
Abstract
Background and Objective
IDH1 (isocitrate dehydrogenase 1) is a potential biomarker and drug target. Genomic and epigenetic data on astrocytoma have demonstrated that the IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Furthermore, recent studies have also indicated that a mutant IDH1 inhibitor induced demethylation of histone H3K9me3 and expression of genes associated with gliogenic differentiation. As the presence of the p.R132H mutation in the IDH1 gene seems to be a more powerful prognostic marker than O6-methylguanine-DNA methyltransferase promoter status, we evaluated the presence of IDH1 mutation in Polish patients with astrocytoma, glioblastoma, oligoastrocytoma, ganglioglioma, oligodendroglioma, and ependymoma.
Methods
The IDH1 mutation status at codon 132 was determined using a mouse monoclonal antibody specific for the R132H mutation, direct sequencing, and Co-amplification at Lower Denaturation Temperature (COLD) polymerase chain reaction (PCR) high-resolution melting-curve analysis (HRM).
Results
Wild-type (WT) IDH1 was detected in cases with a World Health Organization (WHO) grade I astrocytoma. The IDH1 c.G395A; p.R132H mutation was observed in 56 and 94 % of grade II and grade III astrocytoma cases, respectively. Significant differences in the median overall survival were observed in astrocytoma patients grouped on the basis of the presence of IDH1 mutation: survival was 24 months longer in grade II astrocytoma and 12 months longer in glioblastoma. Overall survival was compared between grade II astrocytoma patients with low or high expression of the mutant protein. Interestingly, lower R132H expression correlated with better overall survival.
Conclusion
Our results indicate the usefulness of assessing the R132H IDH1 mutation in glioma patients: the presence or absence of the R132H mutation can help pathologists to distinguish pilocytic astrocytomas (IDH1 WT) from diffuse ones (R132H IDH1/WT). Moreover, low IDH1 p.R132H expression was related to better prognosis. This clinical implication appears to be important for personalization of prognosis and treatment by oncologists.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The Cancer Genome Atlas (TCGA) project included comprehensive genomic characterization of glioblastoma genes and core pathways. This pilot project confirmed that an atlas of changes could be created for specific cancer types. The TCGA research network identified 19 NF1 (neurofibromin 1) somatic mutations, EGFR (epidermal growth factor receptor) alterations, ERBB2 (11 somatic v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2) mutations, and somatic mutations in the PI3K (phosphatidylinositide 3-kinase) complex in glioblastoma [1]. In addition, an integrated genomic analysis of glioblastoma multiforme was performed using sequencing [2]. Parsons et al. [2] found a novel IDH1 (isocitrate dehydrogenase 1) candidate cancer gene in 12 % of glioblastoma multiforme (GBM) patients who had distinct clinical characteristics: younger age and an improved clinical prognosis. Furthermore, the median survival of patients with IDH1 c.G395A; p.R132H was 3.8 years compared with 1.1 years in patients with wild-type (WT) IDH1. Further studies confirmed that all mutations were heterozygous, with one WT allele. Interestingly, all mutations in the IDH1 gene resulted in amino acid substitutions at position 132, an evolutionarily conserved residue located within the isocitrate binding site [3, 4]. Patients with WT IDH1 often had a mutation at codon 172 of the IDH2 (isocitrate dehydrogenase 2) gene. Mutations in both the IDH1 and IDH2 genes reduced the enzymatic catalytic activity of the encoded protein [4]. The IDH1 protein is localized in the cytoplasm and peroxisomes, whereas the IDH2 enzyme is localized in mitochondria [5]. Both IDH1 and IDH2 catalyze the oxidative decarboxylation of isocitrate for alpha-ketoglutarate (alpha-KG) production. Studies using a transformed human embryonic kidney (HEK) 293T cell line expressing IDH1 mutants (R132H, R132C, or 132S) found at least an 80 % reduction in activity compared with that observed for WT IDH1 [6]. The presence of five common mutations at the same codon (132) [4] simplifies the use of several molecular methods, such as direct sequencing and use of an R132H mutation-specific anti-IDH1 antibody, for diagnostic purposes. Recent methylation data from parental, IDH1 WT, and mutant IDH1 astrocytes have indicated an important role of mutant IDH1 in primary human astrocytes that alters specific histone markers, induces extensive DNA hypermethylation, and reshapes the methylome in a fashion that mirrors the changes observed in CpG island methylator phenotype (CIMP)-positive lower-grade gliomas [7]. These data demonstrate that the IDH1 c.G395A; p.R132H mutation is the molecular basis of CIMP in gliomas and represents an advancement in the understanding of oncogenesis and the correlation between genomic and epigenomic changes in gliomas [7]. On the other hand, recent studies on the role of mutant IDH1, in which a selective R132H-IDH1 inhibitor (AGI-5198) delayed growth and promoted differentiation of glioma cells, showed that mutant IDH1 may promote glioma growth through mechanisms beyond its well-characterized epigenetic effects [8].
We assessed the presence of the somatic IDH1 c.G395A; p.R132H mutation in Polish glioma patients. We analyzed mutation status using immunohistochemistry and molecular analyses, and established the prognostic value of IDH1 status in correlation with the clinical outcome.
2 Materials and Methods
2.1 Patient Specimens
We retrospectively reviewed a cohort of 139 patients with histopathologically confirmed glioma diagnosed from 2000 to 2011 at the 10th Military Clinical Hospital and the Franciszek Lukaszczyk Oncology Center (both in Bydgoszcz, Poland). The ages of the patients ranged from 18 to 76 years (average 36 years). Patients were divided into groups according to the histological type of the tumor, whether the tumor was primary (meaning detected primarily) or secondary (meaning a tumor recurrence after previously performed surgery), the average age at diagnosis, and the presence or absence of mutations in the IDH1 gene (Table 1).
2.2 IDH1 Mutation Analysis (Immunohistochemistry, Sanger Sequencing, and COLD PCR HRM)
All samples were analyzed as a blind samples by immunohistochemistry and Co-amplification at Lower Denaturation Temperature (COLD) polymerase chain reaction (PCR) high-resolution melting-curve analysis (HRM). Ten percent of samples were additionally evaluated on the basis of sequencing chromatograms.
IDH1-p.R132H expression was assessed on formalin-fixed, paraffin-embedded (FFPE) tissue, using the immunoperoxidase method based on the two-step visualization system En Vision/HRP (Dako Polska Sp. z o.o., Warsaw, Poland) and the anti-human IDH1 R132H astrocytoma, oligodendroglioma tumor cell marker mouse monoclonal antibody clone H09 (Dianova GmbH, Hamburg, Germany). The signal of the anti-IDH1 R132H antibody was estimated under an Olympus BX41 microscope (magnification 10× and 20×), in accordance with the Dianova instructions [9]. Two clinical pathomorphologists (T.S. and W.J.), who were unaware of the patient characteristics, examined the IDH1 expression in a blinded manner. The percentage of positive cells was defined using a quantitative scale of 1–4. Previous comparison of two IDH1 monoclonal antibodies, DIA-H09 and IMab-1, had shown a higher signal-to-background ratio for DIA-H09 than for IMab-1 [10]. Samples with <10 % positively stained cells were rated as negative, in accordance with previous data [11]. IDH1 expression was marked both in the stroma and in all histologically identifiable tumor cells.
Genomic DNA was isolated automatically from the FFPE tissue using a MagNA DNA isolation kit, in accordance with the manufacturer’s instructions (Roche Applied Science, Penzberg, Germany). All Sanger sequencing was performed bidirectionally, using published primers [6], and was analyzed using Chromas software. HRM analysis was performed on all samples, using the COLD PCR method, HRM guidance, and primers for amplification [12] on a Light Cycler 480 (Roche). Fluorescent melting curves were evaluated using Light Cycler 480 software release 1.5.0 SP4 (Roche), and group sensitivity was set at 0.2 for experiments [12].
2.3 Statistical Analyses
All IDH1 results were expressed as means, using StatSoft software (2009; StatSoft, Inc., Tulsa, OK, USA). Statistica version 9.0 software was used for statistical analyses. For normal distribution, variables were analyzed using the Kolmogorov–Smirnov test with Lillefor’s correction. The association between overall survival and the presence of the IDH1 c.G395A; p.R132H mutation and WT IDH1 based on immunohistochemical analysis was estimated using the Kaplan–Meier method and assessed using the log-rank test. Statistical significance was set at P < 0.05. Overall survival was calculated from the day of the first surgery until death or the end of the follow-up period.
3 Results
3.1 Patient Characteristics
The clinical characteristics of 137 samples derived from glioma patients, principally with astrocytomas, are listed in Table 1. All cases had standard surgery, and the pathological material was FFPE tissue, using the same procedure at the 10th Military Clinical Hospital of Bydgoszcz. We evaluated 87 primary tumors and 48 recurrences in 135 glioma samples; four samples (oligodendroglioma and ependymoma) were not classified as primary tumors or recurrences. The average age at diagnosis was 28.3 years for World Health Organization (WHO) grade I astrocytoma and increased for WHO grade II, III, and IV astrocytomas, up to 43 years. The average age of patients with ganglioglioma and oligoastrocytoma was 32–35 years. The average age at diagnosis of patients with ependymoma and oligodendroglioma was 42.5 years (Table 1).
3.2 Assessment of IDH1 Gene Mutations
IDH1 gene mutations were assessed via immunohistochemistry on all 139 samples (Table 1). In addition, amplicons (including exon 4) were checked using the COLD PCR HRM method (Fig. 1), or selected FFPE tissue samples were sequenced. We analyzed 87 samples that were positive for IDH1 p.R132H, and 52 negative tumor samples (Table 1). IDH1 p.R132H staining was strong in the cytoplasm and weaker in the nucleus (Fig. 2). HRM analysis distinguished two major groups of amplicons: with IDH1 WT status in codon 132, and the R132H mutation. In addition, we found a few different melting curves, which we pooled into a third group: negative for R132H and negative for WT status.

Analysis of IDH1 (isocitrate dehydrogenase 1) mutation status. a and b: A representative IDH1 wild-type (WT) sample (no. 212) from a World Health Organization (WHO) grade II fibrillary astrocytoma, as analyzed using a Sanger sequencing and b immunohistochemistry. c and d: A representative IDH1 c.G395A; p.R132H sample (no. 68) from an oligodendroglioma, as analyzed using c Sanger sequencing and d immunohistochemistry (which was strongly positive for IDH1 with the p.R132H mutation). e Detection of IDH1 WT in a fibrillary astrocytoma sample (no. 212), a gemistocytic astrocytoma sample (no. 224), and a secondary glioblastoma multiforme (GBM) sample (no. 94); and detection of IDH1 p.R132H in a secondary GBM sample (no. 92), a protoplasmatic astrocytoma sample (no. 221), a fibrillary astrocytoma sample (no. 68), and an oligodendroglioma sample (no. 81), using Co-amplification at Lower Denaturation Temperature (COLD) polymerase chain reaction (PCR) with high-resolution melting (HRM).; immunohistochemistry (IHC) magnification ×400. The green and red stars represent wild-type and p.R132H IDH1, respectively; difference plot normalized and temperature (temp)-shifted with melting curve for heterozygous mutation (above the baseline) and IDH1 WT (below the baseline); the baseline (in red) represents the no-template control (NTC)

Immunohistochemical expression of IDH1 (isocitrate dehydrogenase 1) p.R132H in different grades of gliomas, demonstrated using haematoxylin and eosin (H&E) staining (on the left) and immunohistochemical staining with the anti-human IDH1 R132H mouse monoclonal antibody (on the right): a ependymoma; b World Health Organization (WHO) grade I pilocytic astrocytoma; c WHO grade II fibrillary astrocytoma; d WHO grade III anaplastic astrocytoma; e WHO grade IV glioblastoma multiforme; f WHO grade II oligodendroglioma
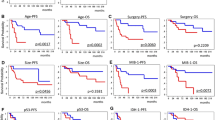
The correlation between expression of the IDH1 R132H mutant protein, as analyzed using an intensity scale in relation to the percentage of positive reactions, is presented in Fig. 4. Interestingly, the median survival of 30 grade II astrocytoma patients with lower IDH1 p.R132H expression of 1+ or 2+ (measured on a scale of 0 to 4, according to the intensity of the immunohistochemical reaction) [Fig. 4a] was 12 months longer than that of the IDH1 p.R132H group, with a score of 3+/4+ (grade II astrocytoma, n = 12). This association was estimated using the Kaplan–Meier method and the log-rank test, and was significant (P < 0.05) [Fig. 4b].
In contrast, the anti-IDH1 R132H antibody did not stain WHO grade I pilocytic astrocytoma and, as expected, no positive staining was observed in WHO grade II ependymoma. Conversely, 100 % positive staining was observed in WHO grade II oligodendroglioma. Representative results of the immunohistochemistry of IDH1 p.R132H in WHO grade I astrocytoma (pilocytic; n = 3), WHO grade II astrocytoma (diffuse, fibrillary, and gemistocytic; n = 75), WHO grade III astrocytoma (astrocytoma and anaplastic; n = 17), and WHO grade IV glioblastoma multiforme (n = 16) are presented in Fig. 2. The average age at diagnosis of astrocytoma ranged from 28.3 years for grade I to 43 years for GBM. IDH1 WT was observed in 100 % of pilocytic astrocytomas. In contrast, the R132H mutation was observed in 94.1 % of the anaplastic astrocytoma samples, all in secondary tumors (Table 1). The IDH1 c.G395A; p.R132H mutation was frequent in tumors with higher WHO grades: grade III astrocytoma (94.1 %), grade II anaplastic oligodendroglioma (76.5 %), and grade III anaplastic oligodendroglioma (75 %). Evaluation of IDH1 status in our GBM group indicated that half of the samples had the R132H mutation and the other half had IDH1 WT; however, the mutation was present in 100 % of the secondary tumors [Table 1].
In selected samples (10 %), IDH1 mutation status was determined using three methods: immunohistochemistry, direct DNA sequencing, and COLD PCR HRM. In mutated samples, we confirmed the presence of a heterozygous c.G395A; p.R132H mutation. In samples with no IDH1 R132H staining, we confirmed the absence of the c.G395A; p.R132H mutation, and DNA sequencing did not reveal the presence of any additional mutations, such as IDH1 R132C, R132G, or R132S in the evaluated 10 % of the entire cohort. Also, we tested whether COLD PCR HRM could be used as a potential screening methodology for quick detection of IDH1 mutations within codon 132. Most of the HRM data were in concordance with immunohistochemistry; in a few cases, we detected the R132H mutation in a DNA sample isolated from the sample analyzed by immunohistochemistry as IDH WT. Also, a few cases had different melting curves, which we pooled into a third group—negative for R132H and negative for WT status—but, unfortunately, Sanger sequencing of those samples was too unclear to provide an unambiguous answer. Figure 1 presents the results of the immunohistochemical analysis, sequencing, and HRM, which yielded consistent results for mutation analysis purposes.
3.3 Correlation Between IDH1 Mutation, R132H Protein Expression, and Clinical Data
Table 1 and Fig. 3 show the patient characteristics regarding the presence or absence of the IDH1 c.G395A; p.R132H mutation in primary and secondary tumors, and overall survival in a cohort of glioma patients according to histology and IDH1 status. The IDH1 c.G395A; p.R132H mutation was associated with longer survival in the grade II astrocytoma, GBM, and pooled groups of patients with WHO grade II glioma: oligoastrocytoma, oligodendroglioma, and ganglioglioma. A grade III astrocytoma with IDH1 WT status was found in only one patient, whereas the p.R132H mutation was detected in 16 secondary tumors (94.1 % of grade III astrocytomas). A Kaplan–Meier survival analysis showed that grade II astrocytoma patients (Fig. 3a) and WHO grade II glioma patients [oligoastrocytoma/oligodendroglioma/ganglioglioma] (Fig. 3c) with the p.R132H IDH1 mutation had significantly longer overall survival (up to 25 months; P = 0.05, log-rank test). Conversely, Kaplan–Meier curves of overall survival (P = 0.05) in secondary GBM indicated an overall survival that was 9 months shorter in the IDH1 WT cohort (Fig. 3b).

Kaplan–Meier curves of overall survival (P = 0.05) according to IDH1 (isocitrate dehydrogenase 1) status in a grade II astrocytoma; b glioblastoma multiforme; and c oligoastrocytoma, oligodendroglioma, and ganglioglioma samples. WT wild-type
Another Kaplan–Meier analysis of grade II astrocytoma patients revealed that those who had low p.R132H IDH1 expression had significantly longer overall survival than did patients with high expression of the protein (42 versus 30 months; P = 0.05, log-rank test) [Fig. 4b]. Although the number of patients was small, a similar trend was observed in the glioblastoma group of patients (data not shown).

Overall survival of grade II astrocytoma patients divided into two subgroups: low (1+/2+) or high (3+/4+) IDH1 (isocitrate dehydrogenase 1) p.R132H expression, as measured using the intensity of the immunohistochemical reaction. a Intensity scale in relation to the percentage of positive reactions; b Kaplan–Meier curves of overall survival between the two groups of grade II astrocytoma patients
4 Discussion
Replacement of arginine at position 132 by histidine is an evolutionarily conserved residue located within the isocitrate binding site [2]. IDH1 c.G395A; p.R132H is more frequent in WHO grades II and III than it is in WHO grade IV (up to 60 and 80 %, respectively) [13], which is in accordance with our data and supports the hypothesis of a role of IDH1 mutation in the early steps of oncogenesis (Table 1). In the same study, only 5–10 % of primary GBM tumors had IDH1 mutated status, versus 50 % in secondary GBM. Our findings were similar to an observation by Ichimura et al. [14] that the p.R132H mutation was present in 53.3 % of secondary GBM cases (Table 1). Those data suggest that IDH1 mutation status can be used as a diagnostic tool to distinguish primary GBM from other malignant astrocytomas.
IDH1 mutation has never been described in WHO grade I astrocytoma, which is in agreement with our observation (Table 1; Fig. 2); the same results (IDH1 WT) were obtained for ependymoma. Conversely, somatic mutation analysis in grade I pilocytic astrocytoma over the past 10 years has indicated the presence of a 3 bp insertion within codon 598 of the BRAF (v-raf murine sarcoma viral oncogene homolog B) gene; this resulted in a constitutively active BRAF [14] and a G-to-C transversion at codon 13 of the KRAS (Kirsten rat sarcoma viral oncogene homolog) gene, which led to increased activation of the Ras pathway [15]. Such a distinct molecular pattern and the presence of the IDH1 c.G395A; p.R132H mutation have practical implications and can be used directly to help pathologists distinguish pilocytic astrocytomas (with IDH1 WT status) from diffuse ones [4, 16]. The presence of the IDH1 c.G395A; p.R132H mutation correlated not only with the WHO grade but also with the frequency of secondary tumors [Table 1] and increased median overall survival (up to 25 months) [Fig. 3a, c].
In the present analysis, we also evaluated the methods of detection of IDH1 c.G395A; p.R132H mutation in gliomas. Recent molecular diagnostic methods have used not only gold-standard Sanger sequencing [17] but also a broad spectrum of molecular biology techniques [18, 19, 20]. Detection of somatic mutations using antibodies specific for the mutation of interest has been applied not only in the case of the IDH1 c.G395A; p.R132H mutation [9] but also in the case of deletions (anti-delE746-A750 antibody) and substitutions (anti-L858R antibody) in the EGFR gene [21]. Although dozens of mutations located in the kinase domain of the EGFR gene need to be evaluated before introduction of targeted therapy in lung cancer patients, a single mutation-specific anti-EGFR antibody does not seem to be an appropriate diagnostic tool; however, it may be the right choice for IDH1 status analysis. We correlated sequencing, HRM, and immunohistochemistry results, and found that they were mostly concordant. Any discrepancies in somatic mutation detection arose from the limit of detection, as was seen in glioblastoma, where immunohistochemistry detected IDH1 R132H expression in 11–25 % cells, while a sequencing chromatogram presented the peak for IDH1 WT. Conversely, because of the fragmentation of the DNA derived from FFPE glioma samples, DNA quality and quantity were low, as the samples had been formalin-fixed since 2000. Sanger sequencing done on 20 % of samples had to be performed at least in duplicate or in triplicate (both strands) to confirm IDH1 status and was diagnostically successful only in 10 % of cases. Moreover, COLD PCR HRM, which was assessed as a potential screening methodology for quick IDH1 diagnostic testing, is a more sensitive method for detection of the IDH1 c.G395A; p.R132H mutation at tumor edges than immunohistochemistry or simple PCR HRM [12]. In fact, we detected mutations in a few cases where IDH1 WT status was stated by immunohistochemistry, which confirms the usefulness of this method and its particular importance during the evaluation of biopsy samples or FFPE tissue with a low percentage of tumor cells. Furthermore, different melting curves that were other than positive for R132H or for WT IDH1 status were detected. We assume that they might represent amplicons with R132C, R132L, R132S, or R132G mutations. However, we found that immunohistochemistry was the most practical method for assessment of IDH1 status in tissue samples. Lastly, the results of expression analyses using immunohistochemistry (Fig. 4) allowed classification of all patients with somatic mutations in the IDH1 gene into two groups: low and high expression of the mutant R132H protein. We observed that patients with low IDH1 mutant protein expression levels in all tumor cells had a better prognosis than did those with high levels of expression of the mutant protein. This type of analysis was performed for the first time here, and suggests that the presence of IDH1 mutations has prognostic value in glioma patients. As the previous comparison by Preusser et al. [12] of DIA-H09 and IMab-1 showed concordance of those two antibodies in 98.9 %, we did not perform the same analysis with another monoclonal antibody.
Moreover, measurement of protein expression levels using immunohistochemistry or real-time PCR can provide additional information that can be used to establish the prognosis. Studies performed in cell lines have shown that IDH1 c.G395A; p.R132H overexpression decreased cell proliferation and Akt phosphorylation and altered cell morphology. Further studies performed in mice have confirmed longer survival in the presence of the mutant protein [22]. The biology of IDH1 WT and IDH1 c.G395A; p.R132H gliomas is different. The c.G395A; p.R132H mutation leads to slow tumor growth and a better outcome; however, in the higher expression range (51–100 %), a better prognosis can still be expected (in comparison with patients with IDH1 WT), albeit with shorter overall survival, than in the group with lower expression of the mutant protein.
5 Conclusion
We evaluated the presence of the R132H mutation as a prognostic biomarker in Polish patients with astrocytoma, glioblastoma, oligoastrocytoma, ganglioglioma, oligodendroglioma, and ependymoma, using immunohistochemistry, Sanger sequencing, and COLD PCR HRM. From the economic point of view, immunohistochemistry seems to be superior to real-time PCR or Sanger sequencing, as there is no need to isolate DNA for further steps. Immunohistochemistry is not only more cost effective but also has a shorter turnover time, especially considering that the low quality of fragmented DNA from FFPE samples often necessitates additional repetition.
The p.R132H IDH1 mutation was observed in 56 % of grade II astrocytomas and in 94 % of grade III astrocytomas. Kaplan–Meier survival analysis showed that grade II astrocytoma patients and WHO grade II glioma patients with the p.R132H IDH1 mutation had significantly longer overall survival, up to 25 months. On the other hand, grade II astrocytoma patients with low p.R132H IDH1 expression had a significantly longer overall survival than did patients with high expression of the mutated protein.
References
Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi:10.1038/nature07385.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–12. doi:10.1126/science.1164382.
Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008;116(6):597–602. doi:10.1007/s00401-008-0455-2.
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–73. doi:10.1056/NEJMoa0808710.
Winkler BS, DeSantis N, Solomon F. Multiple NADPH-producing pathways control glutathione (GSH) content in retina. Exp Eye Res. 1986;43(5):829–47.
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, Xiong Y. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324(5924):261–5. doi:10.1126/science.1170944.
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, Levine R, Heguy A, Viale A, Morris LG, Huse JT, Mellinghoff IK, Chan TA. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–83. doi:10.1038/nature10866.
Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, Kunii K, Pedraza A, Schalm S, Silverman L, Miller A, Wang F, Yang H, Chen Y, Kernytsky A, Rosenblum MK, Liu W, Biller SA, Su SM, Brennan CW, Chan TA, Graeber TG, Yen KE, Mellinghoff IK. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–30. doi:10.1126/science.1236062.
Capper D, Weissert S, Balss J, Habel A, Meyer J, Jager D, Ackermann U, Tessmer C, Korshunov A, Zentgraf H, Hartmann C, von Deimling A. Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol. 2010;20(1):245–54. doi:10.1111/j.1750-3639.2009.00352.x.
Preusser M, Wohrer A, Stary S, Hoftberger R, Streubel B, Hainfellner JA. Value and limitations of immunohistochemistry and gene sequencing for detection of the IDH1-R132H mutation in diffuse glioma biopsy specimens. J Neuropathol Exp Neurol. 2011;70(8):715–23. doi:10.1097/NEN.0b013e31822713f0.
Takano S, Tian W, Matsuda M, Yamamoto T, Ishikawa E, Kaneko MK, Yamazaki K, Kato Y, Matsumura A. Detection of IDH1 mutation in human gliomas: comparison of immunohistochemistry and sequencing. Brain Tumor Pathol. 2011;28(2):115–23. doi:10.1007/s10014-011-0023-7.
Boisselier B, Marie Y, Labussiere M, Ciccarino P, Desestret V, Wang X, Capelle L, Delattre JY, Sanson M. COLD PCR HRM: a highly sensitive detection method for IDH1 mutations. Hum Mutat. 2010;31(12):1360–5. doi:10.1002/humu.21365.
Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, Collins VP. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 2009;11(4):341–7. doi:10.1215/15228517-2009-025.
Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. 2009;28(20):2119–23. doi:10.1038/onc.2009.73.
Sharma MK, Zehnbauer BA, Watson MA, Gutmann DH. RAS pathway activation and an oncogenic RAS mutation in sporadic pilocytic astrocytoma. Neurology. 2005;65(8):1335–6. doi:10.1212/01.wnl.0000180409.78098.d7.
Korshunov A, Meyer J, Capper D, Christians A, Remke M, Witt H, Pfister S, von Deimling A, Hartmann C. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118(3):401–5. doi:10.1007/s00401-009-0550-z.
Crotti L, Lewandowska MA, Schwartz PJ, Insolia R, Pedrazzini M, Bussani E, Dagradi F, George AL Jr, Pagani F. A KCNH2 branch point mutation causing aberrant splicing contributes to an explanation of genotype-negative long QT syndrome. Heart Rhythm. 2009;6(2):212–8. doi:10.1016/j.hrthm.2008.10.044.
Ramalho AS, Lewandowska MA, Farinha CM, Mendes F, Goncalves J, Barreto C, Harris A, Amaral MD. Deletion of CFTR translation start site reveals functional isoforms of the protein in CF patients. Cell Physiol Biochem. 2009;24(5–6):335–46. doi:00025742610.1159/000257426.
Lewandowska MA, Jozwicki W, Zurawski B. KRAS and BRAF mutation analysis in colorectal adenocarcinoma specimens with a low percentage of tumor cells. Mol Diagn Therapy. 2013;17(3):193–203. doi:10.1007/s40291-013-0025-8.
Lewandowska MA, Jozwicki W, Jochymski C, Kowalewski J. Application of PCR methods to evaluate EGFR, KRAS and BRAF mutations in a small number of tumor cells in cytological material from lung cancer patients. Oncol Reports. 2013;30:1045–52. doi:10.3892/or.2013.2579.
Kawahara A, Azuma K, Sumi A, Taira T, Nakashima K, Aikawa E, Abe H, Yamaguchi T, Takamori S, Akiba J, Kage M. Identification of non-small-cell lung cancer with activating EGFR mutations in malignant effusion and cerebrospinal fluid: rapid and sensitive detection of exon 19 deletion E746-A750 and exon 21 L858R mutation by immunocytochemistry. Lung Cancer. 2011;74(1):35–40. doi:10.1016/j.lungcan.2011.02.002.
Bralten LB, Kloosterhof NK, Balvers R, Sacchetti A, Lapre L, Lamfers M, Leenstra S, de Jonge H, Kros JM, Jansen EE, Struys EA, Jakobs C, Salomons GS, Diks SH, Peppelenbosch M, Kremer A, Hoogenraad CC, Smitt PA, French PJ. IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Ann Neurol. 2011;69(3):455–63. doi:10.1002/ana.22390.
Acknowledgments
We thank Dr. Z. Pawlowicz for generating conditions advantageous for our research at The Franciszek Lukaszczyk Oncology Center (Bydgoszcz, Poland). We thank the medical and nursing staff involved in the daily care of the patients, as well as the staff involved in the conduction of this study.
Conflicts of interest
The authors have no conflicts of interest that are directly relevant to the content of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Lewandowska, M.A., Furtak, J., Szylberg, T. et al. An Analysis of the Prognostic Value of IDH1 (Isocitrate Dehydrogenase 1) Mutation in Polish Glioma Patients. Mol Diagn Ther 18, 45–53 (2014). https://doi.org/10.1007/s40291-013-0050-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-013-0050-7