
Abstract
Options for the effective treatment of hereditary optic neuropathies have been a long time coming. The successful launch of the antioxidant idebenone for Leber’s Hereditary Optic Neuropathy (LHON), followed by its introduction into clinical practice across Europe, was an important step forward. Nevertheless, other options, especially for a variety of mitochondrial optic neuropathies such as dominant optic atrophy (DOA), are needed, and a number of pharmaceutical agents, acting on different molecular pathways, are currently under development. These include gene therapy, which has reached Phase III development for LHON, but is expected to be developed also for DOA, whilst most of the other agents (other antioxidants, anti-apoptotic drugs, activators of mitobiogenesis, etc.) are almost all at Phase II or at preclinical stage of research. Here, we review proposed target mechanisms, preclinical evidence, available clinical trials with primary endpoints and results, of a wide range of tested molecules, to give an overview of the field, also providing the landscape of future scenarios, including gene therapy, gene editing, and reproductive options to prevent transmission of mitochondrial DNA mutations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
A definitive treatment for hereditary optic neuropathies is missing; nevertheless, some options are currently available and are under continuous evolution. They range from drugs designed to overcome the metabolic defect and compensate the mitochondrial dysfunction, to gene therapies and stem-cell-based approaches. |
Idebenone, a coenzyme Q10 analogue, is the most studied molecule with antioxidant properties that has shown to improve visual acuity in Leber’s Hereditary Optic Neuropathy (LHON), and hopefully it will be offered soon in other mitochondrial optic neuropathies. |
The ultimate goal for the treatment of hereditary optic neuropathies is the correction of the genetic defect, or the prevention of its transmission to progeny. The delivery of the wild-type gene in LHON patients’ eyes has shown some positive results in terms of visual outcome, but further studies are needed. |
1 Introduction
Hereditary optic neuropathies (HONs) are a heterogeneous group of genetically determined diseases affecting the optic nerve, either as isolated optic atrophy or as part of a syndromic association [1, 2]. The two most common non-syndromic inherited optic neuropathies are Leber’s Hereditary Optic Neuropathy (LHON) and Dominant Optic Atrophy (DOA), accounting for the majority of HONs encountered in clinical practice and responsible for chronic visual impairment in children and young adults [3].
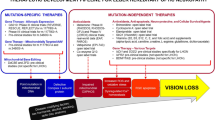
Leber’s Hereditary Optic Neuropathy (LHON) is caused by primary mitochondrial DNA (mtDNA) mutations affecting the mitochondrial respiratory chain complexes [1, 2]. The three most common mtDNA mutations responsible for 90–95% of LHON cases are m.3460G>A (MTND1), m.11778G>A (MTND4), and m.14484T>C (MTND6) mutations, but further rare mtDNA mutations are also described as pathogenic for LHON [4]. Dominant mutations in the nuclear DNA-encoded gene OPA1, are responsible for about 60–70% of DOA cases [1, 2]. OPA1 is a nuclear gene encoding for a protein critical for fusion of the inner mitochondrial membrane (IMM), thus primarily regulating mitochondrial fusion and dynamics, but also involved in mtDNA maintenance and oxidative phosphorylation [5]. LHON and OPA1-related DOA share mitochondrial dysfunction centred on complex I as a key pathogenic mechanism [6]. The common hallmark of HONs, which in the large majority of cases are due to mitochondrial dysfunction as a common pathogenic mechanism, is the selective vulnerability and degeneration of retinal ganglion cells (RGCs) [1, 2, 6, 7]. Understanding the underlying pathogenic mechanisms and natural history of HONs, together with the knowledge of naturally occurring cellular compensatory strategies, are all crucial steps for identifying specific pathways to target in the development of new therapeutic strategies, currently limited mainly to LHON [8, 9]. A milestone in LHON treatment has been the approval of idebenone in 2015 (https://www.ema.europa.eu/en/medicines/human/EPAR/raxone). Idebenone is a synthetic short-chain analogue of ubiquinone with anti-oxidant properties [10], which has shown to increase the rate of visual recovery in LHON patients, especially when given in the acute stage [11, 12]. The use of idebenone in OPA1-related DOA is still under investigations but preliminary data indicate a possible beneficial effect of this therapy [13, 14]. Other pharmaceutical agents, such as EPI-743 [15], elamipretide (mtp-131) [16], oestrogen-related compounds [17], rapamycin [18], and microRNA (miRNA) based [19], by targeting different pathways, have been or are currently under investigation in LHON at clinical [15, 16] and pre-clinical stages [17,18,19]. A significant step forward in the management of LHON is expected from gene therapy, which by intravitreal injection of modified adeno-associated-viral vectors (AAV2) is designed to deliver the wild-type copy of ND4 complex I subunit engineered to be allotopically expressed from the nucleus and imported within mitochondria for functional complementation [20]. Multiple trials, dedicated to acute LHON patients harbouring the m.11778G>A/MTND4 mutation, are concluded or currently ongoing in Europe, USA and China [21]. As far as gene therapy is concerned, in OPA1-related DOA there are ongoing preclinical studies on mouse models [22], with multiple approaches being considered, but to date, there is no active clinical trial in humans. Overall, we will discuss gene therapy and editing as future therapeutic avenues to be developed for any form of HON. Principal molecules and mechanisms are summarised in Fig. 1.

Therapeutic strategies in Leber's Hereditary Optic Neuropathy (LHON). This figure schematically shows the mechanism of action for the different therapeutic approaches in Hereditary Optic Neuropathies
2 Pathogenetic Mechanisms in Mitochondrial Optic Neuropathies
2.1 Respiratory Chain Dysfunction in Mitochondrial Optic Neuropathies
Mitochondria are cytoplasmic organelles, consisting of two membranes separated by an intermembrane space, which surround the mitochondrial matrix, where a number of metabolic pathways take place [23]. Mitochondria are also involved in key cellular homeostatic processes including organelle dynamics [24], interaction with other organelles [25] and controlling the apoptotic death signalling [26], calcium handling [27] and organelle clearing through autophagy [28]. However, the central function of mitochondria is to be the powerhouses of the cell, producing energy through the biochemical process of oxidative phosphorylation (OXPHOS) [29]. OXPHOS redox reactions take place through the mitochondrial respiratory chain culminating in adenosine triphosphate (ATP) synthesis, carried out by five enzymatic complexes embedded in the IMM, typically bent into the cristae, which increase the total available surface. In the mitochondrial respiratory chain, electrons are shuttled from one complex to the other, namely from complex I to III and then IV, providing the energy used to transfer protons across the membrane from the matrix to the intermembrane space. The energy is thus conserved through the resulting electrochemical gradient and eventually used by complex V, as protons are channelled backwards in a controlled way, to catalyse the ATP synthesis from adenosine diphosphate (ADP) and inorganic phosphate [30, 31]. When this fine series of imbricated reactions is affected, either by inherited or acquired causes, the result can be both a reduction in ATP synthesis and excessive generation of reactive oxidative species (ROS), due to lose electrons spilling out from the respiratory chain, which then react with molecular oxygen. Excessive ROS production may damage the respiratory subunits and other mitochondrial enzymes, cause lipid membrane peroxidation activating apoptotic pathways, and even affect mtDNA maintenance resulting into accumulation of mtDNA mutations and multiple deletions. Altogether, these effects can lead to energy failure and eventually cell death [1, 2, 30, 31].
Besides abnormalities in the mitochondrial respiratory activity, other factors may play a role in determining mitochondrial dysfunction, in particular regarding their structure and dynamics within the cell. Mitochondria may indeed exist in two different states: as discrete organelles or as part of an interconnected network [24, 25]. The transition between these two phases depends on opposite processes called fusion and fission, whose regulation is under control of a complex protein machinery, above all the OPA1-gene product, a pro-fusion GTPase, and its cognate partners Mitofusin 1 and 2, and the pro-fission counterpart DRP1 [1, 2, 24, 25]. The correct transport and distribution of mitochondria within the cell sectors where their energy production is most needed, depends on the interaction with citoskeletal proteins, like kinesin and dynein, and any abnormalities in this interplay may cause metabolic imbalance ultimately leading to neurodegeneration [1, 2, 32].
Mitochondrial dysfunction may result from either inherited conditions or by acquired mechanisms. Mitochondrial genetics is peculiar, since any organelle carries multiple copies of its own circular 16,569 bp long DNA (mtDNA), which contains a total of 37 genes, 13 encoding for subunits of the OXPHOS complexes and 24 necessary to the local protein synthesis (2 rRNAs and 22 tRNAs) [1, 2, 30, 31]. All mitochondria are inherited exclusively through the female germline, thus mtDNA mutations are only transmitted along the maternal lineage. Given the large number of mtDNA copies within the same cell, they may carry the same sequence, a condition called homoplasmy, or one or more variant sequences may coexist due to nucleotide changes or rearrangements present at various loads, which configure the conditions defined, respectively, as heteroplasmy or poliplasmy [1, 2, 30, 31]. Despite the genetic independence of nuclear and mitochondrial genomes, the nuclear genome (nDNA) is essential for the correct maintenance and expression of mtDNA, thus mendelian-transmitted mutations of genes encoding for proteins involved in mtDNA replication, transcription and translation, may be responsible for a mitochondrial phenotype, including downstream secondary effects leading to mtDNA instability, resulting in quantitative (depletion) or qualitative (somatic mutations or deletions) defects [1, 2, 30, 31].
Several endogenous and exogenous factors may also interact determining acquired mitochondrial dysfunction [33, 34]. Recently, tobacco smoking has been experimentally proven to cause mitochondrial toxicity and depressive effect on mitochondrial biogenesis in cells from LHON patients and unaffected mutation carriers [35], representing a clear risk factor for developing optic atrophy in patients carrying mtDNA mutations [36,37,38]. Less clear is the impact of alcohol consumption in LHON [36], as it appears to be more relevant than direct toxicity. Alcohol-related nutrient malabsorption via gut irritation and liver dysfunction can possibly lead to vitamin/cofactor deficiency [33, 39]. In fact, deficiencies in Vitamin B12, B1, B2 and folic acid have been implicated as a cause of acquired mitochondrial optic neuropathy [33, 34, 40]. These vitamins act as cofactors to various enzymes in the production of metabolic intermediates—tetrahydrofolate, methionine, homocysteine, cystathionine, cysteine and glutathione—whose malfunctioning results in increased ROS and impinges on mitochondrial function.
Due to similarities between bacteria and mitochondria, several antibiotics such as linezolid, chloramphenicol, fluoroquinolones and macrolides inhibit expression of mitochondrial proteins affecting mitochondrial metabolism in human cells [41, 42]. Nucleoside reverse transcriptase inhibitors, anti-retroviral agents with high affinity for the viral DNA polymerase also cause mitochondrial toxicity by cross-inhibition of the structurally similar human mtDNA polymerase ɤ, responsible for mtDNA replication. This leads to depletion of mtDNA copies and clonal expansion of mutant mtDNA, depressing mitochondrial protein translation and ultimately leading to mitochondrial dysfunction [43, 44].
Other endogenous endocrine mechanisms may directly influence mitochondrial functions like oestrogens, which may play a protective role, thus modifying the severity of mitochondrial defects [45]. By taking advantage of the cytoplasmic transmitochondrial hybrid (cybrid) cell model [46], it has been demonstrated that oestrogens reduce ROS production and limit apoptotic cell death by activating mitochondrial biogenesis in cells carrying LHON mutations, improving the metabolic defect [47]. Activation of mitochondrial biogenesis is a well-known compensatory strategy in mitochondrial diseases [48], particularly well documented in LHON where the unaffected mutation carriers have the highest mtDNA copy number as compared to affected individuals and controls, strongly suggesting that mitochondrial biogenesis modulates LHON penetrance [49].
Mitochondrial homeostasis in cells is enacted by different mechanisms, which may compensate for mitochondrial dysfunction. Unlike nuclear DNA, mtDNA replicates continuously within the cell, adjusting the total mtDNA cellular content to the bioenergetic requirement, thus increasing the mass and number of new mitochondria in a process called mitobiogenesis [50]. Balancing mitobiogenesis, the opposite process, named mitophagy, removes the damaged mitochondria by selective autophagocytosis, which may hamper cell survival [50]. Recent studies investigated mitophagy in LHON, reaching contradictory results; mtDNA LHON mutations seemed to impair mitophagy and turnover in one study, suggesting that pharmacological activation of mitophagy could be a potential therapeutic strategy [51], whereas the opposite scenario was previously reported by another study [52].
2.2 Retinal Ganglion Cell Degeneration
Mitochondrial dysfunction very frequently leads to neurodegeneration selectively targeted to RGCs, whose role is to convey the elaborated transduction of light from the photoreceptors into action potentials via bipolar cells, ultimately to the occipital cortex for vision forming. The axons arising from RGCs run together in the retinal nerve fibre layer (RNFL), then converge in the nasal part of the retina to form the optic nerve head. At this level, about 1.2 million axons pass through the fixed space of the lamina cribrosa, an anatomic “chokepoint”, and exit the eye at its posterior pole organized in bundles forming the optic nerve, which projects to the brain. The RNFL component collecting the axons from the macular region, responsible for central vision, is called the papillomacular bundle [1, 2, 6,7,8,9, 53].
To fully understand the selective vulnerability to energy failure of RGCs and their axons we need to take into account two features: myelination and calibre. Macular RGCs in particular, the most abundant smaller P-type RGCs originating the papillomacular bundle, present a narrower calibre axon, which is not myelinated to avoid blocking the light and allowing retina transparency, instrumental to photoreception. The lack of myelination of these axons implies the absence of saltatory conduction of action potentials, thus requiring higher energy demand, which is supplied by an uneven axonal distribution of mitochondria, preferentially clustering within the unmyelinated retinal and prelaminar sectors, abruptly decreasing in number posteriorly to the lamina cribrosa where myelination begins. Thus, smaller axons comprising the papillomacular bundle are the most vulnerable and represent the primary target of neurodegeneration [1, 2, 53, 54]. Overall, these anatomo-metabolic peculiarities explain why all RGCs and their optic nerve-forming axons have an increased susceptibility to energy depletion and ROS accumulation, both occurring in hereditary and acquired mitochondrial dysfunction [1, 2, 6,7,8,9, 53, 54].
3 Clinical, Genetic and Biochemical Findings in Mitochondrial Optic Neuropathies
3.1 Leber's Hereditary Optic Neuropathy (LHON)
LHON is the most frequent primary mtDNA disorder with a prevalence of 1 in 31,000–50,000 in Northern Europe [55,56,57]. Since the seminal description by Wallace in 1988 [58], currently three common mtDNA missense point mutations are found in over 90% of patients, with some additional rare mutations in the remaining 10%, all invariably affecting subunits of complex I [1, 2, 4]. Complex I (NADH: ubiquinone oxidoreductase), whose function is specifically targeted in LHON, is the largest respiratory complex of the OXPHOS system, resulting from the assembly of about 35–37 nDNA-encoded subunits and 7 mtDNA-encoded subunits. Complex I is the first site of the respiratory chain and transfers electrons from nicotinamide adenine dinucleotide (NADH) to coenzyme Q (ubiquinone, CoQ), generating ubiquinol (CoQH2), which shuttles two electrons to complex III (ubiquinol: ferricytochrome c oxidoreductase). The energy released by electron transfer is coupled to proton translocations, which contribute the generation and maintenance of membrane potential [30, 31]. LHON mutations variably affect Complex I, ultimately resulting in reduced efficiency in ATP synthesis and excessive generation of ROS [1, 2, 6,7,8,9].
Peculiar features of LHON are incomplete penetrance and gender bias [1, 2, 59]. LHON mutations found in the large majority of maternal lineages are homoplasmic and only in a few families the pathogenic mutation may be a recent event, thus heteroplasmic. Overall, in the homoplasmic families only about 50% of the male mutation carriers become affected, and only 10% of the females, highlighting the incomplete penetrance and the male prevalence. Thus, the LHON primary mutation is a necessary but not sufficient factor to developing the disease [60]. It is also well established that recurrent mtDNA polymorphic variants clustered in population-related haplogroups are associated with LHON. In particular, the mtDNA haplogroup J (the sub-haplogroups J1c and J2b) increase the penetrance of LHON mutations, respectively [61,62,63]. It has been reported only recently that rare combinations of individually non-pathogenic missense mtDNA variants may synergistically impact mitochondrial function leading to low-penetrance LHON [64]. Much less understood remains the real contribution of nDNA-modifying variants to LHON penetrance, as reported in small subgroups of individual pedigrees [65, 66].
Male prevalence also remains poorly understood. A long-standing hypothesis proposed the contribution of modifying variants in a gene on chromosome X, under the assumption of a skewed inactivation for affected females [67], but the long series of studies performed to date remained inconclusive and contradictory [68,69,70,71,72]. One exception is a variant (c.157C>T, p. Arg53Trp) in the PRICKLE3 gene on chromosome X, encoding a mitochondrial protein linked to biogenesis of ATPase, which was found to segregate with affected individuals in three Chinese families [66]. As previously mentioned, a different view is provided by the hormonal gender differences, mainly centred on the metabolic regulations of oestrogens directly influencing mitochondria [45], with the experimental evidence that oestrogens increase mitobiogenesis and survival in LHON cell models [47]. As far it concerns how different LHON disease expression is between males and females, there is no systematic study yet, except for the notion that penetrance in females is lowest for the m.14484T>C/MTND6 mutation and highest for the m.3460G>A/MTND1, and the age at disease onset in females is on average later than males, as reported in some large case series [1, 2, 73]. This latter observation may relate to a subset of female LHON patients developing the disease after menopause or delivery [38].
In addition to genetics, environmental factors may also contribute to trigger LHON, in particular tobacco smoking and alcohol drinking are validated risk factors for LHON [36,37,38]. Based on this, it has been proposed [38] and debated [74] that LHON patients may be distinguished in pure or predominantly genetic-driven cases (LHON type I), with classical disease onset at young age between 15 and 30 years, and cases strongly associated with an environmental trigger (LHON type II), with a delayed onset over 35–40 years of age.
The classical clinical presentation of LHON is a subacute, painless loss of central vision in one eye, followed by the involvement of the fellow eye within days/weeks to months. In about 25% of cases, there is simultaneous involvement of both eyes at first presentation. The reduction of visual acuity (VA) associated with impaired colour perception and central scotoma at visual field (VF) testing, hallmark the preferential degeneration of macular RGCs serving the papillomacular bundle. Fundus findings in the asymptomatic mutation carriers and during the early subacute stage include circumpapillary telangiectatic microangiopathy and swelling of the RNFL without leakage at fluorangiography (pseudoedema) [1,2,3]. As the disease progresses, rapid axonal loss leads first to temporal and eventually to diffuse optic atrophy, as the disease reaches the chronic stage at about 1 year after onset [1,2,3, 75]. Optical coherence tomography (OCT) allowed to quantitatively measure the RNFL and RGC changes at the different disease stages, identifying a specific pattern of the natural history of the disease [76,77,78]. The visual prognosis in LHON is usually severe, with most patients progressing to very poor visual acuity associated with a dense central or cecocentral scotoma [1,2,3, 75]. A very recent survey revisiting all available published data on natural history of LHON associated with m.11778G>A/MTND4 mutation, provides a rate of spontaneous recovery of about 11% in patients aged ≥ 15 years [79].
The variability of clinical expression and different propensity to spontaneous visual recovery among LHON patients are key elements to consider while developing and evaluating the efficacy of new therapies. The natural history of the disease is characterised by some early signs signalling the conversion from unaffected to affected [77, 78, 80], followed by a rapid drop in visual acuity that reaches the nadir usually within 4–6 weeks from onset, with quite a variability among patients. After the nadir, some degree of spontaneous visual recovery, due to reduction of the scotoma and/or its fenestrations (islands of vision within the central scotoma), is usually observed over the following years before reaching a plateau. The final outcome of VA may differ from patient to patient. Positive prognostic factors for visual improvement are the m.14484T>C/MTND6 mutation and an early age at onset (< 15 years) for all mutations [1,2,3, 75,76,77,78, 80].
Timing in LHON is a crucial aspect to consider while addressing medical management and assess effectiveness of therapies [3, 21]. An international consensus statement defined three clinical stages of LHON based on the time from onset of visual loss: subacute (less than 6 months), dynamic (6–12 months) and chronic (> 12 months) [75]. Experts strongly agreed that subacute and dynamic stages should be the target for early treatment to preventing further fibers and vision loss. The “sooner the better” paradigm is, thus, key in clinical practice, but unfortunately it is not unusual to have significant diagnostic delays. Concerns are raised about treatment of known mutation carriers who have not yet developed the disease, as there is no available evidence for doing this. The current rule remains not to treat until a clear onset occurs, as demonstrated by VA drop, visual field defect and changes in the fundus appearance. Furthermore, OCT now makes it possible to recognise hallmark changes in the very early stages of onset, even without clinical symptoms, opening new perspectives for future early interventions in LHON [77, 78]. In the future, the possibility of administering a therapy in LHON carriers may be based on a possible “conversion risk score”, which will eventually offer the chance of preventing the onset of the disease.
3.2 OPA1-related Dominant Optic Atrophy (DOA)
DOA has an estimated prevalence of 1 in 25,000 and is currently considered the most common mitochondrial inherited optic neuropathy [1, 2, 81, 82]. About 60–70% of DOA cases are caused by mutations in the optic atrophy 1 gene or OPA1 [83, 84], a nuclear gene that encodes a transmembrane dynamin-related GTPase protein anchored by trans-membrane domain within the IMM and a key player of the mitochondrial dynamics machinery [1, 2, 81, 82, 85, 86]. OPA1-DOA is an autosomal inherited disease and its phenotypic spectrum may be highly variable even within the same family, as also DOA, like LHON, is characterised by incomplete penetrance [1, 2, 82, 85, 86]. The OPA1 gene is localised on chromosome 3q28, includes 31 exons, and the alternative splicing of exons 4, 4b and 5b generates 8 different mRNA variants coding 8 isoforms differently expressed across human tissues [85, 86]. To date, the OPA1-dedicated database (https://opa1.mitodyn.org/) has listed a total of 864 OPA1 gene variants of which over 70% are considered pathogenic variants, and two-thirds of them are located in the dynamin and GTPase protein domains [82, 85, 86]. Most OPA1 mutations are substitutions (64%) or deletions (27%), whereas only a few duplications (5%), insertions (1.4%) and in/del (0.9%) mutations have been annotated up to date. Mutations are mostly family-specific, but some are recurrent. In about 50% of cases, the pathogenic mutations introduce a premature stop codon, leading to the consequent truncation of the open reading frame, which undergoes mRNA decay, ultimately determining the loss of function of the mutant allele. Thus, in these cases, haploinsufficiency is the main disease pathological mechanism [1, 2, 81,82,83,84,85,86,87].
Dominant optic atrophy (DOA) typically begins in the first two decades of life with bilateral visual impairment characterised by temporal disc pallor at fundus examination, centrocecal scotoma, colour vision defect (especially blue-yellow axis) and temporal RNFL thinning at OCT. Its severity is highly variable between families and even amongst individuals belonging to the same family. The natural history is described as a relentless, frequently stable for a long time or slowly progressive optic nerve atrophy, without evidence of spontaneous recovery of vision [1, 2, 82, 88].
Since 2003, the association of optic atrophy with sensorineural deafness or a complex syndrome has been noticed with the specific R445H missense mutation affecting the GTPase domain of OPA1 (numbering refers to isoform 1, NM_015560.2, NP_056375.2) [89, 90]. Subsequently, a multisystemic condition was described in 2008, variously named as “DOA plus” or “DOA+ syndrome” or “OPA1-plus” characterised by optic atrophy, sensorineural deafness, ataxia, chronic progressive external ophthalmoplegia (CPEO) with mitochondrial myopathy, and peripheral neuropathy [91,92,93], but also by spastic paraplegia and multiple sclerosis-like syndrome [93, 94], Behr-like syndrome [95], and syndromic Parkinsonism and dementia [96, 97]. Most patients with “DOA plus” carry missense variants affecting the GTPase domain rather than truncating mutations of OPA1, suggesting that a dominant negative effect might play a role in the pathogenesis of the “plus” phenotype [91,92,93]. Furthermore, besides monoallelic dominant mutations, there has been an increasing recognition of biallelic mutations associated with complex and severe phenotypes [98,99,100,101]. Moreover, compound heterozygous or homozygous OPA1 mutations have been identified in patients with optic atrophy spectrum of complex and severe phenotypes, including Behr syndrome and Leigh-like encephalopathy, highlighting the possibility of truly recessive cases [99,100,101,102,103].
It is now clear that mitochondrial fusion is essential for neuronal growth, function, maintenance and survival, and disruption of mitochondrial function and dynamics has been clearly associated with the OPA1-related optic neuropathy [1, 2, 85, 86]. In fact, DOA affects, as LHON, primarily RGCs within the papillomacular bundle [1, 2, 81,82,83,84,85,86, 104]. Many mechanisms have been proposed to explain DOA-specific RGC loss, including impaired mitochondrial fusion and disruption of mitochondrial cristae morphology, in turn compromising the stability of respiratory complexes, resulting in increased ROS levels and decreased ATP synthesis [105, 106]. Furthermore, it has been shown that OPA1 interacts directly with complexes I, II and III in the IMM and contributes to their assembly and stability. Consequently, the fragmentation of the mitochondrial network results in the release of pro-apoptotic factors, which ultimately trigger irreversible cell death [107].
Despite the growing knowledge on OPA1 functions and related cellular pathways, an effective treatment for DOA is still missing [85, 86]. However, active ongoing progress and encouraging results come from in vitro and pre-clinical studies paving the way to therapeutic approaches to successfully target mitochondrial dysfunction in DOA patients.
3.3 Other Mitochondrial Optic Neuropathies
Clinically phenocopies of LHON or DOA have been described in association with mutations in other nuclear genes, either recessive such as NDUFS2 [108], TMEM126A [109, 110], RNT4IP1 [111, 112], SLC25A46 [113], ACO2 [114], YME1L1 [115], or dominant as AFG3L2 and SPG7 [116,117,118], leading to isolated or syndromic HONs. Also, variable degrees of optic atrophy may be present along with other neurological and extra-neurological symptoms in other mtDNA-based mitochondrial encephalomyopathies, such as mitochondrial encephalopathy with ragged red fibres (MERRF), Leigh syndrome and mitochondrial encephalopathy with lactic acid and stroke-like episodes (MELAS) [1, 2, 6].
4 Therapeutic Options in Mitochondrial Optic Neuropathies
4.1 Antioxidants
Increase in oxidative stress has been invoked as a major consequence of respiratory chain dysfunction, consequently, several antioxidants, bot endogenous molecules and synthetic compounds, have been tested to limit or counterbalance the accumulation of ROS [119]. Coenzyme Q10 (CoQ10) or ubiquinone represents the prototype of endogenous redox agent, shuttling electrons from complexes I and II and those resulting from the oxidation of fatty acids and amino acids, to complex III of the mitochondrial respiratory chain. In doing so, reduced CoQ10 has also antioxidant properties, neutralising ROS. Unfortunately, CoQ10 has a long lipophilic tail that reduces its bioavailability when administered orally and interferes with crossing of membranes [120]. A few randomised controlled clinical trials have tested CoQ10 in mitochondrial diseases, generally with small mitochondrial cohorts characterised by heterogeneous phenotypes, showing some improvement in metabolic parameters, although not clinically significant [121]. Anecdotal use of CoQ10 has also been reported in LHON [122]; nevertheless, it is still widely used in mitochondrial diseases, in association with other nutritional supplements such as cyanocobalamin, folic acid, ascorbic acid, alfa-lipoic acid, acetyl-l-carnitine, creatine monohydrate, riboflavin, the so-called mitochondrial cocktail [123]. Rodriguez and colleagues evaluated the effect of a combination therapy consisting in creatine monohydrate, CoQ10 and lipoic acid, in 16 patients with mitochondrial cytopathies (including 2 patients with LHON and 4 with MELAS), using a randomised, double-blind, placebo-controlled, crossover study design [124]. The mitochondrial cocktail ameliorated surrogate markers of cellular energy dysfunction reducing resting plasmatic lactate and preserving muscular strength [124].
Curcumin, a component of turmeric derived from the root Curcumin longa, and KH176, a ROS-redox modulator [125], are other molecules with antioxidant properties. Curcumin has been tested in a randomised, placebo-controlled trial in LHON patients, even if the results have never been published (www.clinicaltrials.gov NCT00528151).
4.1.1 Idebenone
4.1.1.1 LHON
The most studied molecule in HONs and the only one approved for LHON is idebenone [10]. Idebenone is a synthetic analogue of CoQ10 that shares its antioxidant properties, but has a shorter and less lipophilic tail allowing easier penetration through mitochondrial membranes and blood–brain barrier. Moreover, idebenone activation from its oxidised form occurs in the cytoplasm, independently of mitochondrial function, as it gets activated by the flavoprotein NAD(P)H:quinone oxidoreductase (NQO1) that catalyses a complete two-electron reduction [126]. This step seems crucial as the oxidised form of idebenone has the adverse and potentially toxic property to inhibit complex I [127,128,129]. Thus, the conversion into the reduced form allows idebenone to shuttle the electrons directly to complex III [130], bypassing the complex I dysfunction occurring in LHON, establishing the rational for its use, ultimately preserving energy production. Preclinical studies confirmed a cell-line–specific increase in ATP production and reduced ROS levels in fibroblasts of LHON patients [131] and prevention of RGC loss in a LHON-rotenone–induced mouse model [132]. Orally administered idebenone showed a good bioavailability and linear dose-dependent pharmacokinetics with a significant distribution in the brain and the eye; it is metabolised mainly in the liver and gut, with less than 1% reaching the systemic circulation, and eliminated through urine [133].
The efficacy of idebenone in LHON patients was assessed in a randomised, double-blinded, placebo-controlled study (RHODOS www.clinicaltrials.gov NCT00747487) in 2011. Klopstock and co-authors recruited 82 LHON patients carrying one of the three common mutations within 5 years from onset of vision loss [11]. Patients were randomised and received idebenone 300 mg three times per day or placebo for 24 weeks, with a 2:1 ratio in favour of idebenone. The primary endpoint was best recovery between baseline and week 24. The secondary endpoints were as follows: change in best-corrected visual acuity (BCVA) measured in logMAR using the Early Treatment Diabetic Retinopathy Study (ETDRS) eye chart, and changes in VA of the best eye at baseline and for both eyes independently. Results showed that idebenone was safe and well tolerated. Although the primary end-point did not reach statistical significance, all secondary end-points showed a consistent positive trend in treated patients compared to the placebo group [11], whose VA kept deteriorating as expected by natural history [79]. After the end of the RHODOS study, 58 patients (39 originally randomised to idebenone and 19 to placebo) were enrolled into a follow-up study (RHODOS–OFU www.clinicaltrials.gov NCT01421381), for reassessment of their VA 30 months after discontinuation of idebenone. The primary endpoint was the change in BCVA compared with baseline and week 24 of RHODOS. Results showed that the idebenone group continued to improve even after discontinuation of the drug, while VA did not change for patients in the placebo group [134].
Parallel to the RHODOS study, Carelli and colleagues in 2011 published a retrospective study on 103 LHON patients where 44 patients treated with idebenone within 1 year after visual loss in the second eye at varying doses and a mean duration of 41 months, were compared to 59 untreated patients [12]. Clinically relevant recovery (CRR) was defined as gain of at least two lines on VA charts or a change from “off chart” to “on chart”. In their cohort, the proportion of patients experiencing recovery was higher in the treated group; moreover, earlier visual improvement was associated with prompt start and longer duration of therapy. Other factors positively influencing the rate of recovery were a younger age and the presence of the m.14484T>C/MTND6 mutation, in both treated and untreated patients [12].
In 2015 idebenone (Raxone®) was granted market authorisation in the European Union for treatment of LHON under exceptional circumstances by European Medicine Agency (EMA) in adolescents and adult patients at 900 mg/day divided into 3 doses (https://www.ema.europa.eu/en/medicines/human/EPAR/raxone#authorisation-details-section). To address the still open issues on idebenone therapy optimal timing, duration and target population, an international consensus with experts from Europe and North America was held in Italy in 2016 [75]. Experts agreed that idebenone treatment at 900 mg/day should start as soon as possible in the acute stage (within a year from visual loss onset), while there was not yet sufficient evidence to recommend treatment in chronic patients 1 year after the second eye involvement. In subacute/dynamic patients, treatment at 900 mg/day should be continued for at least 1 year to assess a therapeutic response, evaluated by improvement in VA on ETDRS charts and in mean deviation of automated visual field tests, or until a plateau is reached. Discontinuation should be considered after 1 year once the plateau is reached, or no improvements are observed. Regarding unaffected relatives of LHON patients, currently, treatment with idebenone is not recommended, but lifestyle counselling, especially about quitting smoke and alcohol, is highly suggested [75].
Since idebenone commercial launch, two other multicentre studies were started. The first is an open-label interventional Phase IV study designed to assess the efficacy and safety of idebenone in the long-term treatment of LHON patients (LEROS www.clinicaltrials.gov NCT02774005), having as primary outcome the proportion of eyes with CRR of VA from baseline at 12 months or maintenance of VA better than 1.0 logMAR at Month 12, compared to a matched external natural history control group. The second is an observational, prospective, post-authorisation safety study in patients with LHON treated with idebenone (PAROS www.clinicaltrials.gov NCT02771379), having as primary outcome the assessment of long-term safety profile of idebenone and as secondary outcome its long-term effectiveness, assessed by measuring changes in VA, VF, colour contrast sensitivity and RNFL. Estimated final data collection date for both studies is February 2021.
Moreover, long-term follow-up data of LHON patients treated with idebenone have been collected through an Expanded Access Programme (https://clinicaltrials.gov NCT04381091). This was instrumental for EMA approval and, just recently, the final results were published [135]. Based on these results, CRR was observed in 46.0% of LHON-treated patients, a figure closely matching that previously reported by Carelli et al [12]. Furthermore, time to initial observation in patients with a CRR varied between 2.5 and 26.5 months, with a mean of 9.5 months. These new observations may prompt a consensus to reconvene to discuss the length of time idebenone should be administered to maximise its effect.
The main results and clinical endpoints of the clinical trials in LHON are summarised in Table 1.
4.1.1.2 DOA
The off-label use of idebenone as therapeutic strategy to correct mitochondrial dysfunction and protect RGCs has also been evaluated for OPA1-DOA, as several common features join LHON and DOA [13, 14]. First, both diseases have a similar pattern of axonal neurodegeneration in the optic nerve [1, 2]. Second, the hallmark of LHON is an obvious primary dysfunction of complex I [136], which ultimately also affects OPA1-DOA as evidenced by defective OXPHOS with reduced ATP synthesis when driven by complex I substrates [106]. Third, there is a documented propensity to a chronic increase of ROS production in both [1, 2, 106, 137,138,139].
Thus, encouraging results were initially reported from a small pilot open trial on seven DOA patients with OPA1 haploinsufficiency heterozygous mutations, who were treated for at least 1 year with idebenone [13]. Five of these patients showed improvements in visual function (colour vision, reduction of central scotoma and increased VA) [13]. Since this pilot study, a larger group of DOA patients carrying either OPA1 mutations leading to haploinsufficiency or missense point mutations has been treated with idebenone [14]. These patients received idebenone under the Italian regulation for off-label drug administration with dosage ranging from: 135 to 675 mg/day. Most patients were treated or started treatment before idebenone approval for LHON in 2015. Thus, the dosage in individual cases was modulated to avoid the occurrence of specific clinical side effects or blood exam abnormalities (headache and insomnia, weight changes, age, neutropenia, alteration of liver function indices, hypercholesterolemia, gastrointestinal disturbances) by adjusting to maximal dosage without side effects, as required for off-label drug administration. The key finding of this retrospective comparison between idebenone-treated DOA patients and those untreated considered as the control group, was that treated patients displayed significant occurrence of stabilisation/recovery of VA, four times higher than untreated [14].
Despite the major limitations due to the retrospective nature of the study, these results are encouraging and supportive for the use of idebenone as therapy that may positively change the natural history of DOA posing the basis for future double-blind, placebo-controlled and randomised trials to confirm these observations. In addition, a randomised, placebo-controlled trial of idebenone at 2000 mg/kg/day was carried out in 56 Opa1 mutant mice [140]. As the Opa1 mutant mice showed RGC dendropathy [141], this hallmark was taken as measurable endpoint to assess idebenone effect, besides standard assessment of visual function in mice (optokinetic response). The results of this trial highlighted only a modest effect on Opa1 mutant animals, as revealed by improvement in secondary dendritic length and territory, as well as in optokinetic response, whereas idebenone-treated wild-type mice showed shrinkage in total dendritic length and impaired visual function, raising some concerns on idebenone mechanistic effects [140].
4.1.2 EPI-743
Another quinone analogue is EPI-743, a product of hydrolysis of alpha-tocotrienol (Vitamin E). EPI-743 is orally bioavailable and easily crosses the blood–brain barrier to reach the central nervous system where acts as antioxidant by targeting oxidoreductase enzymes and replenishing glutathione pools [142]. Its efficacy has been evaluated in a small open-label trial (www.clinicaltrials.gov NCT02300753) with five genetically confirmed LHON patients treated with EPI-743 within 90 days from disease onset at a dosage of 100–400 mg/day for at least 1 year. EPI-743 arrested disease progression and reversed vision loss in all but 1 of the 5 patients [15]. A larger set of LHON patients have been treated in the same open-label trial and a final report on the long-term results is expected.
A recent 6-month placebo-controlled trial with EPI-743 followed by an 18-month open-label trial has been carried out on 63 patients with Friedreich’s ataxia [143]. Friedreich’s Ataxia is a recessive complex multisystemic syndrome mostly due to triplet expansions of the frataxin gene, even if, in a minority of cases, missense mutations may combine with triplet expansion by compound heterozygosity. Amongst other neurological symptoms, optic atrophy is observed in about two-thirds of patients [144]. The vision end-point of the trial included low-contrast VA and EPI-743 failed to demonstrate any significant visual benefit; nevertheless, a statistically significant improvement in neurological function and disease progression was observed after 24 months in treated patients compared with the natural history cohort.
4.1.3 Elamipretide
A molecule with different structure and mechanism of action is elamipretide or MTP-131, a tetrapeptide that binds cardiolipin, a phospholipid exclusively targeted to the IMM [145]. Cardiolipin, unlike classical phospholipids with two hydrophobic tails, has two phosphate groups and four acyl side chains, resulting in a conic shape that ensures membrane fluidity and bending, essential for cristae formation in the IMM [146] and OXPHOS functioning, allowing the arrangement of respiratory complexes into super-complexes [147].
Elamipretide, by binding cardiolipin via electrostatic and hydrophobic interactions, prevents conversion of cytochrome c into a peroxidase, preserving its function as simple electron carrier and promoting OXPHOS. In preclinical studies, elamipretide was shown to protect mitochondrial cristae, increase ATP synthesis and reduce ROS production, regardless the type of mitochondrial abnormality [145]. Clinical efficacy has been tested since 2010 with the commercial formulation named Bendavia in different diseases sharing metabolic failure as a common pathogenesis, including heart failure, acute kidney injury, muscular atrophy and neurodegenerative disorders [148].
In 2016, a prospective, randomised, double-masked study with a topical ophthalmic solution of elamipretide in subjects with LHON harbouring the m.11778G>A/MTND4 mutation was carried out in the USA (www.clinicaltrials.gov NCT02693119). In total 12 LHON patients with established vision loss for > 1 year but < 10 years were recruited and treated for 52 weeks. No difference in BCVA was observed, but because of a trend towards improvement, all 12 patients completed an open-label extension with bilateral treatment for a total of at least 84 weeks. In April 2019 a press release stated that the treated patients presented a significant improvement in BCVA, visual field mean deviation, contrast sensitivity and colour discrimination compared to baseline (https://www.prnewswire.com/news-releases/stealth-biotherapeutics-announces-positive-results-for-elamipretide-in-ophthalmic-conditions-300841246.html), but a full scientific, peer-reviewed communication is still missing.
4.2 Antiapoptotic Drugs
Mitochondria play a key role in the intrinsic pathway of apoptosis due to a series of intracellular signals that eventually lead to permeabilisation of the IMM through the opening of a permeability transition pore. This change in mitochondrial permeability allows the release of mitochondrial proteins, including cytochrome c and other activators of apoptosis (apoptosis induction factor, endonuclease G, Smac/DIABLO), into the cytosol, thus activating the cascade of specific enzymes (caspases) responsible for cell degradation and self-destruction [26].
Molecules preventing this cascade of events in RGCs could represent a therapeutic strategy in HONs, under the assumption that the common final pathway for RGC death is mainly apoptotic in nature [1, 2, 5,6,7,8,9]. Cyclosporine A, an inhibitor of the opening of the mitochondrial permeability transition pore, has been tested in 5 LHON patients with unilateral involvement within 6 months from onset at a dosage of 2.5 mg/kg/day for a year (www.clinicaltrials.gov NCT02176733). The primary endpoint was BCVA in the unaffected eye, whereas the secondary endpoints were BCVA in the affected eye, VF, the thickness of GCC and RNFL layers in both eyes. Unfortunately, cyclosporine A failed to prevent the second-eye involvement in these patients [149]. Other molecules targeting different signals along the apoptotic pathways remain good candidates for future consideration.
4.3 Photobiomodulation
Near-infrared light-emitting diode arrays (NIR-LED) is a therapy approach that has been shown to attenuate optic nerve degeneration after acute injury by the mitochondrial toxin, formic acid, generated in the course of methanol intoxication in a rat model [150]. The therapeutic effect of NIR-LED is proposed to be exerted by increasing the amount of cytochrome c oxidase and upregulating expression of genes involved in energy production and antioxidant protection [150]. As the Food and Drug Administration (FDA) has approved this approach for use in humans, an interventional study in LHON to determine effects of short-term phototherapy on pattern electroretinography (PERG) and Photopic Negative Response was set, but failed to recruit a sufficient number of patients (www.clinicaltrials.gov NCT01389817).
4.4 Gene Therapy
Since we are dealing with inherited conditions, the possibility to correct the nDNA or mtDNA defect directly at the molecular level remains the ultimate goal in the treatment of HONs. Importantly, the eye represents a suitable target for gene therapy, as it is easily accessible by intravitreal injection of appropriate viral vectors to deliver the corrected gene to target cells [151]. Furthermore, retinal structure and visual function are measured with multiple and sophisticated technological tools, which may provide convenient endpoints for clinical trials. Finally, at least for LHON and DOA, the natural disease history is sufficiently defined to allow for meaningful clinical trial design [1,2,3, 8, 9, 11, 77, 78, 80, 81].
4.4.1 Gene Therapy Trials in LHON
Gene therapy for disorders associated with mtDNA mutations faces a major challenge represented by the presence of a double membrane to cross in order to deliver the corrected gene inside the mitochondria. Moreover, mtDNA is a multicopy genome with complex genetic rules governing its expression if mutated, in turn determining the genotype-phenotype correlation [1, 2, 8, 9]. Despite some reports exploring the feasibility of direct mitochondrial delivery of nucleic acids [152, 153], this approach remains recognised by most investigators in the field as an unsolved challenge hampering the canonical gene therapy strategies adopted to date for nDNA, mostly based on AAV vectors [151]. One possible route that has been proposed and explored is the expression of nucleus-encoded wild-type mRNAs for mitochondrial proteins, their import and translation within mitochondria to ultimately complement the endogenous mtDNA-encoded mutant genes [154]. However, whether RNA, besides DNA, may be truly imported within mitochondria as yet remains under discussion [155]. For this same reason, the option of editing mtDNA by the powerful Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 technology, based on guide RNA molecules needed for a viable CRISPR/Cas9 system, implies their mitochondrial import, which remains controversial [155]. Just recently, one very promising approach for CRISPR-free mtDNA base editing has been presented, which even if still limited in its editing options seems truly convincing that may efficiently work directly on the mitochondrial genome [156].
A strategy alternative to mitochondrial import of nucleic acids, which was first proposed and tested in yeast [157, 158], is the allotopic expression in the nuclear compartment of a recoded wild-type mtDNA-encoded gene, engineered to target the newly-synthetised cytoplasmic protein for import into the mitochondrial matrix, by using a specific mitochondrial targeting sequence (MTS) [159, 160]. By using this strategy, what is imported within mitochondria is the protein and not the nucleic acid. This strategy was first tested for the common mtDNA mutations affecting, respectively, the ATPase6 subunit gene in neuropathy, ataxia, retinitis pigmentosa (NARP) syndrome [161], and the ND4 subunit gene in LHON [20]. However, the efficiency of mitochondrial protein import seems to depend on which of the 13 mtDNA-encoded subunits is tested, as well as by the arrangements of MTS, and the effective assembly of this wild-type protein within the OXPHOS complexes, in competition with the mutant protein still endogenously produced, has been a matter of debate in the scientific community [162,163,164].
For LHON, a transgene with the wild-type ND4 subunit recoded to obey the genetic code of nDNA, provided with a promoter and MTS packaged into an adeno-associated-virus-(AAV) vector, mediating the nuclear allotopic expression of the corrected mtDNA gene, has been produced by at least three groups, in the USA [20], France [165, 166], and China [167], and recently reviewed by Karaarslan [168]. Preclinical studies on cells [20, 165, 166] and non-genetic animal models of LHON [169,170,171,172,173], targeting the m.11778G>A/MTND4 defect, reported a successful expression and mitochondrial import of the wild-type gene, which rescued the damaged RCGs by intravitreal delivery of the viral vector. A drawback in the preclinical experimental design was the lack of a truly genetic mouse model of LHON, which became available only later, after these studies were published [174]. Thus, the LHON murine model has been set by the allotopic expression of a human ND4 subunit protein carrying the LHON mutation to obtain the eye pathology in mouse, and prevent it by pre-administering the human wild-type ND4 [169,170,171,172,173]. From the point of view of regulatory agencies, this strategy had the obvious advantage of providing evidence that the exact transgene designed for humans was effective in animals. However, this kind of animal modelling of LHON remains open to criticisms, as human mtDNA-encoded ND subunits may be evolutionarily poorly compatible with the remaining endogenous complex I encoded by murine mitochondrial and nuclear genomes. In fact, the construction of xeno-mitochondrial cybrids, combining a human nDNA background with mtDNA derived from different types of primates, highlighted the limitations of integrating genomes evolutionarily distant [175]. Despite the sub-optimal experimental design, these pre-clinical investigations rapidly led to approval and establishment of multiple multicentre clinical trials in humans, some of them still ongoing, in the USA, Europe and China (see Table 2).
In the USA, Guy and colleagues assessed the safety and tolerability of an AAV expressing a normal ND4 complementary DNA (scAAV2-P1ND4v2) on an open-label trial (www.clinicaltrials.gov NCT02161380). LHON patients with m.11778G>A/MTND4 mutation were divided into 3 study arms depending on the stage of the disease: more than 12 months (group 1), less than 12 months (group 2), unilateral or presymptomatic (group 3). Escalating doses of scAAV2-P1ND4v2, namely 1.18 × 10e9 vg (low), 5.81 × 10e9 vg (medium) and 1.0 × 10e11vg (higher), were delivered by unilateral intravitreal injections. Clinical testing included BCVA, VF, OCT, PERG, and neuro-ophthalmic examinations. No serious safety problems were observed at low and medium doses in 14 treated patients (6 from group 1, 6 from group 2 and 2 from group 3), with only two patients presenting an asymptomatic uveitis, which resolved spontaneously [176, 177]. For groups 1 and 2, average visual improvement in the injected eye at 12-month evaluation was 0.24 logMAR, significantly higher than the 0.09 logMAR improvement observed in the fellow eyes. Moreover, in acute patients (group 2) the improvement was significantly higher than that observed in a prior natural history study on 44 acute LHON patients with m.11778G>A/MTND4 mutation [178]. In fact, in that study, at 12 months follow-up, 44% of the worst eyes at baseline had a spontaneous improvement of 3 lines or more, which was observed in up to 66% of treated eyes during the gene therapy trial. Conversely, no injected eyes lost 3 lines of VA, while 22% of the worst eyes at baseline in the natural history group did.
At month 12 after injection, average temporal RNFL thickness was not different from baseline in the treated eyes, while it was significantly reduced in the fellow eyes. However, PERG amplitudes worsened more in treated eyes than in fellow eyes [176].
In China, an initial study on 9 LHON patients carrying the m.11778G>A/MTND4 mutation evaluated the efficacy and safety of a recombinant, replication-defective, AAV type 2 containing a modified DNA sequence encoding the wild-type ND4 protein (rAAV2-ND4) administered by monocular intravitreal injection (www.clinicaltrials.gov NCT01267422). The primary outcome was improvement in BCVA; intraocular pressure, VF, OCT, VEP, liver and kidney function, and antibodies against AAV2 were defined as secondary endpoints. At the 9-month-follow up, improvement in BCVA of the injected eyes by at least 0.3 logMAR was observed in 6 patients, as well as VF defect amelioration, while RNFL thickness remained relatively unchanged [179]. Long-term evaluation at 36 months confirmed the efficacy in 6 patients and determined the safety of rAAV2-ND4, since none of the 9 patients experienced serious adverse events [180]. Interestingly, visual function, as documented by BCVA, VF and VEPs, improved in both treated and untreated eyes; age, disease duration and RNFL thickness did not seem to have a significant effect on the visual outcome. The same group conducted two further multicentric, prospective interventional studies with a single intravitreal injection of rAAV2-ND4 with dose 1 × 10^10 vg/0.05 mL (www.clinicaltrials.gov NCT03153293 and NCT03428178). The first included 142 LHON patients with the m.11778G>A/MTND4 mutation, 69 more than 24 months after onset and 73 less than 24 months prior to onset; BCVA and VF were the primary outcomes and the final results are expected soon. Retrospective studies on the injected patients showed that greater VF index, smaller VF mean deviation and higher BCVA at baseline predicted a better VA prognosis [181], and that baseline BCVA and time between onset and treatment are predictors of rapid visual improvement, in both injected and not-injected eyes [182]. To explain the rapid recovery, within days after treatment, it has been suggested that in some patients a portion of RGCs survive in a “dormant” state that can be activated by gene therapy [179]. The second trial is expected to recruit 120 patients: 20 with onset within 3 months, 20 between 3 and 6 months, 20 between 6 and 12 months, 20 between 12 and 24 months, 20 between 24 and 60 months, and 20 over 60 months. Primary outcome measure is again BCVA, whereas secondary outcomes are VF, visual evoked potentials (VEP), ERG, RNFL, liver and kidney function. Results will be compared before and after treatment at 1, 2, 3, 6 and 12 months.
In Europe and the USA, an open-label Phase I/II clinical trial studied rAAV2/2-ND4 (GS010 LUMEVOQ) was conducted on 15 LHON patients carrying the m.11778G>A/MTND4 mutation with intravitreal delivery in the worse-seeing eye (www.clinicaltrials.gov NCT02064569). The results demonstrated the safety and tolerability of intravitreal injection and observed a preservation of RGCs with a functional visual benefit in a proportion of treated eyes, precisely those treated within 2 years after onset, as compared with those treated later than 2 years [183].
Afterwards, two randomised, double-masked, sham-controlled Phase III clinical studies were conducted to evaluate the clinical efficacy of GS010. They compared the efficacy of unilateral GS010 versus sham injection in a total of 76 LHON patients carrying the m.11778G>A/MTND4 mutation. In RESCUE (www.clinicaltrials.gov NCT02652767), enrolled patients had vision loss duration up to 6 months from onset; in REVERSE (www.clinicaltrials.gov NCT02652780) the vision loss was within the interval 6 months from onset to 1 year. The primary endpoint was the difference in efficacy of GS010 in treated eyes compared to sham-treated eyes based on BCVA measured with the ETDRS 48 weeks post-injection. Secondary endpoints compared BCVA of best-seeing eyes that received GS010 to those receiving sham, and worse-seeing treated eyes compared to sham. The company stated that they obtained, both in REVERSE and RESCUE, a clinically meaningful improvement of BCVA from nadir, corresponding to 25 ETDRS letters equivalent, in GS010-treated eyes (https://www.gensight-biologics.com/category/press-releases/). Interestingly, also for these two trials, RESCUE and REVERSE, as for the Chinese trial, the improvement was surprisingly bilateral, leading to a formal failure of the primary endpoint at the 96-week evaluation. The results of the REVERSE trial are currently in press (VC, CLM personal communication).
To facilitate the interpretation of REVERSE and RESCUE results, the company carried out a retrospective, cross-sectional observational study on subjects with LHON (REALITY www.clinicaltrials.gov NCT03295071). Primary aims were to delineate the natural history of visual function decline and structural changes occurring during the disease, and to understand the impact on quality of life and economics. Interim analysis based on the 15 recruited subjects with mutations in ND4 showed a severe and usually irreversible decline in VA, in contrast to the improvement seen in REVERSE and RESCUE patients (https://www.gensight-biologics.com/category/press-releases/). Notably, in these patients, visual improvement was observed bilaterally, both in GS010- and sham-treated eyes, which persisted at the 96-week evaluation.
In order to find a supporting mechanism for the unexpected bilateral visual improvement, a non-clinical study on primates investigated the biodistribution of GS010 in different tissue samples from monkeys unilaterally injected with GS010. Three months after injection, the presence of GS010 DNA was detected and quantified in both the injected and the contralateral eye, indicating that the expression of the therapeutic gene occurs bilaterally even after unilateral injection. Moreover, the viral DNA was also demonstrated in the anterior segment, retina and optic nerve of the noninjected eye as well as in the optic chiasm, suggesting a possible retrograde, trans-chiasmatic transit of the vector from the injected to the contralateral eye (https://www.gensight-biologics.com/category/press-releases/).
Another ongoing multicentre, double-masked, placebo-controlled trial was designed to evaluate the efficacy and safety of bilateral intravitreal injection of GS010 in LHON patients up to 1 year from vision loss onset (REFLECT www.clinicaltrials.gov NCT03293524). In the active arm, patients received a single intravitreal injection with GS010 in both eyes; in the placebo arm, GS010 was administered to the first affected eye, while the fellow eye received a placebo injection. The primary endpoint was change in BCVA (reported in logMAR) from baseline to 1.5-year assessment in the second-affected/not-yet-affected eyes receiving GS010 compared to placebo. The secondary endpoints included: BCVA at 2 years in the second-affected/not-yet-affected eye compared to both placebo and first-affected eyes treated with GS010; other measures were OCT, contrast sensitivity and quality-of-life scales.
5 Stem Cell Therapy
The Stem Cell Ophthalmology Treatment Study (SCOTS) and its subsequent follow-up SCOTS 2, represent the largest stem cell studies for eye diseases currently available (www.clinicaltrials.gov NCT 01920867 and NCT 03011541), including also DOA and LHON patients [184]. This stem cell therapeutic approach is based on autologous transplantation of bone marrow derived mesenchymal stem cells (BMSCs) [185,186,187] for which neuroprotective effects have been previously reported in a variety of animal models of optic nerve damage, including ocular hypertension induced by episcleral vein ligation, ischaemia–reperfusion, optic tract transection and optic nerve crush [187,188,189,190]. BMSCs are the adult population of mesodermal stromal progenitor cells characterised by high proliferative and differentiation capacities, and able to differentiate into neurons and glial cells [191]. Moreover, the BMSCs supply paracrine support to damaged RGCs via the release of platelet-derived growth factor, a neurotrophic agent for which a neuroprotective effect for RGCs has been reported in an in vivo model of ocular hypertensive glaucoma [192].
SCOTS and SCOTS2 are open label, non-randomised studies (www.clinicaltrials.gov NCT 01920867 and NCT 03011541) including different retinal or optic nerve diseases treated with a combination of injections of autologous BMSCs (retrobulbar, sub-tenon, intravenous, intravitreal and intraocular). The patients’ inclusion criteria are: (1) objective, documented damage to the retina or optic nerve unlikely to improve/progressive; BCVA ≤ 20/40 and/or abnormal visual field (in one or both eyes); (2) at least 3 months’ post-surgical treatment intended to treat any ophthalmologic disease and stable; (3) if under current medical therapy for a retinal or optic nerve disease, stability on that treatment and unlikelihood of having visual function improvement; (4) aged > 18 years. SCOTS2 protocol has three arms, and the decision of which one to provide is based on degree of visual loss, disease causing visual loss, any potential treatment risk factors and patient’s medical status and both eyes are treated.
Arm 1 uses stem cell concentrate provided retrobulbar and sub-tenons followed by intravenous injection; Arm 2 uses stem cell concentrate provided retrobulbar, sub-tenons and intravitreal followed by intravenous injection; Arm 3 treats the eye with better VA using Arm 1 or 2, and the eye with more severe VA impairment using a core pars plana vitrectomy followed by subretinal or intra-optic nerve stem cell concentrate. This is followed by intravenous injection. In 5 LHON patients who underwent SCOTS, there were improvements in VA and peripheral vision. Several of the eyes experienced increases in VA, improvements in visual field were noted, but macular and optic nerve RNFL thickness varied independently from vision improvements [193].
In 2019, the first SCOTS report was published on 6 DOA patients, for which the genetic confirmation of the diagnosis was reported in 4/6 [184]. Of these 6 patients, 83.3% experienced visual improvements in both eyes, which was maintained up to 24 months. Ten eyes (83.3%) experienced gains in VA with a median improvement of 2.125 Snellen lines, or approximately 10.63 letters. Two eyes were considered unchanged. Using logMAR, the average improvement was 29.5%. The authors speculated that mitochondrial transfer and neuroprotective exosome secretions from the BMSC could contribute to the visual improvements observed [184]. Some preclinical studies provided evidence that mitochondrial transfer can take place between BMSC and injured tissue with resultant improvement in ATP production allowing for increased cellular survival [194, 195]. These benefits may vary in response to the type of damage detected by the BMSC, and duration of the proximity of BMSC to tissues [184]. However, the scientific basis of MSCs therapy in hereditary optic neuropathies is still under investigation and validation, and the specific mechanism of action underlying the capability of MSCs to rescue RGCs in DOA remains to be clarified and fully demonstrated. Furthermore, the intrinsic weakness of the trial design, lacking a placebo-controlled arm, lowers the interpretability of the results, as, for example, LHON may spontaneously improve [1, 2].
6 Gene Therapy Approaches at Preclinical Stage
6.1 Gene Therapy for DOA or Other Nuclear Encoded HONs
Gene therapy is strategically aimed at complementing the mutant gene by AAV-mediated expression of the wild type allele when haploinsufficiency is at play, or correcting the pathogenic mutations now that sophisticated endonuclease systems such as CRISPR/Cas9 are available [196], thus, allowing disease treatment at molecular level [86, 197]. This could be a particularly good choice for DOA patients carrying OPA1 haploinsufficiency mutations, whereas there are still points needing better understanding when it comes to missense mutations, where a dominant negative effect is assumed [86]. A first issue relative to OPA1-DOA gene therapy is about the appropriate expression level. In fact, excessive overexpression is deleterious [86], whereas only slight OPA1 overexpression may have therapeutic value, as recently investigated in mouse models [86, 198, 199]. Indeed, a mild increase of isoform 1 protein level protects mice from denervation-induced mitochondrial dysfunction and muscular atrophy, heart and brain damage induced by ischaemia-reperfusion injury and Fas-induced liver apoptosis [199]. In addition, the efficient increase of OPA1 level ameliorates the phenotype of Ndufs4−/− and Cox15sm/sm mice, by improving cristae ultrastructure, complexes and respiratory chain super-complex (RCS) organisation and mitochondrial respiration [198]. A second point needing consideration is, of the 8 human OPA1 isoforms, which should be used as the most efficient to rescue the defective clinical phenotype [86]. Besides isoform 1, which has been used in mice, all eight human isoforms, if slightly overexpressed in cells, have comparable capability to recover mtDNA, cristae and energetics, but not the network dynamics [86, 200]. In this regard, it is now crucial to investigate the profile of OPA1 isoforms expression, OPA1 levels and OPA1 long-/short-forms ratio, in wild-type and mutant human RGCs, to fully understand the specific alterations under pathological conditions, and to choose the isoform with the best therapeutic potential [86]. A third and last issue concerns OPA1 missense mutations, and particularly those associated with the “DOA plus” syndromic phenotype that affects multiple tissues [89,90,91,92,93]. Assuming a dominant negative mode of action of these mutations, just increasing OPA1 levels may not be effective and it will be necessary to develop a gene therapy for multiple tissue targets [86], and efficient gene editing may ultimately be the solution. Encouragingly, in recent years, research has witnessed great development and implementation of delivery strategies for different organs, with tissue-specific AAV serotypes [197].
To date, three OPA1 mouse models recapitulating the human pathology are available to evaluate safety and efficacy of gene therapy, harbouring truncating mutations in exon 8 (c.1051C>T), intron 10 (c.1065+5 G>A), and exon 27 (c.2708-2711delTTAG) [201,202,203], and providing important insights into the disease molecular and cellular pathogenesis. The latter mouse model expressing the common c.2708-2711delTTAG human mutation has been exploited for a first published approach of gene therapy for DOA, where a human isoform1 OPA1 full-length cDNA was incorporated and its expression driven by a CMV promoter [22]. The authors reported RGCs protection after mice intravitreal delivery of this transgene carrying the isoform 1, which prevented their degeneration [22].
A recent study further advanced our understanding of RGCs neurodegeneration due to defective OPA1 in a mouse model by showing that aberrant excess of autophagic activity at the axonal hillock depletes RGC axons of mitochondria, ultimately leading to a dying-back type of axonopathy. This was rescued by genetic inhibition of AMPK, of autophagy and mitophagy, restoring axonal mitochondrial content and restoring vision in the mouse [204].
Currently, multiple approaches, as those just mentioned, are at the pre-clinical experimental stage, pending further evidence of their efficiency and safety in humans; thus, gene therapy is not yet available as a clinical treatment option either for OPA1-related DOA patients or other HONs [191].
6.2 Gene Therapy to Promote Heteroplasmic Shift for mtDNA Mutations and mtDNA Gene Editing
A promising approach for gene therapy applied to mtDNA mutations is the strategy aimed at heteroplasmic shift. In mitochondrial medicine, if the pathogenic mtDNA mutation is heteroplasmic, the clinical expression of the disease often depends on the mutant load and the tissue-specific threshold for reaching a biochemical impairment and OXPHOS dysfunction [30, 31]. Consequently, the elimination of some of the mutant mtDNA molecules that allow the wild-type mtDNAs to overcome the loss by active replication just enough to shift the mutant to wild-type mtDNA ratio below the disease threshold, would theoretically be sufficient to rescue the biochemical defect and prevent the disease manifestation. This may apply to the rare LHON cases with a heteroplasmic mutation, which most frequently applies to the m.3460G>A/MTND1 mutation [1, 2].
After an initial pioneering proof of principle based on the use of restriction enzymes engineered for mitochondrial import [205, 206], several DNA-editing enzymes have now been tested, targeted to specific mtDNA mutations, to lower mutant heteroplasmy levels in both cell cultures and mouse models [207, 208].
Mitochondria-targeted restriction endonucleases (mitoREs) are recombinant enzymes expressed in the cellular nucleus or cytosol that reach mitochondria using localisation signals; once internalised, they recognise specific sequences in the mtDNA, induce double-strand break and can selectively cleave one type of mtDNA in a heteroplasmic cell or mouse line. Their application is limited though by the selective recognition of a pathogenic mutation as target site [209].
Another tested approach is based on Zinc-finger nucleases (ZFNs), heterodimers harbouring a DNA-binding domain that recognises longer target sequences, and a cleavage domain that induces the double‐strand break [210].
Transcription activator-like effector nucleases (TALENs) are another group of DNA-editing enzymes, containing an endonuclease domain and two DNA-binding domains, one binding selectively the mutant mtDNA site and another binding both wild‐type and mutant mtDNA. MitoTALENs, specifically designed to detect a rare LHON mutation (m.14459G>A/MTND6), have successfully shifted heteroplasmy towards wild‐type in patient-derived cybrid cell lines [211]. A limiting factor to mitoTALENs’ clinical application is their large dimension, which make them difficult to deliver by a viral vector.
All these approaches aimed at shifting heteroplasmy, pose the problem of transient mtDNA depletion, in particular if the heteroplasmic load in a given tissue is very high. However, both mitoZFNs and mitoTALENs approaches have been successfully tested in a mouse model carrying the m.5024C>T tRNAAla heteroplasmic pathogenic mutation, demonstrating in vivo the feasibility and efficacy in shifting heteroplasmy, ultimately restoring OXPHOS function [212, 213].
Finally, the powerful CRISPR/Cas9 system, as previously pointed out, remains controversial for mtDNA editing as it is hampered by the major limitation of delivering the guide RNA into the mitochondria [155, 214]. Instead, the next frontier for mtDNA editing is currently propelled by the very recent description of a bacterial toxin (DddA) acting as cytidine deaminase, which was split and fused with a DNA-binding TALE protein, an uracil glycosylase inhibitor and a mitochondrial-targeting signal [156]. The resulting DddA-derived cytosine base editors (DdCBEs) are independent from RNA and are efficiently imported into mitochondria where they catalyse C–G to T–A conversion in the mtDNA. This high selective and very specific mtDNA manipulation approach has potentially revolutionary implications for treatment of mitochondrial disorders, as well as generation of mtDNA-based animal models [215].
6.3 Mitochondrial Replacement by Pronuclear or Spindle Transfer
As far it concerns procreation, preventing transmission of mtDNA mutations to the progeny has been a major challenge since mtDNA is exclusively inherited from the oocyte. Preimplantation genetic diagnosis, performed on cells removed from an eight-cell embryo, is currently offered to women carrying mtDNA mutations to predict the mutation load of embryos [216]. However, the heteroplasmy load in oocytes frequently remains within a risky range, jeopardising the outcome of the procedure, and preimplantation diagnosis does not apply to the homoplasmic mutations, such as in most LHON families [1, 2]. On the contrary, preimplantation diagnosis applies to nuclear encoded defects, such as OPA1-related DOA or other similar conditions.
Mitochondrial replacement therapies (MRTs) are transplantation techniques applied at oocyte or zygote stage to uncouple the inheritance of the nuclear genome and mtDNA. They extract the nuclear DNA from the patient’s egg or embryo, which is then transplanted into a donor cytoplasm containing wild type mtDNA [217]. Three techniques have been tested on animals and human cells so far.
Pronuclear transfer (PNT) applies at zygote stage: the two pronuclei carrying the haploid set of chromosomes, one from the egg and one from the sperm, are transplanted from the zygote to the donor. This technique is associated with relatively high levels of mtDNA carry-over in mice, which are lower in humans; nevertheless, it is an ethical dilemma due to donor embryo destruction [218].
Spindle transfer (ST) was first developed in rhesus monkey and applies at the stage of unfertilised oocyte, where, unlike zygotes, the distribution of mitochondria is uniform. Meiotic spindles are extracted from the oocyte and fused to donor cytoplasts, which are then fertilised and cultured to the blastocyst stage. Blastocyst development and quality was comparable to controls in monkeys [219]. Studies on human oocytes, after artificial parthenogenetic activation, showed normal progression to blastocysts with very low level of mtDNA carryover [220].
Finally, polar body transfer (PBT) consists of one of the two polar bodies, derived from oocyte meiosis and containing a complement of chromosomes, to appropriate oocyte or zygote cytoplasm. It was tested in mice and coupled with PNT or ST, showing normal development of viable offspring, despite reduced lifetime due to apoptotic pressure. Functional capacity of PBT products in humans remains to be determined [221].
The mitochondrial replacement therapy has been approved, under strict requirements, in the UK [222], but the approach of ST was applied on a human case for the first time in 2016 in the USA with the procedure executed in Mexico, where a baby was generated from a mother carrying the m.8993G>A/MTATP6 mutation associated with the NARP/MILS mitochondrial syndromes [223]. This first case has been widely debated [224]. Considering HONs, certainly LHON may be amenable to this approach, as most families are homoplasmic mutant, thus not approachable by the heteroplasmic shift strategy. However, a number of issues on inclusion criteria regarding which patients might be eligible for this procedure, as well as active ethical considerations and uncertainties about long-term unexplored consequences, all contribute to a very limited application of this approach so far.
7 Induced Pluripotent Stem Cells
In 2006, a rapid exponential application of a revolutionary technology to reprogram somatic cells from patients into human induced pluripotent stem cells (hiPSCs) was introduced [225]. These cells can be differentiated into any of the terminal cell types [226], or into tissue organoids reproducing the disease targets in vitro [227]. Protocols are now available to differentiate RGCs [228], as well as eye mini-organs (mini-eye organoid) [229], which may be used as models of disease to unravel the detailed pathogenic mechanisms and test new drugs [227]. The full atlas of single-cell transcriptomes, comparing adult human retinas with mini-eye organoid retinas, has been provided recently, establishing the baseline for any future study of disease conditions [230]. A further proposed development of hiPSC technology is to use them to regenerate lost tissue due to neurodegeneration, ideally reconstituting the RGCs pools and re-growing the optic nerve, reversing blindness in chronic patients without hope [231]. This theoretical scenario is obviously far from clinical application, but represents one of the most spectacular future directions for therapeutic options of HONs. Many issues still need to be solved. One is the quality of hiPSCs that are obtained by reprogramming, as a few studies indicate that during the reprogramming from a single somatic cell, clonal expansion of unwanted heteroplasmic mtDNA variants [232, 233], including some with a pathogenic potential, may occur from the pool of “universal heteroplasmy” [234]. Furthermore, the therapeutic use of hiPSCs with wild-type mtDNA has various challenges in terms of delivery into the right retinal location, the connection with the other retinal cells, and the long-distance growth of the axon to restore the correct signalling to the lateral geniculate nucleus (LGN) [231].
In the field of HONs, a few groups are already working on both LHON [235] and DOA [236]. Yang and colleagues generated hiPSC-derived RGCs and studied, in particular, the axonal transport [235], whereas Chen and colleagues investigated apoptosis in a similar setting, showing that excess apoptosis in OPA1 hiPSCs hampered the differentiation into RGCs [236]. This field is in its infancy and is about to rapidly expand accelerating drug screening and testing therapeutic strategies for HONs.
8 Other Innovative Therapies at Preclinical Stage Contrasting Mitochondrial Dysfunction
8.1 Mitobiogenesis
A compensatory strategy to recover impaired OXPHOS is the activation of mitochondrial biogenesis, which involves increasing mitochondrial mass and mtDNA copy number to raise the total ATP production. This compensatory strategy partially occurs through spontaneous cell rewiring in several mitochondrial diseases and is histologically visible as subsarcolemmal mitochondria proliferation evident at light microscopy and electron microscopy in skeletal muscle, defining the scattered occurrence of the so-called ragged red fibres [30].
The importance of this compensatory mechanism has been highlighted in LHON, where it has been demonstrated that a significantly higher mtDNA content and increased mitochondrial biogenesis is present in both skeletal muscle and blood cells of unaffected LHON mutation carriers, driving the incomplete penetrance [49]. A threshold of > 500 mtDNA copy numbers distinguished the unaffected mutation carriers from the affected individuals, possibly discriminating in the future the probability of a LHON mutation carrier remaining unaffected or undergoing clinical conversion [49]. Therefore, mitochondrial biogenesis is also a druggable target and agents able to activate this cell rewiring could be beneficial in a context of mitochondrial dysfunction.
8.1.1 Oestrogens
The protective role of oestrogens in LHON has been demonstrated, in both cybrids and patient-derived primary fibroblast cell lines [47]. This observation was proposed as partial explanation for gender bias in LHON, a condition where notoriously, males are more prone to develop the disease compared to females. In cybrids, 17b-oestradiol ameliorated the overproduction of ROS and apoptosis rate in galactose medium through the activation of the antioxidant enzyme superoxide dismutase 2, and the promotion of mitochondrial biogenesis with an increase in mtDNA copy numbers [47]. Moreover, a specific oestrogen receptor type β (Erβ) was shown to be present in the mitochondria of human RGCs and its expression is maintained in the surviving cells of LHON patients. The therapeutic implications of these observations may be relevant, since targeting Erβ in RGCs with oestrogen-like molecules could be an effective strategy for young unaffected LHON carriers to prevent the disease onset or reduce disease severity. This strategy has been tested in LHON cybrids and fibroblasts carrying the m.11778G>A/MTND4 mutation using a combination of natural oestrogen-like compounds (phytoestrogens). In fact, in vitro phytoestrogen administration improved cell survival by reducing apoptosis and ROS production, ultimately increasing mitochondrial biogenesis and respiratory competence [17]. Further studies are needed to clinically test phytoestrogens as preventive therapy in LHON by appropriate trials.
8.1.2 Rapamycin
Rapamycin, an approved drug for use in humans, is a specific inhibitor of the mammalian target of rapamycin (mTOR) signalling pathway [237], which has been shown to extend lifespan and reduce disease progression in the Ndufs4 knockout (Ndufs4−/−) mouse, a complex I-based Leigh syndrome mouse model [238]. Furthermore, a study on cybrid cells expressing the m.11778G>A/MTND4 LHON mutation in heteroplasmic state showed that rapamycin promotes mitophagy, resulting in reduction of the mutation levels with partial ATP restoration [239].
More recently, the efficacy of rapamycin and two other molecules, papaverine, a vasodilator, and the GABAA agonist zolpidem were tested on the same Ndufs4−/− mouse, which also develops a LHON-like loss of RGCs leading to rapid vision loss [18]. The treated mice showed significant preservation of the visual function, reduced retinal inflammatory response and enhanced cell survival. All considered, rapamycin seems to ameliorate the phenotypic expression of Leigh syndrome and LHON-like optic atrophy due to profound complex I deficiency. These observations may prompt a repurpose of rapamycin in LHON and maybe other HONs. As for preclinical studies, further clarification of the mechanism by which RGC preservation occurs is needed.
8.1.3 Other Molecules
Over the last decade, drugs that have been considered as boosters of mitochondrial biogenesis include bezafibrate, metformin, rosiglitazone, resveratrol and 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR), which, in most cases, have been shown to activate the transcriptional co-activator PGC-1a, peroxisome proliferator-activated receptors and AMP-activated protein kinase [237]. Amongst the most convincing results available, we consider vitamin B3 and nicotinamide adenine dinucleotide (NAD+) precursors as boosters of mitochondrial biogenesis [240, 241]. Mitochondrial activity is the key regulator of NAD+/NADH ratio, which in turn is central to the redox and metabolic state of cells, given its role in hydrogen carrier for redox enzymes [242]. Oral administration of NAD+ precursors, like niacin, nicotinamide riboside and inositol hexanicotinate, is currently proposed as therapy for mitochondrial disorders to increase mitochondrial population in order to overcome the metabolic defect and ameliorate global mitochondrial function [237, 240, 241].
Another recent interesting scenario was when some microRNAs (miR-181a and miR-181b) were found to specifically regulate the expression of genes involved in mitochondrial homeostasis in the retina by direct targeting of Nrf1. Downregulation of these miRNAs in the Ndufs4−/− mouse model with LHON-like features resulted in preservation of functional RGCs through a co-activation of mitochondrial biogenesis and mitophagy and improving the visual phenotype [243].
8.2 Neuroprotection
Neuroprotection is often an abused denomination; however, several agents have been tested in various animal models of optic neuropathies, especially ischaemic and demyelinating [244]. Nevertheless, it cannot be excluded that they may be applied to HONs in the future. QPI-1007, a chemically modified short interfering RNA (siRNA) that inhibits the expression of the caspase 2 [245], has been administered in patients with non-ischaemic anterior optic neuritis (NAION) but an interim analysis did not warrant enrollment (www.clinicaltrials.gov NCT02341560). Another compound, RPh201, extracted from the mastic gum, was administered in patients with NAION during a placebo-controlled Phase II trial (www.clinicaltrials.gov NCT02045212). They assessed the safety of subcutaneous administration, but no clinical efficacy in terms of VA improvement was detected [246].
Another neuroprotective agent is BN201, a highly soluble small molecule that crosses the blood-brain barrier and provides axonal protection, promoting remyelination and neuronal survival in models of acute optic neuritis, multiple sclerosis and glaucoma [247] (https://bionure.com/the-neuroprotectant-bn201/).
This short selection does not cover the exhaustive field of agents classified as “neuroprotective”, but only highlights the potential cross-contamination of drugs designed for one pathological condition that might be of some use in others, including HONs.
9 Conclusions and Future Directions
In conclusion, we provided an overview on currently available therapeutic options for HONs, but also looking at future developments.
What remains central for mitochondrial HONs is the use of antioxidants, in particular idebenone, for which approval is already set for LHON, but may extend to other conditions such as OPA1-related DOA. However, it would be interesting to clarify the mechanisms and learn how the dependence on NQO1 may impinge on efficacy. As LHON is the paradigm playground for HONs, we expect to better understand the global experience from the multiple trials with gene therapy based on allotopic expression of ND4, as all results are now analysed and published. Certainly, the bilateral improvement poses a number of questions that will necessarily be addressed. Considering LHON as a paradigm for HONs, we may also envisage that ultimately different therapeutic approaches might work in combination, targeting different aspects of the pathogenic mechanisms, as well as using different routes of administration. Idebenone, for example, is thought to partly correct bioenergetics and mitigate oxidative stress. Its administration is systemic, possibly reaching all cell types; thus, besides RGCs, astrocytes, oligodendrocytes, and blood vessel, other cell components are implicated in LHON pathogenesis [1, 2, 8, 9]. The allotopic gene therapy targets the RGCs by highly localised intravitreal administration, without reaching in adequate amount the myelinated portion of the optic nerve. Furthermore, the mechanism targeted by this approach is the direct correction of complex I function, in a percentage of assembled complexes, by inserting the wild-type ND4 subunit. The combination of idebenone and gene therapy may then possibly result in a synergistic and complementary therapeutic effect.
The future seems particularly dynamic for HONs and for the field of mitochondrial medicine in general. With no cure available for over 30 years, there is hope that some of the proposed therapeutic strategies will allow to reduce the disease burden. These include gene therapy and genome editing, reproductive options, as well as drug repurposing and discovery of novel molecules targeting the key pathogenic mechanisms.
References
Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res. 2004;23(1):53–89.
Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies—disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011;30(2):81–114.
La Morgia C, Carbonelli M, Barboni P, Sadun AA, Carelli V. Medical management of hereditary optic neuropathies. Front Neurol. 2014;5:141.
Achilli A, Iommarini L, Olivieri A, Pala M, Hooshiar Kashani B, Reynier P, La Morgia C, Valentino ML, Liguori R, Pizza F, Barboni P, et al. Rare primary mitochondrial DNA mutations and probable synergistic variants in Leber’s hereditary optic neuropathy. PLoS ONE. 2012;7(8):e42242.
Del Dotto V, Fogazza M, Carelli V, Rugolo M, Zanna C. Eight human opa1 isoforms, long and short: What are they for? Biochim Biophys Acta Bioenergy. 2018;1859(4):263–9.
Carelli V, La Morgia C, Valentino ML, Barboni P, Ross-Cisneros FN, Sadun AA. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim Biophys Acta. 2009;1787(5):518–28.
Sadun AA, La Morgia C, Carelli V. Mitochondrial optic neuropathies: our travels from bench to bedside and back again. Clin Exp Ophthalmol. 2013;41(7):702–12.
Yu-Wai-Man P, Votruba M, Burte F, La Morgia C, Barboni P, Carelli V. A neurodegenerative perspective on mitochondrial optic neuropathies. Acta Neuropathol. 2016;132(6):789–806.
Carelli V, La Morgia C, Ross-Cisneros FN, Sadun AA. Optic neuropathies: the tip of the neurodegeneration iceberg. Hum Mol Genet. 2017;26(R2):R139–50.
Gueven N. Idebenone for leber’s hereditary optic neuropathy. Drugs Today (Barc). 2016;52(3):173–81.
Klopstock T, Yu-Wai-Man P, Dimitriadis K, Rouleau J, Heck S, Bailie M, Atawan A, Chattopadhyay S, Schubert M, Garip A, Kernt M, et al. A randomized placebo-controlled trial of idebenone in leber’s hereditary optic neuropathy. Brain. 2011;134(Pt 9):2677–86.
Carelli V, La Morgia C, Valentino ML, Rizzo G, Carbonelli M, De Negri AM, Sadun F, Carta A, Guerriero S, Simonelli F, Sadun AA, et al. Idebenone treatment in leber’s hereditary optic neuropathy. Brain. 2011;134(Pt 9):e188.
Barboni P, Valentino ML, La Morgia C, Carbonelli M, Savini G, De Negri A, Simonelli F, Sadun F, Caporali L, Maresca A, Liguori R, et al. Idebenone treatment in patients with opa1-mutant dominant optic atrophy. Brain. 2013;136(Pt 2):e231.
Romagnoli M, La Morgia C, Carbonelli M, Di Vito L, Amore G, Zenesini C, Cascavilla ML, Barboni P, Carelli V. Idebenone increases chance of stabilization/recovery of visual acuity in opa1-dominant optic atrophy. Ann Clin Transl Neurol. 2020;7(4):590–4.
Sadun AA, Chicani CF, Ross-Cisneros FN, Barboni P, Thoolen M, Shrader WD, Kubis K, Carelli V, Miller G. Effect of epi-743 on the clinical course of the mitochondrial disease leber hereditary optic neuropathy. Arch Neurol. 2012;69(3):331–8.
Rustum Karanjia SGC, Garcia M, Sadun AA. Elamipretide (mtp-131) topical ophthalmic solution for the treatment of leber’s hereditary optic neuropathy. Investig Ophthalmol Visual Sci. 2019;60:2266.
Pisano A, Preziuso C, Iommarini L, Perli E, Grazioli P, Campese AF, Maresca A, Montopoli M, Masuelli L, Sadun AA, d’Amati G, et al. Targeting estrogen receptor beta as preventive therapeutic strategy for leber’s hereditary optic neuropathy. Hum Mol Genet. 2015;24(24):6921–31.
Yu AK, Datta S, McMackin MZ, Cortopassi GA. Rescue of cell death and inflammation of a mouse model of complex 1-mediated vision loss by repurposed drug molecules. Hum Mol Genet. 2017;26(24):4929–36.
Indrieri A, Carrella S, Romano A, Spaziano A, Marrocco E, Fernandez-Vizarra E, Barbato S, Pizzo M, Ezhova Y, Golia FM, Ciampi L, et al. Mir-181a/b downregulation exerts a protective action on mitochondrial disease models. EMBO Mol Med. 2019;11(5):e8734.
Guy J, Qi X, Pallotti F, Schon EA, Manfredi G, Carelli V, Martinuzzi A, Hauswirth WW, Lewin AS. Rescue of a mitochondrial deficiency causing leber hereditary optic neuropathy. Ann Neurol. 2002;52(5):534–42.
Jurkute N, Harvey J, Yu-Wai-Man P. Treatment strategies for leber hereditary optic neuropathy. Curr Opin Neurol. 2019;32(1):99–104.
Sarzi E, Seveno M, Piro-Megy C, Elziere L, Quiles M, Pequignot M, Muller A, Hamel CP, Lenaers G, Delettre C. Opa1 gene therapy prevents retinal ganglion cell loss in a dominant optic atrophy mouse model. Sci Rep. 2018;8(1):2468.
Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. 2018;20(7):745–54.
Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–87.
Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. 2020;21(4):204–24.
Green DR, Galluzzi L, Kroemer G. Cell biology. Metabolic control of cell death. Science. 2014;345(6203):1250256.
De Stefani D, Rizzuto R, Pozzan T. Enjoy the trip: calcium in mitochondria back and forth. Annu Rev Biochem. 2016;85:161–92.
Pickles S, Vigie P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28(4):R170–85.
Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–8.
DiMauro S, Schon EA, Carelli V, Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. 2013;9(8):429–44.
Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, Suomalainen A, Thorburn DR, Zeviani M, Turnbull DM. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2:16080.
Burte F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. 2015;11(1):11–24.
Sadun A. Acquired mitochondrial impairment as a cause of optic nerve disease. Trans Am Ophthalmol Soc. 1998;96:881–923.
Carelli V, Ross-Cisneros FN, Sadun AA. Optic nerve degeneration and mitochondrial dysfunction: genetic and acquired optic neuropathies. Neurochem Int. 2002;40(6):573–84.
Giordano L, Deceglie S, d’Adamo P, Valentino ML, La Morgia C, Fracasso F, Roberti M, Cappellari M, Petrosillo G, Ciaravolo S, Parente D, et al. Cigarette toxicity triggers leber’s hereditary optic neuropathy by affecting mtdna copy number, oxidative phosphorylation and ros detoxification pathways. Cell Death Dis. 2015;6:e2021.
Kirkman MA, Yu-Wai-Man P, Korsten A, Leonhardt M, Dimitriadis K, De Coo IF, Klopstock T, Chinnery PF. Gene-environment interactions in leber hereditary optic neuropathy. Brain. 2009;132(Pt 9):2317–26.
Sadun AA, Carelli V, Salomao SR, Berezovsky A, Quiros PA, Sadun F, DeNegri AM, Andrade R, Moraes M, Passos A, Kjaer P, et al. Extensive investigation of a large brazilian pedigree of 11778/haplogroup j leber hereditary optic neuropathy. Am J Ophthalmol. 2003;136(2):231–8.
Carelli V, d’Adamo P, Valentino ML, La Morgia C, Ross-Cisneros FN, Caporali L, Maresca A, Loguercio Polosa P, Barboni P, De Negri A, Sadun F, et al. Parsing the differences in affected with lhon: genetic versus environmental triggers of disease conversion. Brain. 2016;139(Pt 3):e17.
Ijaz S, Jackson J, Thorley H, Porter K, Fleming C, Richards A, Bonner A, Savovic J. Nutritional deficiencies in homeless persons with problematic drinking: a systematic review. Int J Equity Health. 2017;16(1):71.
Sadun AA. Metabolic optic neuropathies. Semin Ophthalmol. 2002;17(1):29–32.
Duewelhenke N, Krut O, Eysel P. Influence on mitochondria and cytotoxicity of different antibiotics administered in high concentrations on primary human osteoblasts and cell lines. Antimicrob Agents Chemother. 2007;51(1):54–63.
Pacheu-Grau D, Gomez-Duran A, Lopez-Perez MJ, Montoya J, Ruiz-Pesini E. Mitochondrial pharmacogenomics: barcode for antibiotic therapy. Drug Discov Today. 2010;15(1–2):33–9.
Payne BA, Wilson IJ, Hateley CA, Horvath R, Santibanez-Koref M, Samuels DC, Price DA, Chinnery PF. Mitochondrial aging is accelerated by anti-retroviral therapy through the clonal expansion of mtdna mutations. Nat Genet. 2011;43(8):806–10.
Gardner K, Hall PA, Chinnery PF, Payne BA. Hiv treatment and associated mitochondrial pathology: Review of 25 years of in vitro, animal, and human studies. Toxicol Pathol. 2014;42(5):811–22.
Klinge CM. Estrogenic control of mitochondrial function. Redox Biol. 2020;31:101435.
King MP, Attardi G. Human cells lacking mtdna: repopulation with exogenous mitochondria by complementation. Science. 1989;246(4929):500–3.
Giordano C, Montopoli M, Perli E, Orlandi M, Fantin M, Ross-Cisneros FN, Caparrotta L, Martinuzzi A, Ragazzi E, Ghelli A, Sadun AA, et al. Oestrogens ameliorate mitochondrial dysfunction in leber’s hereditary optic neuropathy. Brain. 2011;134(Pt 1):220–34.
Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, Larsson NG. Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci USA. 2002;99(23):15066–71.
Giordano C, Iommarini L, Giordano L, Maresca A, Pisano A, Valentino ML, Caporali L, Liguori R, Deceglie S, Roberti M, Fanelli F, et al. Efficient mitochondrial biogenesis drives incomplete penetrance in leber’s hereditary optic neuropathy. Brain. 2014;137(Pt 2):335–53.
Carelli V, Maresca A, Caporali L, Trifunov S, Zanna C, Rugolo M. Mitochondria: Biogenesis and mitophagy balance in segregation and clonal expansion of mitochondrial DNA mutations. Int J Biochem Cell Biol. 2015;63:21–4.
Sharma LK, Tiwari M, Rai NK, Bai Y. Mitophagy activation repairs leber’s hereditary optic neuropathy-associated mitochondrial dysfunction and improves cell survival. Hum Mol Genet. 2019;28(3):422–33.
Dombi E, Diot A, Morten K, Carver J, Lodge T, Fratter C, Ng YS, Liao C, Muir R, Blakely EL, Hargreaves I, et al. The m.13051g>a mitochondrial DNA mutation results in variable neurology and activated mitophagy. Neurology. 2016;86(20):1921–3.
Sadun AA, Win PH, Ross-Cisneros FN, Walker SO, Carelli V. Leber’s hereditary optic neuropathy differentially affects smaller axons in the optic nerve. Trans Am Ophthalmol Soc. 2000;98:223–32 (discussion 232–225).
Pan BX, Ross-Cisneros FN, Carelli V, Rue KS, Salomao SR, Moraes-Filho MN, Moraes MN, Berezovsky A, Belfort R Jr, Sadun AA. Mathematically modeling the involvement of axons in leber’s hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2012;53(12):7608–17.
Yu-Wai-Man P, Griffiths PG, Brown DT, Howell N, Turnbull DM, Chinnery PF. The epidemiology of leber hereditary optic neuropathy in the north east of England. Am J Hum Genet. 2003;72(2):333–9.
Puomila A, Hamalainen P, Kivioja S, Savontaus ML, Koivumaki S, Huoponen K, Nikoskelainen E. Epidemiology and penetrance of leber hereditary optic neuropathy in Finland. Eur J Hum Genet. 2007;15(10):1079–89.
Mascialino B, Leinonen M, Meier T. Meta-analysis of the prevalence of leber hereditary optic neuropathy mtdna mutations in Europe. Eur J Ophthalmol. 2012;22(3):461–5.
Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ 2nd, Nikoskelainen EK. Mitochondrial DNA mutation associated with leber’s hereditary optic neuropathy. Science. 1988;242(4884):1427–30.
Caporali L, Maresca A, Capristo M, Del Dotto V, Tagliavini F, Valentino ML, La Morgia C, Carelli V. Incomplete penetrance in mitochondrial optic neuropathies. Mitochondrion. 2017;36:130–7.
Harding AE, Sweeney MG, Govan GG, Riordan-Eva P. Pedigree analysis in leber hereditary optic neuropathy families with a pathogenic mtdna mutation. Am J Hum Genet. 1995;57(1):77–86.
Torroni A, Petrozzi M, D’Urbano L, Sellitto D, Zeviani M, Carrara F, Carducci C, Leuzzi V, Carelli V, Barboni P, De Negri A, et al. Haplotype and phylogenetic analyses suggest that one european-specific mtdna background plays a role in the expression of leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am J Hum Genet. 1997;60(5):1107–21.
Carelli V, Achilli A, Valentino ML, Rengo C, Semino O, Pala M, Olivieri A, Mattiazzi M, Pallotti F, Carrara F, Zeviani M, et al. Haplogroup effects and recombination of mitochondrial DNA: novel clues from the analysis of leber hereditary optic neuropathy pedigrees. Am J Hum Genet. 2006;78(4):564–74.
Hudson G, Carelli V, Spruijt L, Gerards M, Mowbray C, Achilli A, Pyle A, Elson J, Howell N, La Morgia C, Valentino ML, et al. Clinical expression of leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am J Hum Genet. 2007;81(2):228–33.
Caporali L, Iommarini L, La Morgia C, Olivieri A, Achilli A, Maresca A, Valentino ML, Capristo M, Tagliavini F, Del Dotto V, Zanna C, et al. Peculiar combinations of individually non-pathogenic missense mitochondrial DNA variants cause low penetrance leber’s hereditary optic neuropathy. PLoS Genet. 2018;14(2):e1007210.
Jiang P, Jin X, Peng Y, Wang M, Liu H, Liu X, Zhang Z, Ji Y, Zhang J, Liang M, Zhao F, et al. The exome sequencing identified the mutation in yars2 encoding the mitochondrial tyrosyl-trna synthetase as a nuclear modifier for the phenotypic manifestation of leber’s hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum Mol Genet. 2016;25(3):584–96.
Yu J, Liang X, Ji Y, Ai C, Liu J, Zhu L, Nie Z, Jin X, Wang C, Zhang J, Zhao F, et al. Prickle3 linked to atpase biogenesis manifested leber's hereditary optic neuropathy. J Clin Invest. 2020;130(9):4935–46.
Bu XD, Rotter JI. X chromosome-linked and mitochondrial gene control of leber hereditary optic neuropathy: evidence from segregation analysis for dependence on x chromosome inactivation. Proc Natl Acad Sci USA. 1991;88(18):8198–202.
Vilkki J, Ott J, Savontaus ML, Aula P, Nikoskelainen EK. Optic atrophy in leber hereditary optic neuroretinopathy is probably determined by an x-chromosomal gene closely linked to dxs7. Am J Hum Genet. 1991;48(3):486–91.
Sweeney MG, Davis MB, Lashwood A, Brockington M, Toscano A, Harding AE. Evidence against an x-linked locus close to dxs7 determining visual loss susceptibility in British and Italian families with Leber hereditary optic neuropathy. Am J Hum Genet. 1992;51(4):741–8.
Pegoraro E, Carelli V, Zeviani M, Cortelli P, Montagna P, Barboni P, Angelini C, Hoffman EP. X-inactivation patterns in female leber’s hereditary optic neuropathy patients do not support a strong x-linked determinant. Am J Med Genet. 1996;61(4):356–62.
Hudson G, Keers S, Yu-Wai-Man P, Griffiths P, Huoponen K, Savontaus ML, Nikoskelainen E, Zeviani M, Carrara F, Horvath R, Karcagi V, et al. Identification of an x-chromosomal locus and haplotype modulating the phenotype of a mitochondrial DNA disorder. Am J Hum Genet. 2005;77(6):1086–91.
Shankar SP, Fingert JH, Carelli V, Valentino ML, King TM, Daiger SP, Salomao SR, Berezovsky A, Belfort R Jr, Braun TA, Sheffield VC, et al. Evidence for a novel x-linked modifier locus for leber hereditary optic neuropathy. Ophthalmic Genet. 2008;29(1):17–24.
Rosenberg T, Norby S, Schwartz M, Saillard J, Magalhaes PJ, Leroy D, Kann EC, Duno M. Prevalence and genetics of leber hereditary optic neuropathy in the Danish population. Invest Ophthalmol Vis Sci. 2016;57(3):1370–5.
Yu-Wai-Man P, Hudson G, Klopstock T, Chinnery PF. Reply: parsing the differences in affected with Lhon: genetic versus environmental triggers of disease conversion. Brain. 2016;139(Pt 3):e18.
Carelli V, Carbonelli M, de Coo IF, Kawasaki A, Klopstock T, Lagreze WA, La Morgia C, Newman NJ, Orssaud C, Pott JWR, Sadun AA, et al. International consensus statement on the clinical and therapeutic management of leber hereditary optic neuropathy. J Neuroophthalmol. 2017;37(4):371–81.
Barboni P, Savini G, Valentino ML, Montagna P, Cortelli P, De Negri AM, Sadun F, Bianchi S, Longanesi L, Zanini M, de Vivo A, et al. Retinal nerve fiber layer evaluation by optical coherence tomography in leber’s hereditary optic neuropathy. Ophthalmology. 2005;112(1):120–6.
Barboni P, Carbonelli M, Savini G, Ramos Cdo V, Carta A, Berezovsky A, Salomao SR, Carelli V, Sadun AA. Natural history of leber’s hereditary optic neuropathy: longitudinal analysis of the retinal nerve fiber layer by optical coherence tomography. Ophthalmology. 2010;117(3):623–7.
Balducci N, Savini G, Cascavilla ML, La Morgia C, Triolo G, Giglio R, Carbonelli M, Parisi V, Sadun AA, Bandello F, Carelli V, et al. Macular nerve fibre and ganglion cell layer changes in acute leber’s hereditary optic neuropathy. Br J Ophthalmol. 2016;100(9):1232–7.
Newman NJ, Carelli V, Taiel M, Yu-Wai-Man P. Visual outcomes in leber hereditary optic neuropathy patients with the m.11778g>a (mtnd4) mitochondrial DNA mutation. J Neuroophthalmol. 2020.
Hwang TJ, Karanjia R, Moraes-Filho MN, Gale J, Tran JS, Chu ER, Salomao SR, Berezovsky A, Belfort R Jr, Moraes MN, Sadun F, et al. Natural history of conversion of leber’s hereditary optic neuropathy: a prospective case series. Ophthalmology. 2017;124(6):843–50.
Newman NJ, Biousse V. Hereditary optic neuropathies. Eye (Lond). 2004;18(11):1144–60.
Lenaers G, Hamel C, Delettre C, Amati-Bonneau P, Procaccio V, Bonneau D, Reynier P, Milea D. Dominant optic atrophy. Orphanet J Rare Dis. 2012;7:46.
Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, et al. Opa1, encoding a dynamin-related gtpase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26(2):211–5.
Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, et al. Nuclear gene opa1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26(2):207–10.
Lenaers G, Reynier P, Elachouri G, Soukkarieh C, Olichon A, Belenguer P, Baricault L, Ducommun B, Hamel C, Delettre C. Opa1 functions in mitochondria and dysfunctions in optic nerve. Int J Biochem Cell Biol. 2009;41(10):1866–74.
Del Dotto V, Fogazza M, Lenaers G, Rugolo M, Carelli V, Zanna C. Opa1: how much do we know to approach therapy? Pharmacol Res. 2018;131:199–210.
Pesch UE, Leo-Kottler B, Mayer S, Jurklies B, Kellner U, Apfelstedt-Sylla E, Zrenner E, Alexander C, Wissinger B. Opa1 mutations in patients with autosomal dominant optic atrophy and evidence for semi-dominant inheritance. Hum Mol Genet. 2001;10(13):1359–68.
Yu-Wai-Man P, Griffiths PG, Burke A, Sellar PW, Clarke MP, Gnanaraj L, Ah-Kine D, Hudson G, Czermin B, Taylor RW, Horvath R, et al. The prevalence and natural history of dominant optic atrophy due to opa1 mutations. Ophthalmology. 2010;117(8):1538–46 (1546 e1531).
Amati-Bonneau P, Odent S, Derrien C, Pasquier L, Malthiery Y, Reynier P, Bonneau D. The association of autosomal dominant optic atrophy and moderate deafness may be due to the r445h mutation in the opa1 gene. Am J Ophthalmol. 2003;136(6):1170–1.
Payne M, Yang Z, Katz BJ, Warner JE, Weight CJ, Zhao Y, Pearson ED, Treft RL, Hillman T, Kennedy RJ, Meire FM, et al. Dominant optic atrophy, sensorineural hearing loss, ptosis, and ophthalmoplegia: a syndrome caused by a missense mutation in opa1. Am J Ophthalmol. 2004;138(5):749–55.
Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissiere A, Campos Y, Rivera H, de la Aleja JG, Carroccia R, Iommarini L, et al. Opa1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain. 2008;131(Pt 2):338–51.
Hudson G, Amati-Bonneau P, Blakely EL, Stewart JD, He L, Schaefer AM, Griffiths PG, Ahlqvist K, Suomalainen A, Reynier P, McFarland R, et al. Mutation of opa1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtdna maintenance. Brain. 2008;131(Pt 2):329–37.
Yu-Wai-Man P, Griffiths PG, Gorman GS, Lourenco CM, Wright AF, Auer-Grumbach M, Toscano A, Musumeci O, Valentino ML, Caporali L, Lamperti C, et al. Multi-system neurological disease is common in patients with opa1 mutations. Brain. 2010;133(Pt 3):771–86.
Verny C, Loiseau D, Scherer C, Lejeune P, Chevrollier A, Gueguen N, Guillet V, Dubas F, Reynier P, Amati-Bonneau P, Bonneau D. Multiple sclerosis-like disorder in opa1-related autosomal dominant optic atrophy. Neurology. 2008;70(13 Pt 2):1152–3.
Marelli C, Amati-Bonneau P, Reynier P, Layet V, Layet A, Stevanin G, Brissaud E, Bonneau D, Durr A, Brice A. Heterozygous opa1 mutations in behr syndrome. Brain. 2011;134(Pt 4):e169 (author reply e170).
Carelli V, Musumeci O, Caporali L, Zanna C, La Morgia C, Del Dotto V, Porcelli AM, Rugolo M, Valentino ML, Iommarini L, Maresca A, et al. Syndromic parkinsonism and dementia associated with opa1 missense mutations. Ann Neurol. 2015;78(1):21–38.
Lynch DS, Loh SHY, Harley J, Noyce AJ, Martins LM, Wood NW, Houlden H, Plun-Favreau H. Nonsyndromic Parkinson disease in a family with autosomal dominant optic atrophy due to opa1 mutations. Neurol Genet. 2017;3(5):e188.
Schaaf CP, Blazo M, Lewis RA, Tonini RE, Takei H, Wang J, Wong LJ, Scaglia F. Early-onset severe neuromuscular phenotype associated with compound heterozygosity for opa1 mutations. Mol Genet Metab. 2011;103(4):383–7.
Bonneau D, Colin E, Oca F, Ferre M, Chevrollier A, Gueguen N, Desquiret-Dumas V, N’Guyen S, Barth M, Zanlonghi X, Rio M, et al. Early-onset behr syndrome due to compound heterozygous mutations in opa1. Brain. 2014;137(Pt 10):e301.
Carelli V, Sabatelli M, Carrozzo R, Rizza T, Schimpf S, Wissinger B, Zanna C, Rugolo M, La Morgia C, Caporali L, Carbonelli M, et al. “Behr syndrome” with opa1 compound heterozygote mutations. Brain. 2015;138(Pt 1):e321.
Nasca A, Rizza T, Doimo M, Legati A, Ciolfi A, Diodato D, Calderan C, Carrara G, Lamantea E, Aiello C, Di Nottia M, et al. Not only dominant, not only optic atrophy: expanding the clinical spectrum associated with opa1 mutations. Orphanet J Rare Dis. 2017;12(1):89.
Spiegel R, Saada A, Flannery PJ, Burte F, Soiferman D, Khayat M, Eisner V, Vladovski E, Taylor RW, Bindoff LA, Shaag A, et al. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous opa1 mutation. J Med Genet. 2016;53(2):127–31.
Rubegni A, Pisano T, Bacci G, Tessa A, Battini R, Procopio E, Giglio S, Pasquariello R, Santorelli FM, Guerrini R, Nesti C. Leigh-like neuroimaging features associated with new biallelic mutations in opa1. Eur J Paediatr Neurol. 2017;21(4):671–7.
Yu-Wai-Man P, Bailie M, Atawan A, Chinnery PF, Griffiths PG. Pattern of retinal ganglion cell loss in dominant optic atrophy due to opa1 mutations. Eye (Lond). 2011;25(5):596–602.
Chevrollier A, Guillet V, Loiseau D, Gueguen N, de Crescenzo MA, Verny C, Ferre M, Dollfus H, Odent S, Milea D, Goizet C, et al. Hereditary optic neuropathies share a common mitochondrial coupling defect. Ann Neurol. 2008;63(6):794–8.
Zanna C, Ghelli A, Porcelli AM, Karbowski M, Youle RJ, Schimpf S, Wissinger B, Pinti M, Cossarizza A, Vidoni S, Valentino ML, et al. Opa1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain. 2008;131(Pt 2):352–67.
Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. Opa1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126(1):177–89.
Gerber S, Ding MG, Gerard X, Zwicker K, Zanlonghi X, Rio M, Serre V, Hanein S, Munnich A, Rotig A, Bianchi L, et al. Compound heterozygosity for severe and hypomorphic ndufs2 mutations cause non-syndromic Lhon-like optic neuropathy. J Med Genet. 2017;54(5):346–56.
Hanein S, Perrault I, Roche O, Gerber S, Khadom N, Rio M, Boddaert N, Jean-Pierre M, Brahimi N, Serre V, Chretien D, et al. Tmem126a, encoding a mitochondrial protein, is mutated in autosomal-recessive nonsyndromic optic atrophy. Am J Hum Genet. 2009;84(4):493–8.
La Morgia C, Caporali L, Tagliavini F, Palombo F, Carbonelli M, Liguori R, Barboni P, Carelli V. First tmem126a missense mutation in an Italian proband with optic atrophy and deafness. Neurol Genet. 2019;5(3):e329.
Angebault C, Guichet PO, Talmat-Amar Y, Charif M, Gerber S, Fares-Taie L, Gueguen N, Halloy F, Moore D, Amati-Bonneau P, Manes G, et al. Recessive mutations in rtn4ip1 cause isolated and syndromic optic neuropathies. Am J Hum Genet. 2015;97(5):754–60.
Charif M, Nasca A, Thompson K, Gerber S, Makowski C, Mazaheri N, Bris C, Goudenege D, Legati A, Maroofian R, Shariati G, et al. Neurologic phenotypes associated with mutations in rtn4ip1 (opa10) in children and young adults. JAMA Neurol. 2018;75(1):105–13.
Abrams AJ, Hufnagel RB, Rebelo A, Zanna C, Patel N, Gonzalez MA, Campeanu IJ, Griffin LB, Groenewald S, Strickland AV, Tao F, et al. Mutations in slc25a46, encoding a ugo1-like protein, cause an optic atrophy spectrum disorder. Nat Genet. 2015;47(8):926–32.
Metodiev MD, Gerber S, Hubert L, Delahodde A, Chretien D, Gerard X, Amati-Bonneau P, Giacomotto MC, Boddaert N, Kaminska A, Desguerre I, et al. Mutations in the tricarboxylic acid cycle enzyme, aconitase 2, cause either isolated or syndromic optic neuropathy with encephalopathy and cerebellar atrophy. J Med Genet. 2014;51(12):834–8.
Hartmann B, Wai T, Hu H, MacVicar T, Musante L, Fischer-Zirnsak B, Stenzel W, Graf R, van den Heuvel L, Ropers HH, Wienker TF, et al. Homozygous yme1l1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation. Elife. 2016;5:e16078.
Klebe S, Depienne C, Gerber S, Challe G, Anheim M, Charles P, Fedirko E, Lejeune E, Cottineau J, Brusco A, Dollfus H, et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain. 2012;135(Pt 10):2980–93.
Caporali L, Magri S, Legati A, Del Dotto V, Tagliavini F, Balistreri F, Nasca A, La Morgia C, Carbonelli M, Valentino ML, Lamantea E, et al. Atpase domain afg3l2 mutations alter opa1 processing and cause optic neuropathy. Ann Neurol. 2020;88(1):18–32.
Charif M, Chevrollier A, Gueguen N, Bris C, Goudenege D, Desquiret-Dumas V, Leruez S, Colin E, Meunier A, Vignal C, Smirnov V, et al. Mutations in the m-aaa proteases afg3l2 and spg7 are causing isolated dominant optic atrophy. Neurol Genet. 2020;6(3):e428.
Orsucci D, Ienco EC, Siciliano G, Mancuso M. Mitochondrial disorders and drugs: what every physician should know. Drugs Context. 2019;8:212588.
Mantle D, Dybring A. Bioavailability of coenzyme q10: an overview of the absorption process and subsequent metabolism. Antioxidants (Basel). 2020;9(5):386.
Glover EI, Martin J, Maher A, Thornhill RE, Moran GR, Tarnopolsky MA. A randomized trial of coenzyme q10 in mitochondrial disorders. Muscle Nerve. 2010;42(5):739–48.
Huang CC, Kuo HC, Chu CC, Kao LY. Rapid visual recovery after coenzyme q10 treatment of leber hereditary optic neuropathy. J Neuroophthalmol. 2002;22(1):66.
Tarnopolsky MA. The mitochondrial cocktail: rationale for combined nutraceutical therapy in mitochondrial cytopathies. Adv Drug Deliv Rev. 2008;60(13–14):1561–7.
Rodriguez MC, MacDonald JR, Mahoney DJ, Parise G, Beal MF, Tarnopolsky MA. Beneficial effects of creatine, coq10, and lipoic acid in mitochondrial disorders. Muscle Nerve. 2007;35(2):235–42.
Beyrath J, Pellegrini M, Renkema H, Houben L, Pecheritsyna S, van Zandvoort P, van den Broek P, Bekel A, Eftekhari P, Smeitink JAM. Kh176 safeguards mitochondrial diseased cells from redox stress-induced cell death by interacting with the thioredoxin system/peroxiredoxin enzyme machinery. Sci Rep. 2018;8(1):6577.
Haefeli RH, Erb M, Gemperli AC, Robay D, Courdier Fruh I, Anklin C, Dallmann R, Gueven N. Nqo1-dependent redox cycling of idebenone: effects on cellular redox potential and energy levels. PLoS ONE. 2011;6(3):e17963.
Ghelli A, Degli Esposti M, Carelli V, Lenaz G. Changes in mitochondrial complex i activity and coenzyme q binding site in leber’s hereditary optic neuropathy (Lhon). Mol Aspects Med. 1997;18(Suppl):S263-267.
Varricchio C, Beirne K, Heard C, Newland B, Rozanowska M, Brancale A, Votruba M. The ying and yang of idebenone: not too little, not too much—cell death in nqo1 deficient cells and the mouse retina. Free Radic Biol Med. 2020;152:551–60.
Jaber SM, Ge SX, Milstein JL, VanRyzin JW, Waddell J, Polster BM. Idebenone has distinct effects on mitochondrial respiration in cortical astrocytes compared to cortical neurons due to differential nqo1 activity. J Neurosci. 2020;40(23):4609–19.
Giorgio V, Petronilli V, Ghelli A, Carelli V, Rugolo M, Lenaz G, Bernardi P. The effects of idebenone on mitochondrial bioenergetics. Biochim Biophys Acta. 2012;1817(2):363–9.
Yu-Wai-Man P, Soiferman D, Moore DG, Burte F, Saada A. Evaluating the therapeutic potential of idebenone and related quinone analogues in leber hereditary optic neuropathy. Mitochondrion. 2017;36:36–42.
Heitz FD, Erb M, Anklin C, Robay D, Pernet V, Gueven N. Idebenone protects against retinal damage and loss of vision in a mouse model of leber’s hereditary optic neuropathy. PLoS ONE. 2012;7(9):e45182.
Kutz K, Drewe J, Vankan P. Pharmacokinetic properties and metabolism of idebenone. J Neurol. 2009;256(Suppl 1):31–5.
Klopstock T, Metz G, Yu-Wai-Man P, Buchner B, Gallenmuller C, Bailie M, Nwali N, Griffiths PG, von Livonius B, Reznicek L, Rouleau J, et al. Persistence of the treatment effect of idebenone in leber’s hereditary optic neuropathy. Brain. 2013;136(Pt 2):e230.
Catarino CB, von Livonius B, Priglinger C, Banik R, Matloob S, Tamhankar MA, Castillo L, Friedburg C, Halfpenny CA, Lincoln JA, Traber GL, et al. Real-world clinical experience with idebenone in the treatment of leber hereditary optic neuropathy. J Neuroophthalmol. 2020.
Baracca A, Solaini G, Sgarbi G, Lenaz G, Baruzzi A, Schapira AH, Martinuzzi A, Carelli V. Severe impairment of complex i-driven adenosine triphosphate synthesis in leber hereditary optic neuropathy cybrids. Arch Neurol. 2005;62(5):730–6.
Tang S, Le PK, Tse S, Wallace DC, Huang T. Heterozygous mutation of opa1 in drosophila shortens lifespan mediated through increased reactive oxygen species production. PLoS ONE. 2009;4(2):e4492.
Yarosh W, Monserrate J, Tong JJ, Tse S, Le PK, Nguyen K, Brachmann CB, Wallace DC, Huang T. The molecular mechanisms of opa1-mediated optic atrophy in drosophila model and prospects for antioxidant treatment. PLoS Genet. 2008;4(1):e6.
Millet AM, Bertholet AM, Daloyau M, Reynier P, Galinier A, Devin A, Wissinguer B, Belenguer P, Davezac N. Loss of functional opa1 unbalances redox state: implications in dominant optic atrophy pathogenesis. Ann Clin Transl Neurol. 2016;3(6):408–21.
Smith TG, Seto S, Ganne P, Votruba M. A randomized, placebo-controlled trial of the benzoquinone idebenone in a mouse model of opa1-related dominant optic atrophy reveals a limited therapeutic effect on retinal ganglion cell dendropathy and visual function. Neuroscience. 2016;319:92–106.
Williams PA, Morgan JE, Votruba M. Opa1 deficiency in a mouse model of dominant optic atrophy leads to retinal ganglion cell dendropathy. Brain. 2010;133(10):2942–51.
Shrader WD, Amagata A, Barnes A, Enns GM, Hinman A, Jankowski O, Kheifets V, Komatsuzaki R, Lee E, Mollard P, Murase K, et al. Alpha-tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. Bioorg Med Chem Lett. 2011;21(12):3693–8.
Zesiewicz T, Salemi JL, Perlman S, Sullivan KL, Shaw JD, Huang Y, Isaacs C, Gooch C, Lynch DR, Klein MB. Double-blind, randomized and controlled trial of epi-743 in friedreich’s ataxia. Neurodegener Dis Manag. 2018;8(4):233–42.
Fortuna F, Barboni P, Liguori R, Valentino ML, Savini G, Gellera C, Mariotti C, Rizzo G, Tonon C, Manners D, Lodi R, et al. Visual system involvement in patients with friedreich’s ataxia. Brain. 2009;132(Pt 1):116–23.
Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014;171(8):2029–50.
Daum G. Lipids of mitochondria. Biochim Biophys Acta. 1985;822(1):1–42.
Mileykovskaya E, Dowhan W. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chem Phys Lipids. 2014;179:42–8.
Karaa A, Haas R, Goldstein A, Vockley J, Weaver WD, Cohen BH. Randomized dose-escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology. 2018;90(14):e1212–21.
Leruez S, Verny C, Bonneau D, Procaccio V, Lenaers G, Amati-Bonneau P, Reynier P, Scherer C, Prundean A, Orssaud C, Zanlonghi X, et al. Cyclosporine a does not prevent second-eye involvement in leber’s hereditary optic neuropathy. Orphanet J Rare Dis. 2018;13(1):33.
Eells JT, Wong-Riley MT, VerHoeve J, Henry M, Buchman EV, Kane MP, Gould LJ, Das R, Jett M, Hodgson BD, Margolis D, et al. Mitochondrial signal transduction in accelerated wound and retinal healing by near-infrared light therapy. Mitochondrion. 2004;4(5–6):559–67.
Garafalo AV, Cideciyan AV, Heon E, Sheplock R, Pearson A, Yu CW, Sumaroka A, Aguirre GD, Jacobson SG. Progress in treating inherited retinal diseases: early subretinal gene therapy clinical trials and candidates for future initiatives. Prog Retin Eye Res. 2019;77:100827.
Yu H, Koilkonda RD, Chou TH, Porciatti V, Ozdemir SS, Chiodo V, Boye SL, Boye SE, Hauswirth WW, Lewin AS, Guy J. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses leber’s hereditary optic neuropathy in a mouse model. Proc Natl Acad Sci USA. 2012;109(20):E1238-1247.
Yu H, Koilkonda RD, Chou TH, Porciatti V, Mehta A, Hentall ID, Chiodo VA, Boye SL, Hauswirth WW, Lewin AS, Guy J. Consequences of zygote injection and germline transfer of mutant human mitochondrial DNA in mice. Proc Natl Acad Sci USA. 2015;112(42):E5689-5698.
Wang G, Shimada E, Zhang J, Hong JS, Smith GM, Teitell MA, Koehler CM. Correcting human mitochondrial mutations with targeted rna import. Proc Natl Acad Sci USA. 2012;109(13):4840–5.
Gammage PA, Moraes CT, Minczuk M. Mitochondrial genome engineering: the revolution may not be crispr-ized. Trends Genet. 2018;34(2):101–10.
Mok BY, de Moraes MH, Zeng J, Bosch DE, Kotrys AV, Raguram A, Hsu F, Radey MC, Peterson SB, Mootha VK, Mougous JD, et al. A bacterial cytidine deaminase toxin enables crispr-free mitochondrial base editing. Nature. 2020;583(7817):631–7.
Gray RE, Law RH, Devenish RJ, Nagley P. Allotopic expression of mitochondrial atp synthase genes in nucleus of Saccharomyces cerevisiae. Methods Enzymol. 1996;264:369–89.
Roucou X, Artika IM, Devenish RJ, Nagley P. Bioenergetic and structural consequences of allotopic expression of subunit 8 of yeast mitochondrial atp synthase. The hydrophobic character of residues 23 and 24 is essential for maximal activity and structural stability of the enzyme complex. Eur J Biochem. 1999;261(2):444–51.
de Grey AD. Mitochondrial gene therapy: an arena for the biomedical use of inteins. Trends Biotechnol. 2000;18(9):394–9.
Zullo SJ. Gene therapy of mitochondrial DNA mutations: a brief, biased history of allotopic expression in mammalian cells. Semin Neurol. 2001;21(3):327–35.
Manfredi G, Fu J, Ojaimi J, Sadlock JE, Kwong JQ, Guy J, Schon EA. Rescue of a deficiency in atp synthesis by transfer of mtatp6, a mitochondrial DNA-encoded gene, to the nucleus. Nat Genet. 2002;30(4):394–9.
Oca-Cossio J, Kenyon L, Hao H, Moraes CT. Limitations of allotopic expression of mitochondrial genes in mammalian cells. Genetics. 2003;165(2):707–20.
Perales-Clemente E, Fernandez-Silva P, Acin-Perez R, Perez-Martos A, Enriquez JA. Allotopic expression of mitochondrial-encoded genes in mammals: achieved goal, undemonstrated mechanism or impossible task? Nucleic Acids Res. 2011;39(1):225–34.
Figueroa-Martinez F, Vazquez-Acevedo M, Cortes-Hernandez P, Garcia-Trejo JJ, Davidson E, King MP, Gonzalez-Halphen D. What limits the allotopic expression of nucleus-encoded mitochondrial genes? The case of the chimeric cox3 and atp6 genes. Mitochondrion. 2011;11(1):147–54.
Bonnet C, Kaltimbacher V, Ellouze S, Augustin S, Benit P, Forster V, Rustin P, Sahel JA, Corral-Debrinski M. Allotopic mrna localization to the mitochondrial surface rescues respiratory chain defects in fibroblasts harboring mitochondrial DNA mutations affecting complex i or v subunits. Rejuvenation Res. 2007;10(2):127–44.
Bonnet C, Augustin S, Ellouze S, Benit P, Bouaita A, Rustin P, Sahel JA, Corral-Debrinski M. The optimized allotopic expression of nd1 or nd4 genes restores respiratory chain complex i activity in fibroblasts harboring mutations in these genes. Biochim Biophys Acta. 2008;1783(10):1707–17.
Pei H, Wan X, Hu W, Dong X, Li B. Construction and detection of a novel type of recombinant human raav2/2-nd4. Eye Sci. 2013;28(2):55–9.
Karaarslan C. Leber’s hereditary optic neuropathy as a promising disease for gene therapy development. Adv Ther. 2019;36(12):3299–307.
Qi X, Sun L, Lewin AS, Hauswirth WW, Guy J. The mutant human nd4 subunit of complex i induces optic neuropathy in the mouse. Invest Ophthalmol Vis Sci. 2007;48(1):1–10.
Ellouze S, Augustin S, Bouaita A, Bonnet C, Simonutti M, Forster V, Picaud S, Sahel JA, Corral-Debrinski M. Optimized allotopic expression of the human mitochondrial nd4 prevents blindness in a rat model of mitochondrial dysfunction. Am J Hum Genet. 2008;83(3):373–87.
Koilkonda RD, Chou TH, Porciatti V, Hauswirth WW, Guy J. Induction of rapid and highly efficient expression of the human nd4 complex i subunit in the mouse visual system by self-complementary adeno-associated virus. Arch Ophthalmol. 2010;128(7):876–83.
Cwerman-Thibault H, Augustin S, Lechauve C, Ayache J, Ellouze S, Sahel JA, Corral-Debrinski M. Nuclear expression of mitochondrial nd4 leads to the protein assembling in complex i and prevents optic atrophy and visual loss. Mol Ther Methods Clin Dev. 2015;2:15003.
Koilkonda R, Yu H, Talla V, Porciatti V, Feuer WJ, Hauswirth WW, Chiodo V, Erger KE, Boye SL, Lewin AS, Conlon TJ, et al. Lhon gene therapy vector prevents visual loss and optic neuropathy induced by g11778a mutant mitochondrial DNA: biodistribution and toxicology profile. Invest Ophthalmol Vis Sci. 2014;55(12):7739–53.
Lin CS, Sharpley MS, Fan W, Waymire KG, Sadun AA, Carelli V, Ross-Cisneros FN, Baciu P, Sung E, McManus MJ, Pan BX, et al. Mouse mtdna mutant model of leber hereditary optic neuropathy. Proc Natl Acad Sci USA. 2012;109(49):20065–70.
Barrientos A, Kenyon L, Moraes CT. Human xenomitochondrial cybrids. Cellular models of mitochondrial complex i deficiency. J Biol Chem. 1998;273(23):14210–7.
Feuer WJ, Schiffman JC, Davis JL, Porciatti V, Gonzalez P, Koilkonda RD, Yuan H, Lalwani A, Lam BL, Guy J. Gene therapy for leber hereditary optic neuropathy: initial results. Ophthalmology. 2016;123(3):558–70.
Guy J, Feuer WJ, Davis JL, Porciatti V, Gonzalez PJ, Koilkonda RD, Yuan H, Hauswirth WW, Lam BL. Gene therapy for leber hereditary optic neuropathy: low- and medium-dose visual results. Ophthalmology. 2017;124(11):1621–34.
Lam BL, Feuer WJ, Schiffman JC, Porciatti V, Vandenbroucke R, Rosa PR, Gregori G, Guy J. Trial end points and natural history in patients with g11778a leber hereditary optic neuropathy: preparation for gene therapy clinical trial. JAMA Ophthalmol. 2014;132(4):428–36.
Zhang Y, Yuan JJ, Liu HL, Tian Z, Liu SW, Li B. Three cases of leber’s hereditary optic neuropathy with rapid increase in visual acuity after gene therapy. Curr Gene Ther. 2019;19(2):134–8.
Yang S, Ma SQ, Wan X, He H, Pei H, Zhao MJ, Chen C, Wang DW, Dong XY, Yuan JJ, Li B. Long-term outcomes of gene therapy for the treatment of leber’s hereditary optic neuropathy. EBioMedicine. 2016;10:258–68.
Zhang Y, Li X, Yuan J, Tian Z, Liu H, Wang D, Li B. Prognostic factors for visual acuity in patients with leber’s hereditary optic neuropathy after raav2-nd4 gene therapy. Clin Exp Ophthalmol. 2019;47(6):774–8.
Liu HL, Yuan JJ, Zhang Y, Tian Z, Li X, Wang D, Du YY, Song L, Li B. Factors associated with rapid improvement in visual acuity in patients with leber's hereditary optic neuropathy after gene therapy. Acta Ophthalmol. 2020;98(6):e730–3.
Vignal C, Uretsky S, Fitoussi S, Galy A, Blouin L, Girmens JF, Bidot S, Thomasson N, Bouquet C, Valero S, Meunier S, et al. Safety of raav2/2-nd4 gene therapy for leber hereditary optic neuropathy. Ophthalmology. 2018;125(6):945–7.
Weiss JN, Levy S. Stem cell ophthalmology treatment study (scots): bone marrow derived stem cells in the treatment of dominant optic atrophy. Stem Cell Investig. 2019;6:41.
Johnson TV, Bull ND, Hunt DP, Marina N, Tomarev SI, Martin KR. Neuroprotective effects of intravitreal mesenchymal stem cell transplantation in experimental glaucoma. Invest Ophthalmol Vis Sci. 2010;51(4):2051–9.
Levkovitch-Verbin H, Sadan O, Vander S, Rosner M, Barhum Y, Melamed E, Offen D, Melamed S. Intravitreal injections of neurotrophic factors secreting mesenchymal stem cells are neuroprotective in rat eyes following optic nerve transection. Invest Ophthalmol Vis Sci. 2010;51(12):6394–400.
Yu S, Tanabe T, Dezawa M, Ishikawa H, Yoshimura N. Effects of bone marrow stromal cell injection in an experimental glaucoma model. Biochem Biophys Res Commun. 2006;344(4):1071–9.
Li N, Li XR, Yuan JQ. Effects of bone-marrow mesenchymal stem cells transplanted into vitreous cavity of rat injured by ischemia/reperfusion. Graefes Arch Clin Exp Ophthalmol. 2009;247(4):503–14.
Zhao T, Li Y, Tang L, Li Y, Fan F, Jiang B. Protective effects of human umbilical cord blood stem cell intravitreal transplantation against optic nerve injury in rats. Graefes Arch Clin Exp Ophthalmol. 2011;249(7):1021–8.
Zwart I, Hill AJ, Al-Allaf F, Shah M, Girdlestone J, Sanusi AB, Mehmet H, Navarrete R, Navarrete C, Jen LS. Umbilical cord blood mesenchymal stromal cells are neuroprotective and promote regeneration in a rat optic tract model. Exp Neurol. 2009;216(2):439–48.
Lopez Sanchez MI, Crowston JG, Mackey DA, Trounce IA. Emerging mitochondrial therapeutic targets in optic neuropathies. Pharmacol Ther. 2016;165:132–52.
Johnson TV, DeKorver NW, Levasseur VA, Osborne A, Tassoni A, Lorber B, Heller JP, Villasmil R, Bull ND, Martin KR, Tomarev SI. Identification of retinal ganglion cell neuroprotection conferred by platelet-derived growth factor through analysis of the mesenchymal stem cell secretome. Brain. 2014;137(Pt 2):503–19.
Weiss JN, Levy S, Benes SC. Stem cell ophthalmology treatment study (scots): bone marrow-derived stem cells in the treatment of leber’s hereditary optic neuropathy. Neural Regen Res. 2016;11(10):1685–94.
Liu K, Ji K, Guo L, Wu W, Lu H, Shan P, Yan C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res. 2014;92:10–8.
Liu K, Guo L, Zhou Z, Pan M, Yan C. Mesenchymal stem cells transfer mitochondria into cerebral microvasculature and promote recovery from ischemic stroke. Microvasc Res. 2019;123:74–80.
Doudna JA. The promise and challenge of therapeutic genome editing. Nature. 2020;578(7794):229–36.
Naso MF, Tomkowicz B, Perry WL 3rd, Strohl WR. Adeno-associated virus (aav) as a vector for gene therapy. BioDrugs. 2017;31(4):317–34.
Civiletto G, Varanita T, Cerutti R, Gorletta T, Barbaro S, Marchet S, Lamperti C, Viscomi C, Scorrano L, Zeviani M. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 2015;21(6):845–54.
Varanita T, Soriano ME, Romanello V, Zaglia T, Quintana-Cabrera R, Semenzato M, Menabo R, Costa V, Civiletto G, Pesce P, Viscomi C, et al. The opa1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015;21(6):834–44.
Del Dotto V, Mishra P, Vidoni S, Fogazza M, Maresca A, Caporali L, McCaffery JM, Cappelletti M, Baruffini E, Lenaers G, Chan D, et al. Opa1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep. 2017;19(12):2557–71.
Alavi MV, Bette S, Schimpf S, Schuettauf F, Schraermeyer U, Wehrl HF, Ruttiger L, Beck SC, Tonagel F, Pichler BJ, Knipper M, et al. A splice site mutation in the murine opa1 gene features pathology of autosomal dominant optic atrophy. Brain. 2007;130(Pt 4):1029–42.
Davies VJ, Hollins AJ, Piechota MJ, Yip W, Davies JR, White KE, Nicols PP, Boulton ME, Votruba M. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet. 2007;16(11):1307–18.
Sarzi E, Angebault C, Seveno M, Gueguen N, Chaix B, Bielicki G, Boddaert N, Mausset-Bonnefont AL, Cazevieille C, Rigau V, Renou JP, et al. The human opa1delttag mutation induces premature age-related systemic neurodegeneration in mouse. Brain. 2012;135(Pt 12):3599–613.
Zaninello M, Palikaras K, Naon D, Iwata K, Herkenne S, Quintana-Cabrera R, Semenzato M, Grespi F, Ross-Cisneros FN, Carelli V, Sadun AA, et al. Inhibition of autophagy curtails visual loss in a model of autosomal dominant optic atrophy. Nat Commun. 2020;11(1):4029.
Srivastava S, Moraes CT. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum Mol Genet. 2001;10(26):3093–9.
Bayona-Bafaluy MP, Blits B, Battersby BJ, Shoubridge EA, Moraes CT. Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc Natl Acad Sci USA. 2005;102(40):14392–7.
Bacman SR, Gammage PA, Minczuk M, Moraes CT. Manipulation of mitochondrial genes and mtdna heteroplasmy. Methods Cell Biol. 2020;155:441–87.
Zekonyte U, Bacman SR, Moraes CT. DNA-editing enzymes as potential treatments for heteroplasmic mtdna diseases. J Intern Med. 2020;287(6):685–97.
Bacman SR, Moraes CT. Transmitochondrial technology in animal cells. Methods Cell Biol. 2007;80:503–24.
Gammage PA, Rorbach J, Vincent AI, Rebar EJ, Minczuk M. Mitochondrially targeted zfns for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol Med. 2014;6(4):458–66.
Hashimoto M, Bacman SR, Peralta S, Falk MJ, Chomyn A, Chan DC, Williams SL, Moraes CT. Mitotalen: a general approach to reduce mutant mtdna loads and restore oxidative phosphorylation function in mitochondrial diseases. Mol Ther. 2015;23(10):1592–9.
Gammage PA, Viscomi C, Simard ML, Costa ASH, Gaude E, Powell CA, Van Haute L, McCann BJ, Rebelo-Guiomar P, Cerutti R, Zhang L, et al. Genome editing in mitochondria corrects a pathogenic mtdna mutation in vivo. Nat Med. 2018;24(11):1691–5.
Bacman SR, Kauppila JHK, Pereira CV, Nissanka N, Miranda M, Pinto M, Williams SL, Larsson NG, Stewart JB, Moraes CT. Mitotalen reduces mutant mtdna load and restores trna(ala) levels in a mouse model of heteroplasmic mtdna mutation. Nat Med. 2018;24(11):1696–700.
Loutre R, Heckel AM, Smirnova A, Entelis N, Tarassov I. Can mitochondrial DNA be crisprized: pro and contra. IUBMB Life. 2018;70(12):1233–9.
Ledford H. Scientists make precise gene edits to mitochondrial DNA for first time. Nature. 2020;583(7816):343.
Richardson J, Irving L, Hyslop LA, Choudhary M, Murdoch A, Turnbull DM, Herbert M. Concise reviews: assisted reproductive technologies to prevent transmission of mitochondrial DNA disease. Stem Cells. 2015;33(3):639–45.
Wolf DP, Mitalipov N, Mitalipov S. Mitochondrial replacement therapy in reproductive medicine. Trends Mol Med. 2015;21(2):68–76.
Hyslop LA, Blakeley P, Craven L, Richardson J, Fogarty NM, Fragouli E, Lamb M, Wamaitha SE, Prathalingam N, Zhang Q, O’Keefe H, et al. Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature. 2016;534(7607):383–6.
Tachibana M, Amato P, Sparman M, Woodward J, Sanchis DM, Ma H, Gutierrez NM, Tippner-Hedges R, Kang E, Lee HS, Ramsey C, et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature. 2013;493(7434):627–31.
Paull D, Emmanuele V, Weiss KA, Treff N, Stewart L, Hua H, Zimmer M, Kahler DJ, Goland RS, Noggle SA, Prosser R, et al. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature. 2013;493(7434):632–7.
Wang T, Sha H, Ji D, Zhang HL, Chen D, Cao Y, Zhu J. Polar body genome transfer for preventing the transmission of inherited mitochondrial diseases. Cell. 2014;157(7):1591–604.
Russell OM, Gorman GS, Lightowlers RN, Turnbull DM. Mitochondrial diseases: hope for the future. Cell. 2020;181(1):168–88.
Zhang J, Liu H, Luo S, Lu Z, Chavez-Badiola A, Liu Z, Yang M, Merhi Z, Silber SJ, Munne S, Konstantinidis M, et al. Live birth derived from oocyte spindle transfer to prevent mitochondrial disease. Reprod Biomed Online. 2017;34(4):361–8.
Reardon S. Genetic details of controversial “three-parent baby” revealed. Nature. 2017;544(7648):17–8.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76.
Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2017;16(2):115–30.
Kruczek K, Swaroop A. Pluripotent stem cell-derived retinal organoids for disease modeling and development of therapies. Stem Cells. 2020;38(10):1206–15.
Jin ZB, Gao ML, Deng WL, Wu KC, Sugita S, Mandai M, Takahashi M. Stemming retinal regeneration with pluripotent stem cells. Prog Retin Eye Res. 2019;69:38–56.
Fligor CM, Huang KC, Lavekar SS, VanderWall KB, Meyer JS. Differentiation of retinal organoids from human pluripotent stem cells. Methods Cell Biol. 2020;159:279–302.
Cowan CS, Renner M, De Gennaro M, Gross-Scherf B, Goldblum D, Hou Y, Munz M, Rodrigues TM, Krol J, Szikra T, Cuttat R, et al. Cell types of the human retina and its organoids at single-cell resolution. Cell. 2020;182(6):1623–40 (e1634).
Laha B, Stafford BK, Huberman AD. Regenerating optic pathways from the eye to the brain. Science. 2017;356(6342):1031–4.
Kang E, Wang X, Tippner-Hedges R, Ma H, Folmes CD, Gutierrez NM, Lee Y, Van Dyken C, Ahmed R, Li Y, Koski A, et al. Age-related accumulation of somatic mitochondrial DNA mutations in adult-derived human ipscs. Cell Stem Cell. 2016;18(5):625–36.
Perales-Clemente E, Cook AN, Evans JM, Roellinger S, Secreto F, Emmanuele V, Oglesbee D, Mootha VK, Hirano M, Schon EA, Terzic A, et al. Natural underlying mtdna heteroplasmy as a potential source of intra-person hipsc variability. EMBO J. 2016;35(18):1979–90.
Payne BA, Wilson IJ, Yu-Wai-Man P, Coxhead J, Deehan D, Horvath R, Taylor RW, Samuels DC, Santibanez-Koref M, Chinnery PF. Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet. 2013;22(2):384–90.
Yang TC, Yarmishyn AA, Yang YP, Lu PC, Chou SJ, Wang ML, Lin TC, Hwang DK, Chou YB, Chen SJ, Yu WK, et al. Mitochondrial transport mediates survival of retinal ganglion cells in affected lhon patients. Hum Mol Genet. 2020;29(9):1454–64.
Chen J, Riazifar H, Guan MX, Huang T. Modeling autosomal dominant optic atrophy using induced pluripotent stem cells and identifying potential therapeutic targets. Stem Cell Res Ther. 2016;7:2.
Garone C, Viscomi C. Towards a therapy for mitochondrial disease: an update. Biochem Soc Trans. 2018;46(5):1247–61.
Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, Oh K, et al. Mtor inhibition alleviates mitochondrial disease in a mouse model of leigh syndrome. Science. 2013;342(6165):1524–8.
Dai Y, Zheng K, Clark J, Swerdlow RH, Pulst SM, Sutton JP, Shinobu LA, Simon DK. Rapamycin drives selection against a pathogenic heteroplasmic mitochondrial DNA mutation. Hum Mol Genet. 2014;23(3):637–47.
Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J, Viscomi C, et al. Nad(+)-dependent activation of sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014;19(6):1042–9.
Pirinen E, Auranen M, Khan NA, Brilhante V, Urho N, Pessia A, Hakkarainen A, Kuula J, Heinonen U, Schmidt MS, Haimilahti K, et al. Niacin cures systemic nad(+) deficiency and improves muscle performance in adult-onset mitochondrial myopathy. Cell Metab. 2020;31(6):1078–90 (e1075).
Rajman L, Chwalek K, Sinclair DA. Therapeutic potential of nad-boosting molecules: the in vivo evidence. Cell Metab. 2018;27(3):529–47.
Schondorf DC, Ivanyuk D, Baden P, Sanchez-Martinez A, De Cicco S, Yu C, Giunta I, Schwarz LK, Di Napoli G, Panagiotakopoulou V, Nestel S, et al. The nad+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in ipsc and fly models of parkinson’s disease. Cell Rep. 2018;23(10):2976–88.
Levin LA. Neuroprotection in optic neuropathy. Asia Pac J Ophthalmol (Phila). 2018;7(4):246–50.
Solano EC, Kornbrust DJ, Beaudry A, Foy JW, Schneider DJ, Thompson JD. Toxicological and pharmacokinetic properties of qpi-1007, a chemically modified synthetic sirna targeting caspase 2 mrna, following intravitreal injection. Nucleic Acid Ther. 2014;24(4):258–66.
Rath EZ, Hazan Z, Adamsky K, Solomon A, Segal ZI, Levin LA. Randomized controlled phase 2a study of rph201 in previous nonarteritic anterior ischemic optic neuropathy. J Neuroophthalmol. 2019;39(3):291–8.
Villoslada P, Vila G, Colafrancesco V, Moreno B, Fernandez-Diez B, Vazquez R, Pertsovskaya I, Zubizarreta I, Pulido-Valdeolivas I, Messeguer J, Vendrell-Navarro G, et al. Axonal and myelin neuroprotection by the peptoid bn201 in brain inflammation. Neurotherapeutics. 2019;16(3):808–27.
Acknowledgements
Our research on hereditary optic neuropathies, over the last ten years, has been supported by the Italian Ministries of Health and of Research (to VC, CLM); Telethon—Italy [Grant numbers GGP06233, GGP10005, GGP11182, GGP14187] (to VC); ‘Programma di ricerca Regione-Università 2010–2012’ [Grant number PRUa1RI-2012-008] (to VC); the patient-led organisations (IFOND, UMDF, MITOCON, The Poincenot Family, and the Gino Galletti Foundation) and patient's donations (to VC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding has been received for the preparation of this manuscript.
Conflict of interest
VC acts as scientific consultant in boards of GenSight Biologics, Stealth BioTherapeutics, Santhera Pharmaceuticals, and Chiesi. VC received speaker honoraria from Chiesi and an unrestricted research grant from Stealth BioTherapeutics. PB, MC, VC, CLM, MR, received speaker’s honoraria and travel support from Santhera Pharmaceuticals. PB received speaker honoraria from GenSight Biologics, Visufarma and Omkron. CLM received consulting fees from Regulatory Pharmanet and Chiesi Pharmaceuticals. GA, PB, MC, VC, CLM are investigators in clinical trials from Santhera Pharmaceuticals and GenSight Biologics. MR serves as Study Coordinator in Santhera Pharmaceuticals and GenSight Biologics sponsored trials. None of these activities are in any way related to the writing of this study.
Ethics approval
Not applicable.
Consent
Not applicable.
Data availability
Not applicable. No datasets were generated or analysed during the current study.
Author contributions
All authors contributed to the study conception and design. Material preparation and data collection were performed by GA, MR, MC, PB and CLM. The first draft of the manuscript was written by GA, MR, VC and CLM. VC and CLM reviewed the manuscript. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Amore, G., Romagnoli, M., Carbonelli, M. et al. Therapeutic Options in Hereditary Optic Neuropathies. Drugs 81, 57–86 (2021). https://doi.org/10.1007/s40265-020-01428-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-020-01428-3