
Abstract
Cystic fibrosis is the most common inherited condition in the Caucasian population and is associated with significantly reduced life expectancy. Recent advances in treatment have focussed on addressing the underlying cause of the condition, the defective production, expression and function of the cystic fibrosis transmembrane conductance regulator (CFTR) protein. Several drugs with different modes of action have produced promising results in clinical trials, and some have been incorporated into routine clinical care for specific patients in many countries worldwide. Further trials continue to explore the safety and efficacy of these drugs in the youngest age groups and to search for more effective therapies to treat the most common disease-causing gene mutations in an ever-expanding drug pipeline. As evidence mounts for the early onset of disease in young patients, the prospect of introducing disease-modifying therapy in early life becomes more pertinent, although the cost implications of these expensive drugs are significant. In this review, we summarise these new therapy advances and review those currently being explored in clinical trials.

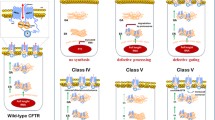
(reprinted from De Boeck and Amaral [14] with permission from Elsevier)
Similar content being viewed by others

References
CFF. 2017 Cystic Fibrosis Foundation Patient Registry Highlights. Cystic Fibrosis Foundation. 2017. https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2017-Cystic-Fibrosis-Foundation-Patient-Registry-Highlights.pdf Accessed 13 Sept 2018.
The Clinical and Functional TRanslation of CFTR (CFTR2). US CF Foundation, Johns Hopkins University, The Hospital for Sick Children. 2011. https://cftr2.org/. Accessed 2 Oct 2018.
Sly PD, Gangell CL, Chen L, Ware RS, Ranganathan S, Mott LS, et al. Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med. 2013;368(21):1963–70. https://doi.org/10.1056/nejmoa1301725.
Olivier AK, Yi Y, Sun X, Sui H, Liang B, Hu S, et al. Abnormal endocrine pancreas function at birth in cystic fibrosis ferrets. J Clin Investig. 2012;122(10):3755–68. https://doi.org/10.1172/jci60610.
Vilozni D, Bentur L, Efrati O, Minuskin T, Barak A, Szeinberg A, et al. Spirometry in early childhood in cystic fibrosis patients. Chest. 2007;131(2):356–61. https://doi.org/10.1378/chest.06-1351.
Davies JC, Cunningham S, Alton EWFW, Innes JA. Lung clearance index in CF: a sensitive marker of lung disease severity. Thorax. 2008;63(2):96. https://doi.org/10.1136/thx.2007.082768.
Subbarao P, Stanojevic S, Brown M, Jensen R, Rosenfeld M, Davis S, et al. Lung clearance index as an outcome measure for clinical trials in young children with cystic fibrosis. A pilot study using inhaled hypertonic saline. Am J Respir Crit Care Med. 2013;188(4):456–60. https://doi.org/10.1164/rccm.201302-0219oc.
Belessis Y, Dixon B, Hawkins G, Pereira J, Peat J, MacDonald R, et al. Early cystic fibrosis lung disease detected by bronchoalveolar lavage and lung clearance index. Am J Respir Crit Care Med. 2012;185(8):862–73. https://doi.org/10.1164/rccm.201109-1631oc.
Nasr SZ, Sakmar E, Christodoulou E, Eckhardt BP, Streetman DS, Strouse PJ. The use of high resolution computerized tomography (HRCT) of the chest in evaluating the effect of tobramycin solution for inhalation in cystic fibrosis lung disease. Pediatr Pulmonol. 2010;45(5):440–9. https://doi.org/10.1002/ppul.21188.
Rosenow T, Oudraad MC, Murray CP, Turkovic L, Kuo W, de Bruijne M, et al. Reply: Excess risk of cancer from computed tomography scan is small but not so low as to be incalculable. Am J Respir Crit Care Med. 2015;192(11):1397–9. https://doi.org/10.1164/rccm.201508-1574le.
Wielputz MO, Eichinger M, Puderbach M. Magnetic resonance imaging of cystic fibrosis lung disease. J Thorac Imaging. 2013;28(3):151–9. https://doi.org/10.1097/rti.0b013e31828d40d4.
Davies JC, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med. 2016;4(2):107–15. https://doi.org/10.1016/s2213-2600(15)00545-7.
Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology—drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157–67. https://doi.org/10.1056/nejmra035092.
De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med. 2016;4(8):662–74. https://doi.org/10.1016/s2213-2600(16)00023-0.
Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95(7):1005–15.
Sawczak V, Getsy P, Zaidi A, Sun F, Zaman K, Gaston B. Novel approaches for potential therapy of cystic fibrosis. Curr Drug Targets. 2015;16(9):923–36.
Veit G, Avramescu RG, Chiang AN, Houck SA, Cai Z, Peters KW, et al. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Biol Cell. 2016;27(3):424–33. https://doi.org/10.1091/mbc.e14-04-0935.
Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–72. https://doi.org/10.1056/nejmoa1105185.
Konstan MW, Plant BJ, Elborn JS, Rodriguez S, Munck A, Ahrens R, et al. Efficacy response in CF patients treated with ivacaftor: post-hoc analysis. Pediatr Pulmonol. 2015;50(5):447–55. https://doi.org/10.1002/ppul.23173.
Flume PA, Wainwright CE, Elizabeth Tullis D, Rodriguez S, Niknian M, Higgins M, et al. Recovery of lung function following a pulmonary exacerbation in patients with cystic fibrosis and the G551D-CFTR mutation treated with ivacaftor. J Cyst Fibros. 2018;17(1):83–8. https://doi.org/10.1016/j.jcf.2017.06.002.
Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187(11):1219–25. https://doi.org/10.1164/rccm.201301-0153oc.
McKone EF, Borowitz D, Drevinek P, Griese M, Konstan MW, Wainwright C, et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp- CFTR mutation: a phase 3, open-label extension study (PERSIST). Lancet Respir Med. 2014;2(11):902–10. https://doi.org/10.1016/s2213-2600(14)70218-8.
De Boeck K, Munck A, Walker S, Faro A, Hiatt P, Gilmartin G, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13(6):674–80. https://doi.org/10.1016/j.jcf.2014.09.005.
Fidler MC, Sullivan JC, Boj SF, Vries R, Munck A, Higgins M, et al. WS18.5 Evaluation of the contributions of splicing and gating defects to dysfunction of G970R-CFTR [abstract]. J Cyst Fibros. 2017;16:S31. https://doi.org/10.1016/s1569-1993(17)30259-x.
Rosenfeld M, Wainwright CE, Higgins M, Wang LT, McKee C, Campbell D, et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to < 24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single-arm study. Lancet Respir Med. 2018;6(7):545–53. https://doi.org/10.1016/s2213-2600(18)30202-9.
Rowe SM, Heltshe SL, Gonska T, Donaldson SH, Borowitz D, Gelfond D, et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med. 2014;190(2):175–84. https://doi.org/10.1164/rccm.201404-0703oc.
Sawicki GS, McKone EF, Pasta DJ, Millar SJ, Wagener JS, Johnson CA, et al. Sustained benefit from ivacaftor demonstrated by combining clinical trial and cystic fibrosis patient registry data. Am J Respir Crit Care Med. 2015;192(7):836–42. https://doi.org/10.1164/rccm.201503-0578oc.
Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros. 2014;13(1):29–36. https://doi.org/10.1016/j.jcf.2013.06.008.
Moss RB, Flume PA, Elborn JS, Cooke J, Rowe SM, McColley SA, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med. 2015;3(7):524–33. https://doi.org/10.1016/s2213-2600(15)00201-5.
Dekkers JF, Wiegerinck CL, de Jonge HR, Bronsveld I, Janssens HM, de Winter-de Groot KM, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19(7):939–45. https://doi.org/10.1038/nm.3201.
Durmowicz AG, Lim R, Rogers H, Rosebraugh CJ, Chowdhury BA. The US Food and Drug Administration’s experience with ivacaftor in cystic fibrosis. Establishing efficacy using in vitro data in Lieu of a clinical trial. Ann Am Thorac Soc. 2018;15(1):1–2. https://doi.org/10.1513/annalsats.201708-668ps.
Vertex. HIGHLIGHTS OF PRESCRIBING INFORMATION KALYDECO. Vertex Pharmaceuticals. 2018. https://pi.vrtx.com/files/uspi_ivacaftor.pdf. Accessed 18 Sept 2018.
EMA. European Medicines Agency EPAR summary for the public Kalydeco. European Medicines Agency. 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/002494/WC500130744.pdf. Accessed 18 Sept 2018.
ATRG. Kayldeco public summary. Australian Government Department of Health. 2018. https://www.ebs.tga.gov.au/servlet/xmlmillr6?dbid = ebs/PublicHTML/pdfStore.nsf&docid = D76D18E21A98A81FCA2582AD00424213&agid = (PrintDetailsPublic)&actionid = 1. Accessed 18 Sept 2018.
CADTH. Kalydeco Common Drug Review. Canadian Agency for Drugs and Technologies in Health (CADTH). 2015. https://www.cadth.ca/sites/default/files/cdr/clinical/SR0379_KalydecoCF_CL_Report.pdf. Accessed 18 Sept 2018.
FDA. Full Prescribing Information for Orkambi. Food and Drug Administration. 2015. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/206038Orig1s000lbl.pdf. Accessed 28 Aug 2018.
FDA. Full Prescribing Information for Kalydeco Food and Drug Administration. 2017. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/203188s019lbl.pdf. Accessed 28 Aug 2018.
EMA. Kalydeco: EPAR-product information. European Medicines Agency. 2017. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002494/WC500130696.pdf. Accessed 28 Aug 2018.
Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA. 2011;108(46):18843–8. https://doi.org/10.1073/pnas.1105787108.
Vermeulen F, Proesmans M, Boon M, Havermans T, De Boeck K. Lung clearance index predicts pulmonary exacerbations in young patients with cystic fibrosis. Thorax. 2014;69(1):39–45. https://doi.org/10.1136/thoraxjnl-2013-203807.
Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67(1):12–8. https://doi.org/10.1136/thoraxjnl-2011-200393.
Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014;2(7):527–38. https://doi.org/10.1016/s2213-2600(14)70132-8.
Wainwright CE, Elborn JS, Ramsey BW. Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–31. https://www.nejm.org/doi/full/10.1056/NEJMoa1409547.
Elborn JS, Ramsey BW, Boyle MP, Konstan MW, Huang X, Marigowda G, et al. Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: a pooled analysis. Lancet Respir Med. 2016;4(8):617–26. https://doi.org/10.1016/s2213-2600(16)30121-7.
Konstan MW, McKone EF, Moss RB, Marigowda G, Tian S, Waltz D, et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet Respir Med. 2017;5(2):107–18. https://doi.org/10.1016/s2213-2600(16)30427-1.
Milla CE, Ratjen F, Marigowda G, Liu F, Waltz D, Rosenfeld M, et al. Lumacaftor/ivacaftor in patients aged 6–11 years with cystic fibrosis and homozygous for F508del-CFTR. Am J Respir Crit Care Med. 2017;195(7):912–20. https://doi.org/10.1164/rccm.201608-1754oc.
Ratjen F, Hug C, Marigowda G, Tian S, Huang X, Stanojevic S, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. 2017;5(7):557–67. https://doi.org/10.1016/s2213-2600(17)30215-1.
Mcnamara J, Mccolley SA, Owen CA, Liu F, Tian S, Waltz D, Marigowda G, Sawicki GS. A 2-part, phase 3 single-arm study to evaluate the safety and pharmacokinetics (PK) of lumacaftor/ivacaftor (LUM/IVA) combination therapy in patients (pts) aged 2–5 years with cystic fibrosis (CF) homozygous for the F508del-CFTR mutation [abstract]. J Cyst Fibros. 2018;17(S3):S2–3.
Cholon DM, Quinney NL, Fulcher ML, Esther CR Jr, Das J, Dokholyan NV, et al. Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Sci Transl Med. 2014;6(246):246ra96. https://doi.org/10.1126/scitranslmed.3008680.
Vertex. Vertex Announces Reimbursement Agreement in Australia for ORKAMBI® (lumacaftor/ivacaftor) for people with cystic fibrosis ages six years and older with two copies of the F508del mutation. Vertex Pharmaceuticals. 2018. https://investors.vrtx.com/news-releases/news-release-details/vertex-announces-reimbursement-agreement-australia-orkambir. Accessed 13 Sept 2018.
Taylor-Cousar JL, Munck A, McKone EF, van der Ent CK, Moeller A, Simard C, et al. Tezacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med. 2017;377(21):2013–23. https://doi.org/10.1056/nejmoa1709846.
Donaldson SH, Pilewski JM, Griese M, Cooke J, Viswanathan L, Tullis E, et al. Tezacaftor/ivacaftor in subjects with cystic fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med. 2018;197(2):214–24. https://doi.org/10.1164/rccm.201704-0717oc.
Rowe SM, Daines C, Ringshausen FC, Kerem E, Wilson J, Tullis E, et al. Tezacaftor–ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med. 2017;377(21):2024–35. https://doi.org/10.1056/nejmoa1709847.
Tullis E, Colombo C, Davies J, McKee C, DeSouza C, Walktz D, Savage J, Fisher M, Shilling R, Moskowitz S, Robertson S, Tian S, Taylor-Cousar JL, Rowe SM, editor. Preliminary safety and efficacy of triple combination CFTR modulator regimens in CF [abstract]. North American Cystic Fibrosis Conference; 2017; Indianapolis; Breaking Science Presentation.
Vertex. Vertex selects two next-generation correctors, VX-659 and VX-445, to advance into phase 3 development as part of two different triple combination regimens for people with cystic fibrosis. Vertex Pharmaceuticals. 2018. http://investors.vrtx.com/releasedetail.cfm?releaseid=1055958. Accessed 28 Aug 2018.
Bhatt P, Kwok I, Bailey V, Chin J, Bresilla C, Dasgupta A, Cole B, Krouse M. In vitro efficacy of combination FDL169/FDL176 is greater than tezacaftor/ivacaftor [abstract]. Pediatr Pulmonol. 2017;52(Supplement):S218.
De Boeck KVBE, van der Ent C, Verhulst S, Weersink E, Conrath K, Kanters D, Namour F, de Kock H, Van de Steen O. GLPG1837 in subjects with cystic fibrosis and S1251S mutation: results from a phase IIa study (SAPHIRA2) [abstract]. Pediatr Pulmonol. 2016;51(Supplement 45):S288.
Davies J, Bell SC, Van Koningsbruggen-Rietschel S, McKone EF, Macgregor G, Drevinek P, et al. WS13.6 GLPG1837 in subjects with cystic fibrosis (CF) and the G551D mutation: results from a phase II study (SAPHIRA1) [abstract]. J Cyst Fibros. 2017;16:S24–5. https://doi.org/10.1016/s1569-1993(17)30236-9.
Galapagos. ALBATROSS with GLPG2222 shows positive clinical results in CF patients. Galapagos. 2017. http://hugin.info/133350/R/2150681/825566.pdf. Accessed 28 Aug 2018.
Bell S, De Boeck K, Drevinek P, Plant B, Barry P, Elborn S, de Kock H, Loyau S, Muller K, Vandebriel L, Kanters D. GLP2222 in subjects with cystic fibrosis and the F508del/class III mutation on stable treatment with ivacaftor: results from a phase II study (ALBATROSS) [abstract]. J Cyst Fibros. 2018;17(S3):S2.
Molinski SV, Ahmadi S, Ip W, Ouyang H, Villella A, Miller JP, et al. Orkambi(R) and amplifier co-therapy improves function from a rare CFTR mutation in gene-edited cells and patient tissue. EMBO Mol Med. 2017;9(9):1224–43. https://doi.org/10.15252/emmm.201607137.
Flume P, Sawicki G, Pressler T, Schwarz C, Fajac I, Layish D, Bialek P, Wilson S, Kang L, Mclaughlin B, Scafidi S, Lee P-S, Gilmartin G. Phase 2 initial results evaluating PTI-428, a novel CFTR amplifier, in patients with cystic fibrosis [abstract]. J Cyst Fibros. 2018;17(S3):S1–2.
Beumer W, Swildens J, Henig N, Anthonijsz H, Biasutto P, Leal T, et al. WS01.2 QR-010, an RNA therapy, restores CFTR function using in vitro and in vivo models of ΔF508 CFTR. J Cyst Fibros. 2015;14:S1. https://doi.org/10.1016/s1569-1993(15)30002-3.
Elborn S, Bouisset F, Boff M, Checchio T, Perquin J, Lamontagne N, Montgomery S, Henig N. A first-in-human, phase 1b, dose-escalation study of QR-010, a novel antisense oligonucleotide administered in subjects with cystic fibrosis homozygous for the F508del CFTR mutation [abstract]. Pediatr Pulmonol. 2017;52(Supplement 47):S289.
Zainal Abidin N, Haq IJ, Gardner AI, Brodlie M. Ataluren in cystic fibrosis: development, clinical studies and where are we now? Expert Opin Pharmacother. 2017;18(13):1363–71. https://doi.org/10.1080/14656566.2017.1359255.
Anderson WF, Gorini L, Breckenridge L. Role of ribosomes in streptomycin-activated suppression. Proc Natl Acad Sci USA. 1965;54(4):1076–83.
Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349(15):1433–41. https://doi.org/10.1056/nejmoa022170.
Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008;372(9640):719–27. https://doi.org/10.1016/s0140-6736(08)61168-x.
Sermet-Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, Mogenet A, et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med. 2010;182(10):1262–72. https://doi.org/10.1164/rccm.201001-0137oc.
Kerem E, Viviani L, Zolin A, MacNeill S, Hatziagorou E, Ellemunter H, et al. Factors associated with FEV1 decline in cystic fibrosis: analysis of the ECFS Patient Registry. Eur Respir J. 2014;43(1):125–33. https://doi.org/10.1183/09031936.00166412.
PTC Therapeutics. PTC therapeutics announces results from pivotal phase 3 clinical trial of ataluren in patients living with nonsense mutation cystic fibrosis. PTC therapeutics. 2017. http://ir.ptcbio.com/releasedetail.cfm?ReleaseID=1015471. Accessed 28 Aug 2018.
CFF. Drug Development Pipeline. Cystic Fibrosis Foundation. https://www.cff.org/Trials/Pipeline. Accessed 28 Aug 2018.
Eloxx. Eloxx Pipeline. Eloxx Pharmaceuticals. 2018. http://www.eloxxpharma.com/pipeline/. Accessed 28 Aug 2018.
Mott LS, Park J, Murray CP, Gangell CL, de Klerk NH, Robinson PJ, et al. Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax. 2012;67(6):509–16. https://doi.org/10.1136/thoraxjnl-2011-200912.
Cheah E, Venuti E, McKay K, Gaskin K. ePS05.6 Cessation of pancreatic enzyme replacement therapy (PERT) after initiation of therapy with ivacaftor; a case series. J Cyst Fibros. 2015;14:S51. https://doi.org/10.1016/s1569-1993(15)30165-x.
Dilokthornsakul P, Hansen RN, Campbell JD. Forecasting US ivacaftor outcomes and cost in cystic fibrosis patients with the G551D mutation. Eur Respir J. 2016;47(6):1697–705. https://doi.org/10.1183/13993003.01444-2015.
Dilokthornsakul P, Patidar M, Campbell JD. Forecasting the long-term clinical and economic outcomes of lumacaftor/ivacaftor in cystic fibrosis patients with homozygous phe508del mutation. Value Health. 2017;20(10):1329–35. https://doi.org/10.1016/j.jval.2017.06.014.
Vertex. FDA approves KALYDECO® (ivacaftor) for more than 900 people ages 2 and older with cystic fibrosis who have certain residual function mutations. Vertex Pharmceuticals. 2017. https://investors.vrtx.com/news-releases/news-release-details/fda-approves-kalydecor-ivacaftor-more-900-people-ages-2-and. Accessed 18 Sept 2018.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
RD, CE, DH, and IM have no conflicts of interest and have received no funding in relation to this manuscript. JCD has performed clinical trial leadership, advisory board and educational roles for the following companies for which fees have been paid to Imperial College London or Royal Brompton Hospital: Vertex Pharmaceuticals, PTC, Proteostasis, Galapagos, Abbvie, Chiesi, Eloxx, Ionis, Enterprise Therapeutics, Pulmocide, Novartis, Algipharma. JCD has received no funding in relation to this manuscript.
Funding
No sources of funding were used to conduct this study or prepare this manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dobra, R., Edmondson, C., Hughes, D. et al. Potentiators and Correctors in Paediatric Cystic Fibrosis Patients: A Narrative Review. Pediatr Drugs 20, 555–566 (2018). https://doi.org/10.1007/s40272-018-0315-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40272-018-0315-z