
Abstract
Schizophrenia (SCZ) is a severe psychiatric disorder, which lacks a unifying neuropathology. However, reproducible molecular alterations exist, including RNA expression changes affecting GABAergic and other neuronal signaling in cerebral cortex, myelination, and other cellular functions. Yet, for the large majority of RNAs altered in the SCZ brain, the underlying transcriptional and post-transcriptional disease-associated mechanisms remain unclear. Here, we provide an update on epigenetic regulators of gene expression that are potentially affected in some cases with SCZ, including DNA cytosine methylation, histone modifications and histone variants, and chromosomal loop formations facilitating long-range interactions of gene promoters with distal enhancer elements. Exploration of chromatin structure and function, in combination with transcriptome and genome sequencing, is likely to critically advance insight into the molecular mechanisms of disease in specific cases with SCZ.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Schizophrenia (SCZ) is a severe psychiatric disorder defined by symptoms such as delusions, hallucinations, and thought disorder, reducing the lifespan of an affected individual on average by 15 years, with cardiovascular risk as the chief cause of increased mortality [1]. Since the second half of the last century, antipsychotic drugs have been the mainstay of treatment but, even now, the majority of schizophrenia patients still suffer from a severe illness that substantially impacts quality of life and increases the risk for suicide, while experiencing incomplete response to treatment [2, 3]. Therefore, there is a pressing need to develop new treatment options in schizophrenia that ideally should go beyond the molecular targets of the currently available antipsychotics, which are primarily aimed at monoaminergic receptor systems [4, 5]. However, rational drug development in schizophrenia is greatly impeded by the lack of a unifying neuropathology and a complex genetic risk architecture, which does not appear to converge onto narrowly defined signaling pathways or molecular mechanisms.
It is now generally accepted that many individuals on the schizophrenia spectrum are affected by dysregulated gene expression influencing widespread areas of the cerebral cortex and other brain regions, thereby pointing to molecular and cellular alterations in the oligodendrocyte lineage [6–11], and compromised inhibitory and excitatory neurotransmission due to altered expression of ligand-gated ion receptors, reuptake transporters, metabolic enzymes, etc. [12–24]. It is largely unclear whether these transcriptional changes are directly related to the underlying etiology or are secondary events in the pathophysiology of disease.
These molecular findings in the postmortem brains of subjects with SCZ often serve as a starting point and, when followed up by experimental work in preclinical and translational laboratories, bear the potential to critically advance insight into the neurobiology of psychosis. For example, a more recent key advance in the pathophysiology of SCZ concerns molecular defects in cortical inhibitory GABAergic circuitry, which ultimately are thought to contribute to cognitive defects by altering synchronization of electrical activity across widespread cortical areas [25–28]. Remarkably, the empirical framework for this hypothesis was originally rooted in a large body of postmortem literature reporting downregulated gene expression for the rate-limiting GABA synthesis enzyme, 67Kda glutamic acid decarboxylase (GAD67), in multiple subtypes of cortical interneurons, including those commonly defined as fast-spiking parvalbumin+, and low-threshold spiking somatostatin + cells [15, 18, 29–33]. Indeed, it is now thought that 30–40 % of individuals with SCZ are affected by robust deficits in GABAergic gene expression in the prefrontal cortex (PFC) [34].
While the aforementioned RNA expression changes in the SCZ postmortem brain have had a considerable impact by reshaping current models of the pathophysiology of the disease, mechanistically the disease-associated molecular defects remain incompletely understood. Transcriptional mechanisms are tightly linked to epi- (Greek for over, above) genetic regulation of chromatin structure and function, including several types of DNA cytosine methylation and an estimated number of more than 100 site- and residue-specific post-translational histone modifications. Importantly, nucleosomes as the elementary unit of chromatin (comprising 146 base pairs of genomic DNA wrapped around an octamer of the four core histones, H2A, H2B, H3, and H4), various histone and DNA methylation markings, and other molecular architectures inside the nucleus (Fig. 1) remain stable for a prolonged period after death. Thus, nucleosomal organization, including epigenetic decorations, is maintained in postmortem brain tissue, which typically is exposed to 5–30 (or more) hours of autolysis time before being safely stored in a freezer [35, 36]. Unsurprisingly, there is a rapidly increasing number of studies that have charted—either locus-specific or even genome-wide—disease-associated changes in DNA cytosine methylation and hydroxymethylation, residue-specific histone methylation and acetylation, and various other types of epigenetic markings in human brain tissue or in some of its surrogates such as the olfactory epithelium. Here, we provide a concise update on epigenetic studies in the SCZ brain and discuss the resulting implications for the neurobiology and treatment of the disease.

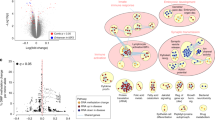
Epigenetic mechanisms potentially relevant for transcriptional dysregulation in schizophrenia. Schematic illustration of some of the general principles of epigenetic regulation. Gene (green) poised for transcription by polymerase II (Pol II) initiation complex, with nucleosome-free interval at transcription start site (TSS). The distal enhancer sequence (blue), which on the linear genome could be separated by hundreds of kilobases from a gene, is in a loop-like structure moved in close proximity to its promoter target. The subset of heterochromatic portions of the genome (red), including silenced genes, are organized inside the nucleus in a non-random fashion and are bordering the nuclear envelope and the periphery of the nucleolus (intranuclear organelle involved in ribosomal gene expression and assembly). A small subset of representative histone variants and histone H3 site-specific lysine (K) residues at the N-terminal tail (K4, K9, K27, K36, K79) and H4K20 residue are shown as indicated, together with panel of mono- and trimethyl, or acetyl, modifications, which differentiate between active promoters, transcribed gene bodies, and repressive chromatin, as indicated. DNA cytosines that are hydroxymethylated at the C5 position are in the nervous system most prominent at active promoters and gene bodies, while methylated cytosines are positioned around repressed promoters and in constitutive heterochromatin, and within the body of some of the actively transcribed genes. Also shown are the cohesin complex, which tethers together promoter–enhancer and other types of chromosomal loopings, and multiple members of the BAF nucleosome sliding/chromatin remodeling complex as critical regulators for RNA polymerase access and mobility at transcription start sites and active genes. See the text for details, including implications for the SCZ brain
DNA Cytosine Methylation
Two related but functionally very different types of DNA modifications, methylation (m) and hydroxymethylation (hm) of cytosine at the carbon 5 position in CpG dinucleotides (DNA sites where a cytosine nucleotide occurs next to a guanine nucleotide), provide the bulk of the epigenetic modifications in vertebrate DNA [37]. In addition, there are chemical intermediates resulting from mC5 and hmC5 synthesis and breakdown (He, et al. 2011, Ito, et al. 2011). The majority of DNA methylation is found at sites of CpG dinucleotides, particularly at sequences enriched with CpGs. In the cerebral cortex, however, up to 25 % of mC5 in the brain is found at non-CpG sites [38]. The hmC5 mark is concentrated toward the 5′ end of genes and the proximal-most portions of transcriptional units, and broadly correlates with local gene expression levels [39, 40]. In the brain, one of the tissues with the highest levels of hmC5, the mark is enriched in many active genes [41] and may play a role in the regulation of intron/exon boundaries and splicing events of neuron-specific gene transcripts [42]. On the other hand, less than 3 % of methyl-cytosine (mC5) markings are positioned around the 5′ end of genes [43]. The classical concept of the transcriptional regulatory role of DNA methylation, which has also guided many brain-related studies, is that DNA methylation at transcription start sites and proximal gene promoters functions as a negative regulator of transcription [44]. Furthermore, it is now generally accepted that DNA methylation, just like the other types of epigenetic markings discussed below, undergoes highly dynamic regulation in the developing brain and maintains the potential for bi-directional regulation and reversibility in the adult brain, particularly in the context of experience, exposure to drugs, and learning.
Some of the early epigenetic studies in SCZ brains were focused on DNA methylation of candidate gene promoters. Examples include hypermethylation of the REELIN promoter, hypomethylation of COMT, and hypermethylation of SOX10. REELIN encodes a glycoprotein that is critically important for cortical inhibitory GABAergic circuitry and, more generally, neuronal migration and connectivity formation [45, 46]. COMT encodes catechyl-O-methyltransferase, which is a key regulator of monoamine signaling [47]. SOX10 is a transcription factor gene that is important for oligodendrocyte maturation and myelination [48]. Some of the promoter DNA methylation changes that were found to be associated with corresponding alterations in gene expression in the cerebral cortex from small cohorts of SCZ subjects were independently replicated [49]. There is a general consensus that between-group differences (SCZ vs. control) show at best only subtle changes between cases and controls [50•, 51, 52]. This is not surprising, given the considerable degree of heterogeneity in terms of disease etiology, genetic risk architecture, and between-subject variability in exposure to various drugs that are known to affect brain DNA methylation levels, including alcohol [53], nicotine [54, 55], and stimulants [56, 57]. Genetic variation is likely a major factor for between-subject variability in brain DNA methylation. According to recent genome-scale studies conducted in the cerebral and cerebellar cortex, methylation of several hundred CpG-enriched sequences is significantly affected by single nucleotide polymorphisms (SNPs) and variants separated from the CpG site by more than one megabase [58, 59]. An even larger number of genetic polymorphisms have been linked to gene expression differences, including many SNP-based haplotypes within promoters and around the 5′ ends of annotated transcripts [60••]. Therefore, a firm conclusion on whether or not subgroups of SCZ subjects are defined by gene-specific DNA methylation changes in the cerebral cortex or other brain regions will require studies with large cohorts comprising hundreds of specimens and integrative analyses with DNA methylation mapping, genome sequencing, and careful evaluation of drug exposure and other confounding factors.
Histone Modifications
Epigenetic regulation of chromatin by virtue of histone modifications is extremely complex, with an estimated number of more than 100 residue-specific post-translational modifications (histone PTMs) molding and shaping the epigenome of a vertebrate cell [61]. These include, but are not limited to, mono- (me1), di- (me2)-, and tri- (me3) methylation, acetylation, and crotonylation, polyADP-ribosylation, and small-protein (ubiquitin, SUMO) modification of specific lysine residues, as well as arginine (R) methylation and “citrullination”, serine (S) phosphorylation, tyrosine (T) hydroxylation, etc. [61–63]. An epigenetic histone code, or a combinatorial set of multiple types of histone PTMs, is thought to define promoters, gene bodies, enhancer and repressor elements, and other regulatory sequences [64•]. It is important to emphasize that histone PTMs rarely occur in isolation; instead, multiple histone PTMs appear to be co-regulated and, as a group, they define the aforementioned chromatin states [65]. Many active promoters, for example, are defined by high levels of histone H3 lysine 4 methylation in combination with various histone lysine acetylation markings [64•]. Likewise, repressive histone PTMs, including the trimethylated forms of H3K9, H3K27, and H4K20, potentially co-localize to some of the same loci in the genome. There is evidence for coordinated and sequential regulation of histone PTMs. To mention one example, phosphorylation of histone H3 at the serine (S)10 position often serves as a trigger for subsequent acetylation of neighboring lysine residues histone H3 lysine 9 (H3K9) and lysine 14 (H3K14) in the context of transcriptional activation, while at the same time blocking repression-associated methylation of H3K9 [66]. The brain is no exception to this type of coordinated histone PTM regulation, and it has been observed, for example, in striatal neurons after exposure to antipsychotic drugs that are acting as dopamine D2 receptor antagonists [67].
There is a rapidly increasing literature on histone PTM alterations in the SCZ brain, with the majority of studies exploring facilitative or repressive markings at candidate gene promoters that are important for GABAergic or glutamatergic signaling in the PFC and other cortical areas. For example, the GAD1(GAD67) GABA synthesis enzyme gene promoter shows in the SCZ cortex a shift from facilitative histone acetylation and H3K4 methylation toward repressive histone methylation markings [68, 69]. Interestingly, such histone PTM changes have been reported in animals exposed to suboptimal postnatal care and parenting [70] or antimitotic drugs during prenatal development [71], which would be consistent with the neurodevelopmental hypothesis of schizophrenia. These findings may be relevant for future treatments of SCZ because, in the adult cerebral cortex, promoter-bound histone PTM at GAD1 and various other genes are altered after exposure to antipsychotic and mood-stabilizing drugs [69, 72–74].
In addition to these pioneering histone PTM studies on candidate gene promoters in the SCZ brain, early genome-scale findings have been reported. In one pilot study on dissociated olfactory epithelium in four SCZ subjects, 22 genes showed altered expression in conjunction with changes in histone H3K4 and H3K27 methylation. Brain tissue is usually obtained postmortem. In vivo studies have consequently relied on peripheral blood, which has limits when comparing tissue-specific epigenetic changes. In an elegant approach, the authors addressed this challenge by using (olfactory) nervous tissue, which is more complementary to postmortem brain tissue [75].
Histone Variants
Histone variants—well known examples of which include H3.3, H2A.Z, and H2A.X—differ from the canonical histones only at very few amino acid positions but could play an important role in replication-independent assembly of nucleosomes and chromatin fibers [76]. Several histone variants robustly affect nucleosome stability and compaction [77]. One model postulates that during the process of gene expression, RNA polymerase and the transcriptional activation and elongator complexes destabilize nucleosomes, which, in turn, promote nucleosome remodeling and variant histone incorporation, which then further potentiate or stabilize the process of gene expression [78, 79]. Transcriptome changes in SCZ could include altered expression of histone H2B type 1D (HIST1H2BD) and other variants [80] that are positioned in the SCZ susceptibility locus on chromosome 6p22.1 [81].
Chromatin-Bound RNAs (CBRs)
The process of gene expression results in the nascent RNA molecule emerging from genomic DNA. In contrast, CBR applies to an RNA species that regulates chromatin function. CBRs and nascent RNA transcripts are not mutually exclusive. According to some estimates, up to 2–3 % of the nucleic acid content in chromatin is contributed by polyadenylated RNAs [82]. Among the best-known examples of a CBR is provided by the X-chromosome Inactive Transcript (XIST) [83, 84].
Perhaps one of the most illustrative and complex examples, as it pertains to a CBR with a critical role for normal brain development and function, includes chromosome 15q11–13—a highly regulated locus, which is subject to genomic imprinting (parent-of-origin-specific gene expression) and is responsible for a range of neurodevelopmental syndromes, including Prader–Willi and Angelman [85]. Furthermore, DNA structural variants within this locus could contribute to the genetic risks of schizophrenia and bipolar disorder, further emphasizing that this locus is broadly relevant for a range of neuropsychiatric disease [85]. Of note, a very large non-coding (nc) RNA arises from 15q11–13, covering 1 Mb in the mouse and 600 kb in humans, with 148 exons and introns [86]. This long SNPRN-UBE3A ncRNA, which normally is highly expressed on the paternal chromosome but not on the maternal chromosome, includes clusters of smaller ncRNAs, which are thought to modulate nucleolar functions in neurons, and an antisense transcript, UBE3A-AS, which suppresses UBE3A sense transcription of the same gene on the paternal chromosome [85]. There is evidence that the SNPRN-UBE3A ncRNA, and the smaller RNAs derived from it, produce an “RNA cloud” in cis (at the site of the genomic locus), which contributes to lasting decondensation of this locus on the paternal chromosome, including epigenetic decoration with open chromatin-bound histone modifications and loss of repressive chromatin-associated histone and DNA methylation [85, 87]. UBE3A, which is also known as E6-AP, encodes a ubiquitin ligase, which regulates RING-1B, a component of a repressive chromatin remodeling complex PRC1 [88]. Because PRC1 is a key regulator for genome-wide repressive histone (H3K27) methylation, dysregulated expression of long SNPRN-UBE3A ncRNA may affect orderly activity of the PRC1 complex in the developing brain [89–91], perhaps resulting in chromatin defects across widespread portions of neuronal or glial genomes, with serious implications for brain function and behavior.
Chromatin Remodeling and Nucleosome Positioning
Chromatin-remodeling complexes, according to their classical definition, regulate sliding and mobility of nucleosomes, powered by ATP hydrolysis, thereby regulating gene expression and RNA polymerase II access at transcription start sites [92]. Examples of well-known chromatin remodelers with a critical role in brain development include the BAF (SWI/SNF) complex and the CHD family of proteins [92]. Interestingly, mutations in numerous members of the BAF complex and multiple CHD proteins have now been linked to psychiatric disease, including SCZ [92–94].
Higher-Order Chromatin
Epigenetic decoration of nucleosomes, including the DNA and histone PTM and histone variants described, does not adequately portray chromatin architecture. This is because chromosomal arrangements in the interphase nucleus show a non-random organization. For example, loci at sites of active gene expression are more likely to be clustered together and positioned toward a central position within the nucleus, while heterochromatin and silenced loci move more toward the nuclear periphery [95, 96]. Chromosomal loopings, in particular, are highly regulated structures and are associated with transcriptional regulation and the process of gene expression. Chromosomal loopings enable regulatory enhancer or silencer elements—which, on the linear genome, are potentially positioned hundred of kilobases further up- or downstream from their target gene—to interact directly with that specific promoter [97–99]. The regulation of higher-order chromatin is certainly of critical importance for human health, including orderly brain development and function. For example, Cornelia de Lange syndrome (CdLS)—with an estimated incidence of 1:10–30,000 live births among the more frequent genetic disorders, as per http://ghr.nlm.nih.gov —is associated with neuropsychiatric symptoms, including psychosis [100]. CdLS (including Online Mendelian Inheritance of Man (OMIM) 122470 and 300590) involves causative mutations in the cohesin complex—a multisubunit protein, which includes, among others, nipped B-like protein (NIPBL), structural maintenance of chromosomal proteins SMC1A and SMC3, and histone deacetylase HDAC8 [101, 102]. Cohesin is thought to form ring-like structures bringing together DNA segments from different locations, and could provide a structural foundation for chromosomal loop formations, including promoter–enhancer loopings for cell-type specific gene expression programmes [103]. Importantly, chromosomal loop formations and 3-dimensional genome architectures are to some degree preserved in postmortem brain tissue [104•], and early findings indeed suggest that dysregulated gene expression in the SCZ brain could be associated with weakening of longer-range promoter–enhancer loopings [105••].
Conclusion and Outlook
Molecular alterations in SCZ include altered DNA methylation and histone PTM at a subset of genes. While none of the reported postmortem findings are expected to be consistently present across all cases diagnosed with the disorder, epigenetic dysregulation of gene promoter chromatin could contribute to alterations in GABAergic circuitry and other signaling defects of the cerebral cortex in SCZ. Larger studies are now necessary, ideally in hundreds of postmortem brain specimens, to undertake comprehensive mapping of neuronal and non-neuronal epigenomes, including DNA methylation, histone PTM, and chromosomal loop mapping in diseased and control brains. Ideally, this should be undertaken with whole-genome sequencing of the same cases, because both candidate gene studies and genome-scale surveys point to a connection between the epigenetic alterations in the diseased brain and the genetic risk architecture of the underlying psychiatric disorder [69, 106, 107]. There can be little doubt that epigenomic exploration of the brain and other tissues will provide critical insights into the molecular pathology of specific cases diagnosed with SCZ and other psychiatric disease.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Hennekens CH, Hennekens AR, Hollar D, Casey DE. Schizophrenia and increased risks of cardiovascular disease. Am Heart J. 2005;150(6):1115–21.
Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209–23.
Swartz MS, Perkins DO, Stroup TS, Davis SM, Capuano G, Rosenheck RA, et al. Effects of antipsychotic medications on psychosocial functioning in patients with chronic schizophrenia: findings from the NIMH CATIE study. Am J Psychiatry. 2007;164(3):428–36.
Taly A. Novel approaches to drug design for the treatment of schizophrenia. Expert Opin Drug Disc. 2013;8(10):1285–96.
Kim DH, Stahl SM. Antipsychotic drug development. Curr Top Behav Neurosci. 2010;4:123–39.
Martins-de-Souza D, Gattaz WF, Schmitt A, Rewerts C, Marangoni S, Novello JC, et al. Alterations in oligodendrocyte proteins, calcium homeostasis and new potential markers in schizophrenia anterior temporal lobe are revealed by shotgun proteome analysis. J Neural Transm. 2009;116(3):275–89.
Regenold WT, Phatak P, Marano CM, Gearhart L, Viens CH, Hisley KC. Myelin staining of deep white matter in the dorsolateral prefrontal cortex in schizophrenia, bipolar disorder, and unipolar major depression. Psychiatry Res. 2007;151(3):179–88.
Katsel P, Davis KL, Haroutunian V. Variations in myelin and oligodendrocyte-related gene expression across multiple brain regions in schizophrenia: a gene ontology study. Schizophr Res. 2005;79(2–3):157–73.
Aston C, Jiang L, Sokolov BP. Microarray analysis of postmortem temporal cortex from patients with schizophrenia. J Neurosci Res. 2004;77(6):858–66.
Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci U S A. 2001;98(8):4746–51.
Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T, Jones PB, et al. Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet. 2003;362(9386):798–805.
Duncan CE, Webster MJ, Rothmond DA, Bahn S, Elashoff M, Shannon WC. Prefrontal GABA(A) receptor alpha-subunit expression in normal postnatal human development and schizophrenia. J Psychiatr Res. 2010;44(10):673–81.
Charych EI, Liu F, Moss SJ, Brandon NJ. GABA(A) receptors and their associated proteins: implications in the etiology and treatment of schizophrenia and related disorders. Neuropharmacology. 2009;57(5–6):481–95.
Woo TU, Kim AM, Viscidi E. Disease-specific alterations in glutamatergic neurotransmission on inhibitory interneurons in the prefrontal cortex in schizophrenia. Brain Res. 2008;1218:267–77.
Akbarian S, Huang HS. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res Rev. 2006;52(2):293–304.
Guidotti A, Auta J, Davis JM, Dong E, Grayson DR, Veldic M, et al. GABAergic dysfunction in schizophrenia: new treatment strategies on the horizon. Psychopharmacology. 2005;180(2):191–205.
Dracheva S, Elhakem SL, McGurk SR, Davis KL, Haroutunian V. GAD67 and GAD65 mRNA and protein expression in cerebrocortical regions of elderly patients with schizophrenia. J Neurosci Res. 2004;76(4):581–92.
Hashimoto T, Bazmi HH, Mirnics K, Wu Q, Sampson AR, Lewis DA. Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am J Psychiatry. 2008;165(4):479–89.
Benes FM. Amygdalocortical circuitry in schizophrenia: from circuits to molecules. Neuropsychopharmacology. 2010;35(1):239–57.
Beneyto M, Kristiansen LV, Oni-Orisan A, McCullumsmith RE, Meador-Woodruff JH. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology. 2007;32(9):1888–902.
Meador-Woodruff JH, Healy DJ. Glutamate receptor expression in schizophrenic brain. Brain Res Brain Res Rev. 2000;31(2–3):288–94.
Hemby SE, Ginsberg SD, Brunk B, Arnold SE, Trojanowski JQ, Eberwine JH. Gene expression profile for schizophrenia: discrete neuron transcription patterns in the entorhinal cortex. Arch Gen Psychiatry. 2002;59(7):631–40.
Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron. 2000;28(1):53–67.
Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P. Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J Neurosci. 2002;22(7):2718–29.
Uhlhaas PJ, Singer W. Neuronal dynamics and neuropsychiatric disorders: toward a translational paradigm for dysfunctional large-scale networks. Neuron. 2012;75(6):963–80.
Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35(1):57–67.
Zhang Y, Behrens MM, Lisman JE. Prolonged exposure to NMDAR antagonist suppresses inhibitory synaptic transmission in prefrontal cortex. J Neurophysiol. 2008;100(2):959–65.
Fatemi SH, Stary JM, Earle JA, Araghi-Niknam M, Eagan E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res. 2005;72(2–3):109–22.
Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney Jr WE, et al. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry. 1995;52(4):258–66.
Konradi C, Yang CK, Zimmerman EI, Lohmann KM, Gresch P, Pantazopoulos H, et al. Hippocampal interneurons are abnormal in schizophrenia. Schizophr Res. 2011;131(1–3):165–73.
Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57(11):1061–9.
Woo TU, Walsh JP, Benes FM. Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psychiatry. 2004;61(7):649–57.
Thompson Ray M, Weickert CS, Wyatt E, Webster MJ. Decreased BDNF, trkB-TK + and GAD67 mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J Psychiatry Neurosci. 2011;36(3):195–203.
Volk DW, Matsubara T, Li S, Sengupta EJ, Georgiev D, Minabe Y, et al. Deficits in transcriptional regulators of cortical parvalbumin neurons in schizophrenia. Am J Psychiatry. 2012;169(10):1082–91.
Huang HS, Matevossian A, Jiang Y, Akbarian S. Chromatin immunoprecipitation in postmortem brain. J Neurosci Methods. 2006;156(1–2):284–92.
Ernst C, McGowan PO, Deleva V, Meaney MJ, Szyf M, Turecki G. The effects of pH on DNA methylation state: In vitro and post-mortem brain studies. J Neurosci Methods. 2008;174(1):123–5.
Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324(5929):929–30.
Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148(4):816–31.
Jin SG, Wu X, Li AX, Pfeifer GP. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011;39(12):5015–24.
Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29(1):68–72.
Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151(7):1417–30.
Khare T, Pai S, Koncevicius K, Pal M, Kriukiene E, Liutkeviciute Z, et al. 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat Struct Mol Biol. 2012;19(10):1037–43.
Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466(7303):253–7.
Sharma RP, Grayson DR, Guidotti A, Costa E. Chromatin, DNA methylation and neuron gene regulation: the purpose of the package. J Psychiatry Neurosci. 2005;30(4):257–63.
Abdolmaleky HM, Cheng KH, Russo A, Smith CL, Faraone SV, Wilcox M, et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet. 2005;134B(1):60–6.
Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A. 2005;102(26):9341–6.
Abdolmaleky HM, Cheng KH, Faraone SV, Wilcox M, Glatt SJ, Gao F, et al. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum Mol Genet. 2006;15(21):3132–45.
Iwamoto K, Bundo M, Yamada K, Takao H, Iwayama-Shigeno Y, Yoshikawa T, et al. DNA methylation status of SOX10 correlates with its downregulation and oligodendrocyte dysfunction in schizophrenia. J Neurosci. 2005;25(22):5376–81.
Wockner LF, Noble EP, Lawford BR, Young RM, Morris CP, Whitehall VL, et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl Psychiatry. 2014;4:e339.
Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82(3):696–711. This is the first study to profile DNA methylation changes on a genome-wide scale in the postmortem schizophrenic brain.
Tochigi M, Iwamoto K, Bundo M, Komori A, Sasaki T, Kato N, et al. Methylation status of the reelin promoter region in the brain of schizophrenic patients. Biol Psychiatry. 2008;63(5):530–3.
Siegmund KD, Connor CM, Campan M, Long TI, Weisenberger DJ, Biniszkiewicz D, et al. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PloS One. 2007;2(9):e895.
Marutha Ravindran CR, Ticku MK. Changes in methylation pattern of NMDA receptor NR2B gene in cortical neurons after chronic ethanol treatment in mice. Brain Res Mol Brain Res. 2004;121(1–2):19–27.
Satta R, Maloku E, Zhubi A, Pibiri F, Hajos M, Costa E, et al. Nicotine decreases DNA methyltransferase 1 expression and glutamic acid decarboxylase 67 promoter methylation in GABAergic interneurons. Proc Natl Acad Sci U S A. 2008;105(42):16356–61.
Satta R, Maloku E, Costa E, Guidotti A. Stimulation of brain nicotinic acetylcholine receptors (nAChRs) decreases DNA methyltransferase 1 (DNMT1) expression in cortical and hippocampal GABAergic neurons of Swiss albino mice. Society for Neuroscience Abstract. 2007.
Numachi Y, Shen H, Yoshida S, Fujiyama K, Toda S, Matsuoka H, et al. Methamphetamine alters expression of DNA methyltransferase 1 mRNA in rat brain. Neurosci Lett. 2007;414(3):213–7.
Numachi Y, Yoshida S, Yamashita M, Fujiyama K, Naka M, Matsuoka H, et al. Psychostimulant alters expression of DNA methyltransferase mRNA in the rat brain. Ann N Y Acad Sci. 2004;1025:102–9.
Numata S, Ye T, Hyde TM, Guitart-Navarro X, Tao R, Wininger M, et al. DNA methylation signatures in development and aging of the human prefrontal cortex. Am J Hum Genet. 2012;90(2):260–72.
Zhang D, Cheng L, Badner JA, Chen C, Chen Q, Luo W, et al. Genetic control of individual differences in gene-specific methylation in human brain. Am J Hum Genet. 2010;86(3):411–9.
Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478(7370):519–23. This is a comprehensive study on gene expression profiles across the lifespan of the prefrontal cortex, using hundreds of postmortem brain specimens.
Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146(6):1016–28.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705.
Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14(11):1025–40.
Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12(1):7–18. This paper provides an excellent overview on the assocation of specific types of histone modifications with chromatin structures and function.
Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–12.
Nowak SJ, Corces VG. Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet. 2004;20(4):214–20.
Li J, Guo Y, Schroeder FA, Youngs RM, Schmidt TW, Ferris C, et al. Dopamine D2-like antagonists induce chromatin remodeling in striatal neurons through cyclic AMP-protein kinase A and NMDA receptor signaling. J Neurochem. 2004;90(5):1117–31.
Tang B, Dean B, Thomas EA. Disease- and age-related changes in histone acetylation at gene promoters in psychiatric disorders. Transl Psychiatry. 2011;1:e64.
Huang HS, Matevossian A, Whittle C, Kim SY, Schumacher A, Baker SP, et al. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci. 2007;27(42):11254–62.
Zhang TY, Hellstrom IC, Bagot RC, Wen X, Diorio J, Meaney MJ. Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. J Neurosci. 2010;30(39):13130–7.
Mackowiak M, Bator E, Latusz J, Mordalska P, Wedzony K. Prenatal MAM administration affects histone H3 methylation in postnatal life in the rat medial prefrontal cortex. Eur Neuropsychopharmacol. 2014;24(2):271–89.
Kurita M, Holloway T, Garcia-Bea A, Kozlenkov A, Friedman AK, Moreno JL, et al. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci. 2012;15(9):1245–54.
Costa E, Chen Y, Dong E, Grayson DR, Kundakovic M, Maloku E, et al. GABAergic promoter hypermethylation as a model to study the neurochemistry of schizophrenia vulnerability. Expert Rev Neurother. 2009;9(1):87–98.
Aoyama Y, Mouri A, Toriumi K, Koseki T, Narusawa S, Ikawa N, et al. Clozapine ameliorates epigenetic and behavioral abnormalities induced by phencyclidine through activation of dopamine D1 receptor. Int J Neuropsychopharmacol. 2013:1-15.
Kano S, Colantuoni C, Han F, Zhou Z, Yuan Q, Wilson A, et al. Genome-wide profiling of multiple histone methylations in olfactory cells: further implications for cellular susceptibility to oxidative stress in schizophrenia. Mol Psychiatry. 2013;18(7):740–2.
Woodcock CL. Chromatin architecture. Curr Opin Struct Biol. 2006;16(2):213–20.
Jin C, Felsenfeld G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007;21(12):1519–29.
Sutcliffe EL, Parish IA, He YQ, Juelich T, Tierney ML, Rangasamy D, et al. Dynamic histone variant exchange accompanies gene induction in T cells. Mol Cell Biol. 2009;29(7):1972–86.
Bintu L, Kopaczynska M, Hodges C, Lubkowska L, Kashlev M, Bustamante C. The elongation rate of RNA polymerase determines the fate of transcribed nucleosomes. Nat Struct Mol Biol. 2011;18(12):1394–9.
Sanders AR, Goring HH, Duan J, Drigalenko EI, Moy W, Freda J, et al. Transcriptome study of differential expression in schizophrenia. Hum Mol Genet. 2013;22(24):5001–14.
Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe'er I, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460(7256):753–7.
Rodriguez-Campos A, Azorin F. RNA is an integral component of chromatin that contributes to its structural organization. PloS One. 2007;2(11):e1182.
Brockdorff N. Noncoding RNA, and Polycomb recruitment. RNA. 2013;19(4):429–42.
Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–6.
Leung KN, Chamberlain SJ, Lalande M, LaSalle JM. Neuronal chromatin dynamics of imprinting in development and disease. J Cell Biochem. 2011;112(2):365–73.
Le Meur E, Watrin F, Landers M, Sturny R, Lalande M, Muscatelli F. Dynamic developmental regulation of the large non-coding RNA associated with the mouse 7C imprinted chromosomal region. Dev Biol. 2005;286(2):587–600.
Xin Z, Allis CD, Wagstaff J. Parent-specific complementary patterns of histone H3 lysine 9 and H3 lysine 4 methylation at the Prader-Willi syndrome imprinting center. Am J Hum Genet. 2001;69(6):1389–94.
Zaaroor-Regev D, de Bie P, Scheffner M, Noy T, Shemer R, Heled M, et al. Regulation of the polycomb protein Ring1B by self-ubiquitination or by E6-AP may have implications to the pathogenesis of Angelman syndrome. Proc Natl Acad Sci U S A. 2010;107(15):6788–93.
Vogel T, Stoykova A, Gruss P. Differential expression of polycomb repression complex 1 (PRC1) members in the developing mouse brain reveals multiple complexes. Dev Dyn. 2006;235(9):2574–85.
Tarabykin V, Britanova O, Fradkov A, Voss A, Katz LS, Lukyanov S, et al. Expression of PTTG and prc1 genes during telencephalic neurogenesis. Mech Dev. 2000;92(2):301–4.
Golden MG, Dasen JS. Polycomb repressive complex 1 activities determine the columnar organization of motor neurons. Genes Dev. 2012;26(19):2236–50.
Ronan JL, Wu W, Crabtree GR. From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet. 2013;14(5):347–59.
Vogel-Ciernia A, Wood MA. Neuron-specific chromatin remodeling: a missing link in epigenetic mechanisms underlying synaptic plasticity, memory, and intellectual disability disorders. Neuropharmacol. 2013.
Koga M, Ishiguro H, Yazaki S, Horiuchi Y, Arai M, Niizato K, et al. Involvement of SMARCA2/BRM in the SWI/SNF chromatin-remodeling complex in schizophrenia. Hum Mol Genet. 2009;18(13):2483–94.
Cremer T, Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet. 2001;2(4):292–301.
Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, et al. A three-dimensional model of the yeast genome. Nature. 2010;465(7296):363–7.
Wood AJ, Severson AF, Meyer BJ. Condensin and cohesin complexity: the expanding repertoire of functions. Nat Rev Genet. 2010;11(6):391–404.
Gaszner M, Felsenfeld G. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat Rev Genet. 2006;7(9):703–13.
Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503(7475):290–4.
Moss JF, Oliver C, Berg K, Kaur G, Jephcott L, Cornish K. Prevalence of autism spectrum phenomenology in Cornelia de Lange and Cri du Chat syndromes. Am J Ment Retard. 2008;113(4):278–91.
Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012;489(7415):313–7.
Gervasini C, Parenti I, Picinelli C, Azzollini J, Masciadri M, Cereda A, et al. Molecular characterization of a mosaic NIPBL deletion in a Cornelia de Lange patient with severe phenotype. Eur J Med Gene. 2013;56(3):138–43.
Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467(7314):430–5.
Mitchell AC, Bharadwaj R, Whittle C, Krueger W, Mirnics K, Hurd Y, et al. The genome in three dimensions: a new frontier in human brain research. Biol Psychiatry. 2013. This paper provides detailed protocols to map 3-dimensional genome architecture and chromosomal conformatins in postmortem brain specimens.
Bharadwaj R, Jiang Y, Mao W, Jakovcevski M, Dincer A, Krueger W, et al. Conserved chromosome 2q31 conformations are associated with transcriptional regulation of GAD1 GABA synthesis enzyme and altered in prefrontal cortex of subjects with schizophrenia. J Neurosci. 2013;33(29):11839–51. This is the first paper to map chromosomal loop formations in postmortem brain of subjects with schizophrenia.
Shulha HP, Cheung I, Whittle C, Wang J, Virgil D, Lin CL, et al. Epigenetic signatures of autism: trimethylated H3K4 landscapes in prefrontal neurons. Arch Gen Psychiatry. 2012;69(3):314–24.
Bundo M, Toyoshima M, Okada Y, Akamatsu W, Ueda J, Nemoto-Miyauchi T, et al. Increased L1 retrotransposition in the neuronal genome in schizophrenia. Neuron. 2014;81(2):306–13.
Acknowledgment
Work in the authors’ laboratory is funded by the National Institutes of Health and the Brain & Behavior Research Foundation.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Schahram Akbarian declares no conflicts of interest. Tobias Halene declares no conflicts of interest. Cyril Peter declares no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the author.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Halene, T.B., Peter, C.J. & Akbarian, S. Epigenetic Dysregulation in the Schizophrenic Brain. Curr Behav Neurosci Rep 1, 86–93 (2014). https://doi.org/10.1007/s40473-014-0007-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40473-014-0007-0
Keywords
- 3-dimensional genome
- Chromatin-bound RNAs
- Chromatin remodeling
- Chromosomal loop formation
- CpG
- DNA cytosine methylation
- DNA modification
- Enhancer
- Epigenetic
- GABAergic
- GAD1
- Higher-order chromatin
- Histone modification
- Histone variants
- Hydroxymethylation
- Methylation
- Nucleosome positioning
- Postmortem
- Post-translational modification
- Risk architecture
- Schizophrenia
- Silencer
- Transcriptome