in the Canadian registry of children receiving palivizumab (CARESS) for respiratory syncytial virus prevention. PLoS One. 2015;10(8):e134711.
Resch B. Product review on the monoclonal antibody palivizumab for prevention of respiratory syncytial virus infection. Hum Vaccin Immunother. 2017;13:2138–49.
Jappe U. Allergic reactions to oncology biologics. Immunology Section of EAACI Congress; Munich, Germany. 2018. BM Task Force for AllergoOncology.
Vultaggio A, Petroni G, Pratesi S, Nencini F, Cammelli D, Ferraro A, et al. How the immune system responds to therapeutic biological agents. J Int Med Res. 2016;44:38–42.
Lenz HJ. Management and preparedness for infusion and hypersensitivity reactions. Oncologist. 2007;12:601–9.
Ring J, Beyer K, Biedermann T, Bircher A, Duda D, Fischer J, et al. Guideline for acute therapy and management of anaphylaxis. S2 guideline of DGAKI, AeDA, GPA, DAAU, BVKJ, ÖGAI, SGAI, DGAI, DGP, DGPM, AGATE and DAAB. Allergo J Int. 2014; 23:96–112.
Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6:56.
Funding
Funding of the Federal Ministry for Science and Education (BMBF), German Center for Lung Research, is gratefully acknowledged.
Funding
Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A. Gülsen: Data collection, design of tables and figures, analysis and interpretation of data, writing and drafting of the manuscript; B. Wedi: major role in revising the manuscript; U. Jappe: concept of the manuscript, acquisition of data, design of Figure 1, writing and revising the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
A. Gülsen, B. Wedi, and U. Jappe declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gülsen, A., Wedi, B. & Jappe, U. Hypersensitivity reactions to biologics (part I): allergy as an important differential diagnosis in complex immune-derived adverse events. Allergo J Int 29, 97–125 (2020). https://doi.org/10.1007/s40629-020-00126-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40629-020-00126-6