
Abstract
Hepatitis C virus (HCV)-infected patients often suffer from liver cirrhosis, which can be complicated by renal impairment. Therefore, in this review we describe the treatment possibilities in HCV patients with hepatic and renal impairment. Cirrhosis alters the structure of the liver, which affects drug-metabolizing enzymes and drug transporters. These modifications influence the plasma concentration of substrates of drugs metabolized/transported by these enzymes. The direct-acting antivirals (DAAs) are substrates of, for example, cytochrome P450 enzymes in the liver. Most DAAs are not studied in HCV-infected individuals with decompensated cirrhosis, and therefore awareness is needed when these patients are treated. Most DAAs are contraindicated in cirrhotic patients; however, patients with a Child-Pugh score of B or C can be treated safely with a normal dose sofosbuvir plus ledipasvir or daclatasvir, in combination with ribavirin. Patients with renal impairment (glomerular filtration rate [GFR] <90 mL/min) or who are dependent on dialysis often tolerate ribavirin treatment poorly, even after dose adjustments. However, most DAAs can be used at the normal dose because DAAs are not renally excreted. To date, grazoprevir plus elbasvir is the preferred DAA regimen in patients with renal impairment as data are pending for sofosbuvir patients with GFR <30 mL/min (as for ledipasvir and velpatasvir). However, sofosbuvir has been used in a small number of patients with severe renal impairment and, based on these trials, we recommend sofosbuvir 400 mg every day when no other DAA regimen is available. Ledipasvir and velpatasvir are not recommended in patients with severe renal impairment.
Similar content being viewed by others


All drugs used in hepatitis C virus (HCV) treatment can be used in patient with compensated liver cirrhosis (Child-Pugh score A). |
All drugs used in HCV treatment can be used in patients with moderate renal insufficiency (glomerular filtration rate [GFR] ≥30 mL/min). |
In patients with GFR ≤29 mL/min or advanced liver disease, HCV drugs might be contraindicated or dosage adjustments may be necessary. |
1 Introduction
Chronic hepatitis C virus (HCV)-related liver cirrhosis is the leading cause of liver transplantation in many countries [1–3]. Eventually, 15–30 % of chronically infected HCV patients develop liver cirrhosis [4, 5]. Symptoms of decompensated cirrhosis are portal hypertension (with increased risk for variceal bleedings), ascites, hepatic encephalopathy, and hepato-renal syndrome. In addition, cirrhotic patients have an enhanced risk of hepatocellular carcinoma, which is an important cause of mortality [6, 7].
HCV is associated with both renal and hepatic impairment, and care must be taken when prescribing direct-acting antivirals (DAAs) in these patients. The drugs described in this review are ribavirin and the novel DAAs.
Impaired kidney or liver function may result in altered drug concentrations, causing either toxicity or subtherapeutic levels, because these organs are mainly responsible for metabolizing and excreting drugs. For instance, patients with reduced renal function have a decreased ability to eliminate water-soluble agents [8] and patients with impaired liver function have reduced expression of drug-metabolizing enzymes and thus reduced metabolizing capacity [6].
There is only limited information on the pharmacokinetics, safety, efficacy, and dosage in these special populations. Moreover, this information is often difficult to find and not presented in a comprehensive manner. Therefore, the aim of this review is to give an overview of the pharmacokinetics, efficacy, and safety of drugs used for HCV treatment in patients with renal or hepatic impairment and to provide dose recommendations for prescribing these drugs in these special populations.
2 Methods
An extensive search was performed using PubMed (1946 to October 2015) and EMBASE (1947 to October 2015) to identify peer-reviewed studies containing information on pharmacokinetics, efficacy, and safety in patients with impaired renal or hepatic function and HCV medication. Search terms used included generic and brand names. Various general search terms were also used describing impaired renal and hepatic function, e.g., ‘end stage renal disease’ (ESRD), ‘dialysis’, ‘cirrhosis’, and ‘hepatic impairment’. Google, Google Scholar, and ClinicalTrials.gov were used to identify conference papers and abstracts. All searches were performed in the English language. Additional articles and primary sources were identified with citation snowballing. Lastly, the summary of product characteristics (SmPC) approved by the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) prescribing information were main sources of information for this review.
This review focuses on the novel DAAs, e.g., simeprevir, paritaprevir, asunaprevir, grazoprevir, daclatasvir, ombitasvir, ledipasvir, elbasvir, velpatasvir, sofosbuvir, and dasabuvir. To date, velpatasvir is not yet licensed. The included DAAs are used in international guidelines [9, 10] or were submitted for registration up to November 2015 by the EMA and/or FDA. Ribavirin is also discussed because it is still a component of the therapy for cirrhotic patients. We omitted telaprevir and boceprevir from the review as their current use is limited. Additionally, we do not describe peginterferon-α as we believe it should not be used in patients with cirrhosis or renal impairment.
3 Pharmacokinetics
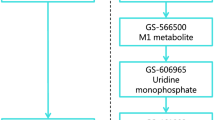
This section summarizes the pharmacokinetics of drugs used in HCV treatment, in both healthy subjects and in patients with impaired renal or hepatic function: DAAs (protease inhibitors [PIs], NS5A inhibitors, NS5B polymerase inhibitors, and fixed-dose regimens) and other antivirals (ribavirin). The clinical consequences and dosage recommendations based on these observations are summarized in Tables 1 and 2. Figure 1 gives an overview of the hepatic and renal metabolism of these drugs.

Overview of the hepatic or non-enzymatic metabolism of drugs used for the treatment of hepatitis C: cytochrome P450 enzymes involved and biliary and/or renal excretion of drug (metabolites). Asterisk The site of metabolism is unknown but two metabolizing pathways are involved: (1) a reversible phosphorylation pathway; and (2) a degradative pathway involving deribosylation and amide hydrolysis. Plus or minus Sofosbuvir is extensively metabolized in the liver in the active metabolite GS-461203, followed by dephosphorylation which results in the inactive compound GS-331007. ASV asunaprevir, CYP cytochrome P450, DCV daclatasvir, DSV dasabuvir, EBV elbasvir, GRZ grazoprevir, LDV ledipasvir, OBV ombitasvir, PTV paritaprevir, RBV ribavirin, SIM simeprevir, SOF sofosbuvir, VPV velpatasvir
3.1 Protease Inhibitors
3.1.1 Simeprevir
Simeprevir is a second-wave, first-generation PI and is prescribed at a dose of 150 mg/day. Simeprevir is highly bound to plasma proteins (>99.9 %), and is a substrate of various drug transporters such as P-glycoprotein (P-gp), organic anion-transporting polypeptide (OATP) 1B1, OATP1B2, OATP2B1, and multidrug resistance protein (MRP2), and different cytochrome P450 (CYP) enzymes (intestinal CYP3A4, CYP2C19, and CYP2C8). The plasma concentration of simeprevir was two- to three-fold higher in HCV-infected patients than in healthy subjects [11].
Compared with healthy individuals, simeprevir steady-state area under the plasma concentration–time curve (AUC) was 2.4- and 5.2-fold higher in Child-Pugh score B (CP-B) and score C (CP-C) patients, respectively. Therefore, the manufacturer recommends that simeprevir should not be used in CP-C patients and that caution should be taken in CP-B patients [11]. Another trial reported similar results: non-HCV CP-B patients had twofold increased exposure compared with healthy individuals and CP-C patients had twofold higher exposure to simeprevir than CP-B patients [12]. After a dose of 150 mg, Sekar et al. [13] observed equal exposure and protein binding between non-HCV Child-Pugh score A (CP-A) and CP-B subjects.
The steady-state AUC of simeprevir increased (62 %) in patients with severe renal impairment (Glomerular Filtration Rate [GFR]: 15–29 mL/min). This may indicate that exposure may increase in patients with severe renal impairment and ESRD (GFR ≤15 mL/min). Thus caution is needed in these patients. However, the label states that simeprevir can be used by patients with all grades of renal impairment. At last, simeprevir is not removed by dialysis [11].
3.1.2 Asunaprevir
Asunaprevir is a PI that has activity against multiple genotypes. It is used at a dose of 100 mg twice daily which is metabolized by the liver (CYP3A4) and mainly excreted through the biliary system. Asunaprevir is 98.8 % bound to serum proteins [14, 15].
The pharmacokinetics of asunaprevir were studied in non-HCV infected subjects with CP-A/B/C and compared with healthy volunteers; they were comparable in CP-A subjects and controls. Maximum concentration (C max) and AUC increased 10- and 5-fold in CP-B subjects and 23- and 32-fold in CP-C subjects, respectively. Therefore, it is not recommended that CP-B/C patients be treated with asunaprevir. Protein binding in all groups was >99.5 % and the unbound fraction was ± 0.004 [14].
Asunaprevir was studied in non-HCV subjects dependent on dialysis compared with healthy controls. Protein binding, C max, AUC, and trough concentration (C trough) were not affected by dialysis [16]. Comparable results were presented in an open-label study in HCV-uninfected subjects with normal (GFR >90 mL/min), mild (GFR 50–89 mL/min), moderate (GFR 30–49 mL/min), or severe renal disease (GFR <30 mL/min) or patients dependent on dialysis. Subjects received asunaprevir, daclatasvir, and beclabuvir (NS5B inhibitor). ESRD subjects had slightly decreased asunaprevir concentrations. Subjects with moderate and severe renal impairment had increased C max (65 and 100 %, respectively) and AUC (50 and 76 %, respectively) values, respectively, compared with controls [17].
3.2 NS5A Inhibitors
3.2.1 Daclatasvir
Daclatasvir is an NS5A inhibitor that is administered at a dosage of 60 mg/day. Daclatasvir is highly bound to plasma proteins (99 %). It is hepatically metabolized (CYP3A4) and is a substrate of P-gp. Biliary excretion is the major route of elimination.
Compared with healthy volunteers, C max and AUC (total daclatasvir = unbound and bound drug) values were lower in non-HCV patients with CP-A/B/C after a single dose of daclatasvir 30 mg. However, there was no influence on the unbound fraction of daclatasvir when CP-B/C patients were compared with HCV-infected controls [18, 19].
Patients with mild, moderate, severe, or end-stage renal disease had increased unbound daclatasvir AUCs of 18, 39, 51, and 20 %, respectively, compared with normal renal function. A similar trend was seen in total daclatasvir exposure. Although the exposure was affected, the authors concluded that no dose adjustments are necessary in patients with renal impairment and that these differences are within the high inter-individual variability of daclatasvir pharmacokinetics [18, 20].
The steady-state pharmacokinetics of daclatasvir 60 mg have been studied in combination with asunaprevir and beclabuvir in patients with moderate and severe renal impairment, showing increased exposure of daclatasvir (C max 35 and 45 %, and AUC 50 and 65 %, respectively). Patients undergoing dialysis had comparable pharmacokinetic parameters with healthy subjects [17].
3.3 NS5B Polymerase Inhibitors
3.3.1 Sofosbuvir
Sofosbuvir is an NS5B polymerase inhibitor that is administered at 400 mg/day. Sofosbuvir is intracellulair metabolized and forms the active metabolite GS-461203, followed by dephosphorylation resulting in the inactive compound GS-331007. GS-331007 is primarily renally excreted (78 % of the administered dose). Sofosbuvir is a substrate of P-gp and breast cancer resistance protein (BCRP) and is 61–65 % bound to plasma proteins. GS-331007 is minimally bound to plasma proteins [21, 22].
In a study of the pharmacokinetic properties of sofosbuvir, the steady-state AUC of sofosbuvir 400 mg following 7-day dosing in CP-B and CP-C patients increased 126 and 143 %, respectively, relative to control subjects. The GS-331007 AUC was slightly increased: 18 and 9 % [23]. Lawitz et al. [24] reported increased C max and AUC values of sofosbuvir of 80 and 130 %, respectively, in patients with hepatic impairment (CP-B and CP-C) compared with non-cirrhotic controls. The pharmacokinetics of GS-331007 were similar in these three groups.
In patients with mild, moderate, and severe renal insufficiency sofosbuvir, AUC values were elevated by 61, 107, and 171 % compared with controls. GS-331007 AUC values was 55, 88, and 451 % higher in these patients. Administration before and after dialysis influenced the exposure to GS-331007 as it is removed during dialysis. After 4 h of dialysis, 18 % of the administered dose had been removed [23, 25]. A study comparing sofosbuvir 400 mg every day or only on the day of dialysis showed that there was no accumulation of sofosbuvir or GS-331007 in both treatment groups [26]. Gane et al. treated patients with severe renal impairment with daily sofosbuvir 200 mg and low-dose ribavirin. Compared with historical controls the patients had comparable sofosbuvir exposure and fourfold higher GS-331007 concentrations [27]. A recently presented study of ten patients describing the steady-state pharmacokinetics of sofosbuvir in patients with a GFR of <30 mL/min (mean creatinine clearance 26.2 mL/min) showed comparable results. Exposure to GS-331007 and sofosbuvir increased 6- and 1.4-fold, respectively, compared with patients with normal renal function [28].
The manufacturer does not recommend using sofosbuvir in patients with severe renal impairment or ESRD, since studies are still ongoing (NCT01958281 [29]). The main issue might be the increased exposure to GS-331007 (AUC 451 %). This is caused by decreased clearance of GS-331007. However, increased exposure of GS-331007 is not associated with increased toxicity [30].
Several small studies and case reports have shown that both low-dose (200 mg) and normal-dose (400 mg) sofosbuvir were overall well-tolerated [26, 31]. Pending more definite results of ongoing studies, we recommend patients be treated with sofosbuvir 400 mg/day (GFR <30 mL/min or ESRD) in case there is no safer DAA option available. We base this advice on a number of arguments. First, accumulation of sofosbuvir does not take place in patients dependent on dialysis, suggesting that a standard dosage of 400 mg/day will produce similar concentrations of active intracellular metabolites independent of renal function [26]. Secondly, (interim) analyses of small studies show that sofosbuvir at standard doses is well-tolerated in these patients groups. Lastly, data are available for the sustained virologic response (SVR) at week 12 (SVR12) of patients treated with half-dose sofosbuvir, which varied from 40 to 90 % [28, 32]. Patients treated with sofosbuvir 400 mg/day reached SVR12 in 60–100 % of cases [28, 33]. These results suggest that a reduced dose of the prodrug sofosbuvir may result in lower concentrations of active intracellular metabolites.
3.4 Fixed-Dose Regimens
3.4.1 Ledipasvir/Sofosbuvir
Ledipasvir is an NS5A inhibitor available in a fixed-dose tablet with sofosbuvir containing sofosbuvir 400 mg and ledipasvir 90 mg. The metabolism of ledipasvir is unknown but unchanged ledipasvir is mainly found in feces, indicating biliary excretion. It is a substrate of P-gp and BCRP and it is >99.8 % bound to plasma proteins [21].
No relevant differences were seen in between the pharmacokinetics of control patients with normal hepatic function and CP-C patients after a dose of ledipasvir 60 mg [21]. Single and multiple doses of ledipasvir 30 mg (in combination with 200 mg of the investigational PI vedroprevir) resulted in a reduction of the C max (36 %) and an extended elimination half-life (t ½) in CP-C patients (84.4 vs. 45.7 h in healthy subjects). The free fraction of ledipasvir increased in patients with severe hepatic impairment (0.21 vs. 0.11 % in healthy subjects). No significant changes were seen between CP-B patients and control subjects [34].
No pharmacokinetic differences were observed between healthy subjects and patients with severe renal impairment, although no safety data are available for patients with GFR <30 mL/min or ESRD (Sect. 4.4.1) [21, 35].
The pharmacokinetics of sofosbuvir in patients with impaired renal and hepatic function are discussed in Sect. 3.3.1.
3.4.2 Velpatasvir/Sofosbuvir
Velpatasvir is a novel NS5A inhibitor that will probably be licensed in a fixed-dose tablet with sofosbuvir (100 mg/400 mg) [36, 37]. Velpatasvir is primarily metabolized by the liver and excreted through the biliary system. Velpatasvir is substrate of P-gp and OATPs, and strong inducers or inhibitors of CYP influence the plasma concentration of velpatasvir, suggesting it is a substrate of CYP enzymes [38, 39].
Non-HCV subjects with CP-B and CP-C received a single dose of velpatasvir 100 mg and the AUC from time zero to infinity (AUC∞) was comparable with subjects with normal hepatic function: AUC∞ decreased 17 % and increased 14 %, respectively. However, C max in both groups decreased ~50 % and the unbound fraction increased with decreasing hepatic function [40].
A study in HCV-uninfected subjects with GFR <30 mL/min showed that renal insufficiency had a modest influence on the pharmacokinetics of velpatasvir (single dose of 100 mg). C max was increased 11 % and AUC∞ increased 50 % [41]. Further studies are ongoing and the results are still pending (NCT02185794) [42].
3.4.3 Grazoprevir/Elbasvir
Grazoprevir (PI) and elbasvir (NS5A inhibitor) are newly licensed in the USA and data from phase III studies were recently published. Grazoprevir is a substrate of CYP3A4, P-gp, and OATPs and prescribed in a dosage of 100 mg/day [43]. Exposure to grazoprevir was approximately one- to two-fold higher in HCV patients than in healthy controls [44–46].
Elbasvir is prescribed in a dose of 50 mg/day. It is a substrate of CYP3A4, P-gp, and OATP [47]. Both elbasvir and grazoprevir are highly hepatically metabolized and less than 1 % is renally extracted [43, 48].
Jacobson et al. [49] presented pharmacokinetic data of grazoprevir plus elbasvir in HCV patients with CP-B. CP-B patients received grazoprevir 50 mg and elbasvir 50 mg and healthy controls received normal-dose grazoprevir and elbasvir. Despite the reduced dose, grazoprevir AUC and C trough values were increased 30 and 73 %, respectively, compared with controls. Elbasvir exposure was comparable between these two groups [49]. However, the fixed-dose combination is only available in a dose of grazoprevir 100 mg and elbasvir 50 mg; therefore, and due to a lack of safety and efficacy data, the combination is contraindicated for CP-B and CP-C patients [50].
Pharmacokinetic data are available in non-HCV patients with GFR <30 mL/min and in patients dependent on dialysis. Dialysis did not influence the steady-state pharmacokinetics of both grazoprevir and elbasvir. Grazoprevir was slightly removed by dialysis (<0.5 %) and elbasvir was not removed. Subjects with GFR <30 mL/min (not on dialysis) had increased grazoprevir and elbasvir exposure. AUC and C trough values of grazoprevir were elevated 65 and 60 % compared with controls (GFR >80 mL/min). Elbasvir pharmacokinetics showed similar results: AUC was 86 % higher and C trough was 107 % higher. The unbound fraction of grazoprevir was comparable between the three treatment groups. The unbound fraction of elbasvir was below the limit of detection [32, 50].
3.4.4 Paritaprevir/Ritonavir, Ombitasvir, and Dasabuvir
The fixed-dose combination of paritaprevir (75 mg), ritonavir (50 mg), and ombitasvir (12.5 mg) is administered as two tablets once daily with or without dasabuvir 250 mg twice daily.
Paritaprevir is a second-generation PI, which is a substrate of CYP3A4/5, P-gp, OATP1B1, and OATP1B3. Ritonavir is added to improve the pharmacokinetics of paritaprevir by inhibiting CYP3A4 (‘boosting’). Paritaprevir itself also inhibits various drug transporters and is 97–98.6 % bound to plasma proteins. After hepatic metabolism, paritaprevir is excreted through the biliary system. CP-C patients had 3.2- and 9.5-fold higher C max and AUC values than control subjects [51, 52]. Paritaprevir is contraindicated in CP-B/C patients. The unbound fraction was 1.1 % in subjects with normal hepatic function and 0.78, 0.75, and 1.2 % by patients with CP-A, CP-B, and CP-C, respectively.
In patients with mild, moderate, and severe renal insufficiency, the AUC of paritaprevir increased by 19, 33, and 45 %. C max was comparable with control subjects [51].
Ombitasvir is an NS5A inhibitor and highly metabolized: only 8.9 % of the unchanged drug is excreted, and a total of 13 metabolites were identified. Amide hydrolysis and oxidative metabolism are responsible for its biotransformation. Ombitasvir is >99.9 % bound to plasma proteins and biliary excretion is the major elimination pathway. In CP-C patients, ombitasvir reduced the AUC and C max values by 68 and 54 %, respectively. The unbound fraction of ombitasvir increased from ~0.020 % in control subjects and CP-A/B patients to 0.047 % in CP-C patients [51, 52]. Ombitasvir exposure was not affected by any degree of renal insufficiency [51].
Dasabuvir is an NS5B polymerase inhibitor and a substrate of CYP2C8, CYP3A4, P-gp, BCRP, and organic cation transporter (OCT) 1. Dasabuvir is hepatically metabolized into seven metabolites, of which M1 accounts for 21 % of the administered dose. However, unchanged dasabuvir accounts for 60 % of the exposure. Dasabuvir is >99.5 % and M1 94.5 % bound to plasma proteins. The AUC values of dasabuvir and M1 were equal in healthy controls and CP-A patients. CP-B patients had reduced dasabuvir and M1 AUC values (16 and 57 %, respectively). CP-C patients had elevated AUCs for dasabuvir and M1: 325 and 77 %, respectively [52, 53]. Dasabuvir unbound fractions were lower in patients with CP-A, CP-B, and CP-C: 0.29, 0.28, and 0.42 %, respectively (control subjects: 0.61 %). The unbound fraction of M1 in control subjects was 5.8 % and it was 5.1, 5.4, and 6.8 % in CP-A, CP-B, and CP-C patients [52]. Due to the elevated AUC of dasabuvir (and M1) in CP-C patients, dasabuvir is contraindicated in these patients.
The AUC of dasabuvir decreased in patients with mild (21 %), moderate (37 %), and severe (50 %) renal insufficiency. As exposure slightly declines in patients with renal impairment, no dose adjustments are required in these patients [53].
To conclude, paritaprevir/ritonavir plus ombitasvir with or without dasabuvir can be used safely in patients with any stage of renal impairment. Due to a recent FDA announcement, the label for this combination regimen has been updated, stating that paritaprevir/ritonavir and ombitasvir with or without dasabuvir is contraindicated for both CP-B and CP-C patients. These changes have been made based on results from post-marketing surveillance showing liver decompensation and liver failure in patients with advanced cirrhosis (CP-B/C) (n = 26) after 1–4 weeks of treatment [54, 55].
3.5 Other Antivirals
3.5.1 Ribavirin
Ribavirin is a guanine analog with activity against a range of RNA and DNA viruses. Ribavirin is always prescribed as part of a combination therapy. In general practice, ribavirin is administered in a weight-based dose (<75 kg = 1000 mg/day; ≥75 kg = 1200 mg/day), although this may differ by genotype and commercial product [56, 57]. The t ½ of ribavirin is ~300 h and approximately 61 % of the administered dose is renally excreted, of which 17 % is unchanged ribavirin. The site of metabolism is unknown but two metabolizing pathways are involved: (1) a reversible phosphorylation pathway; and (2) a degradative pathway involving deribosylation and amide hydrolysis [57]. It is notable that the ratio of whole blood:plasma is 60:1 and the volume of distribution (V d) is 5000L, which is caused by the extensive accumulation of ribavirin in the erythrocytes [56, 57].
The SmPC states that the pharmacokinetics of ribavirin are similar in control subjects and patients with CP-A/B/C and therefore no dose adjustments were deemed necessary in patients with cirrhosis [56]. By contrast, a single-dose study described an increased C max with increasing severity of cirrhosis (the AUC was not significantly different between those patient groups) [58].
Patients with moderate or severe renal impairment had 20–30 % higher ribavirin exposure despite adjusted daily doses of 600 and 400 mg, respectively. ESRD patients had 20 % lower ribavirin plasma exposure when given 200 mg daily than did subjects with GFR >80 mL/min receiving the standard dose [56].
Brennan et al. [59] studied steady-state plasma concentrations in patients with renal impairment. Data were hard to interpret because many dose adjustments were necessary due to toxicity in patients with moderate and severe renal insufficiency. At week 12 of treatment, these patients had 36 and 25 % higher AUCs with adjusted daily doses of 600 and 400 mg, respectively, compared with control subjects. The apparent total clearance of ribavirin from plasma (CL/F) was 20.0 L/h in patients with normal renal function but decreased in patients with renal insufficiency, ranging from 5 to 6 L/h [59]. In a single-dose study, increased AUC and decreased clearance were linearly correlated with the severity of renal dysfunction (single dose of 400 mg) [60].
Taking into account the information from the literature and our clinical experience with ribavirin [61, 62], we recommend a weight-based loading dose of ribavirin followed by 200 mg/day in patients with severe renal dysfunction or ESRD. We also recommend alternating 200 and 400 mg/day in patients with moderate renal function. Steady-state plasma concentrations of ribavirin are directly achieved using a loading dose, which is necessary due to its long t ½. Ribavirin is not removed by dialysis and these patients often have lower hemoglobin levels. Caution is needed due to accumulation of ribavirin in the erythrocytes causing hemolysis. If available, therapeutic drug monitoring can be helpful to individualize treatment with ribavirin in patients with impaired and/or variable renal function.
4 Efficacy and Safety
HCV therapy consists of combinations of drugs, and therefore efficacy and safety are mostly evaluated in patients using combination therapy, making data hard to interpret for only one drug. Efficacy and safety data are shown in Table 3 for CP-A/B/C patients. Table 4 presents the data for patients with mild, moderate, severe, and end-stage renal disease; only multiple-dose studies performed in HCV patients are included.
4.1 Protease Inhibitors
4.1.1 Simeprevir
Adverse events (AEs) were retrospectively reported in 22 % of CP-A/B HCV genotype 1 patients (n = 119) treated with simeprevir and sofosbuvir ± ribavirin. SVR was reached in 78 % of the CP-A/B patients (n = 84), of whom 29 % were CP-B patients [63]. Another retrospective study, in which CP-B/C patients were treated with sofosbuvir and simeprevir, reported that 9 % of patients discontinued due to AEs (CP-A = 1 % discontinued). These patients were hospitalized more often than CP-A patients. Furthermore, 91 % of the CP-A patients reached SVR versus 73 % of the CP-B/C patients [64]. Both the EMA and FDA have warned of possible safety issues with CP-B/C patients because simeprevir safety data are lacking [11, 65].
In an observational study including ESRD patients with or without dialysis (n = 17) treated with simeprevir and sofosbuvir, AEs were reported in 23 %. No patient discontinued due to an AE [33]. Trials describing treatment with a reduced dose of sofosbuvir and simeprevir are discussed in Sect. 4.3.1 [66, 67].
4.1.2 Asunaprevir
An open-label, randomized, uncontrolled trial with daclatasvir 30 mg, asunaprevir 200 mg, and beclabuvir 75 mg twice daily reported SVR rates of ~94 % in naive cirrhotic patients. Treatment-experienced patients had SVR rates ≥87 %. Ribavirin increased treatment response. Nine patients experienced a serious AE (SAE) and three patients discontinued the study due to AEs. The authors concluded that most AEs were caused by ribavirin and that there were no significant differences between cirrhotic and non-cirrhotic patients [68]. In a trial including genotype 1- and 4-infected cirrhotic patients (METAVIR score F3/4; n = 223), an SVR of 84 % was reported after treatment with daclatasvir and asunaprevir. Pooled analyses of four phase II/III studies showed that SVR was reached in 84 % of genotype 1b cirrhotic patients (n = 229). No meaningful differences in safety were described between cirrhotic and non-cirrhotic patients. Overall, most reported AEs were headache, fatigue, nausea, and diarrhea (≥10 %) [69].
An SVR of 96 % was reached in dialysis-dependent genotype 1 patients when treated with daclatasvir 60 mg and asunaprevir 100 mg (n = 21). Of these patients, 97 % experienced an AE. Anemia (29 %) and nasopharyngitis (29 %) were the most commonly reported AEs [70].
4.2 NS5A Inhibitors
4.2.1 Daclatasvir
The safety and efficacy of daclatasvir in cirrhotic patients was studied in combination with asunaprevir (described in Sect. 4.1.2) or sofosbuvir ± ribavirin. In combination with sofosbuvir 400 mg, an SVR of 83 % was reached in CP-A/B/C patients (phase III trial). SVR rates in CP-C patients were lower: 56 % (compared with CP-A/B patients: 93 %). In this trial, anemia (20 %), fatigue (18 %), and nausea (17 %) were the most commonly reported AEs, of which 18 % were grade 3–4 [71]. Another open-label, phase III study included patients with cirrhosis/advance fibrosis (genotype 3) who were treated with sofosbuvir 400 mg and daclatasvir 60 mg (n = 50). These patients most commonly reported insomnia (30 %), headache (24 %), and fatigue (20 %) [72]. No SVR was reported in this preliminary analysis.
Daclatasvir, in combination with asunaprevir and beclabuvir, was overall well-tolerated by patients with renal impairment. SAEs were reported in 67 % of the patients and SVR was ≥96 % (n = 21) [70].
4.3 NS5B Polymerase Inhibitors
4.3.1 Sofosbuvir
No SAEs were reported in a multiple-dose study where 400 mg of sofosbuvir was administered to HCV-infected CP-B/C patients (n = 17) [24]. Sofosbuvir-containing regimens were in general well-tolerated in patients with advanced cirrhosis. Fatigue, nausea, headache, and anemia were the most frequently reported AEs (≥10 %) [73, 74]. SVR rates varied from 29 to 91 %, depending on genotype, Child-Pugh score, and other DAAs (Table 3) [63, 64, 73, 74].
Sofosbuvir was well-tolerated in dialysis-dependent patients treated with sofosbuvir 200 mg daily or 400 mg every day or on the day of dialysis [26]. However, as discussed, the SVR12 of patients treated with half-dose sofosbuvir varied from 40 to 90 % [28, 32, 66, 67]. Two other recently presented studies (n = 17 and 10) concluded that full-dose sofosbuvir (400 mg/day) had good tolerability and was safe in patients with GFR <30 mL/min with and without dialysis. SVR12 was reached in 60–100 % of the patients [28, 33]. There was no evidence of an elevated risk of sofosbuvir-related toxicity. No cardiac toxicity was reported [28]. A longitudinal, observational cohort reported SVR rates of 85 % (n = 18), 81 % (n = 63), and 88 % (n = 168) in patients with severe, moderate, and mild renal insufficiency, respectively, who were treated with sofosbuvir-containing regimens [31].
4.4 Fixed-Dose Regimens
4.4.1 Ledipasvir/Sofosbuvir
A phase II study with ledipasvir and sofosbuvir ± ribavirin reported AEs in 98 % of the CP-B (n = 59) and 100 % of the CP-C (n = 49) patients. Of these patients, 22 and 35 %, respectively, experienced an SAE. SVR was reached in ≥82 and ≥91 % of the CP-B and CP-C patients, respectively [75]. A randomized, double-blind, placebo-controlled trial reported >96 % SVR. These patients had a CP score of 5–7. Asthenia (52 %) and headache (34 %) were the most common AEs in this trial [76]. Studies in patients with renal impairment and ledipasvir are still ongoing (NCT01958281 [29]).
4.4.2 Velpatasvir/Sofosbuvir
A randomized, double-blind, placebo-controlled trial described the use of velpatasvir in HCV patients with CP-B in combination with sofosbuvir (genotype 1–6). Different treatment regimens were used (Table 3) and SVRs ≥83 % were reported. Overall, this combination was well-tolerated. The most reported AEs were fatigue, nausea, and headache (≥20 %). In total, nine patients discontinued treatment due to an AE [77]. No studies have been published yet describing the safety of velpatasvir in HCV patients with renal insufficiency.
4.4.3 Grazoprevir/Elbasvir
The safety and efficacy of grazoprevir plus elbasvir ± ribavirin in CP-A patients was studied in a phase II trial. SVR was reached in more than 90 % of the patients (n = 253). More discontinuations due to AEs (2 vs. 0 %) and drug-related AEs (71 vs. 54 %) were seen in the patients treated with grazoprevir, elbasvir, and ribavirin. The regimen was well-tolerated by the patients [78]. Another phase II trial showed that a reduced dose of grazoprevir (50 mg) and normal-dose elbasvir was well-tolerated in CP-B patients (n = 30) despite the increased exposure. Fatigue (30 %) was the most reported AE and end of treatment (EOT) response was 100 % (n = 25/30) [49].
As described in Sect. 3.4.3, increased exposure to grazoprevir and elbasvir was reported in patients with a GFR <30 mL/min without dialysis. A phase III study presented safety data for grazoprevir 100 mg and elbasvir 50 mg in patients with GFR ≤29 mL/min (n = 111). High rates of AEs were reported (76 %), but these were comparable with the placebo group (84 %). SAEs and laboratory abnormalities were also comparable between groups. Taking these results in combination with high SVR rates (~95 %), it was concluded that this regimen is safe and effective for the use in patients with renal insufficiency [48].
4.4.4 Paritaprevir/Ritonavir, Ombitasvir, and Dasabuvir
The fixed-dose combination of paritaprevir/ritonavir, ombitasvir, and dasabuvir plus ribavirin in CP-A patients was studied by Poordad et al. [79]. Of these patients, 91% reported AEs, 6 % of whom had an SAE. Only 2 % of patients discontinued the study due to AEs. More AEs were seen during 24-week treatment than during 12-week treatment (phase III trial) and SVR rates were approximately 94 % (n = 380). As discussed in Sect. 3.4.4, the label of this combination regimen was adjusted due to information that became available during post-marketing surveillance.
During a phase I trial, this combination was studied in patients with severe renal insufficiency (n = 20). An interim analysis shows that EOT response was 100 % (n = 14/20). All patients completed the trial but experienced AEs such as anemia (40 %), fatigue (30 %), nausea (25 %), and diarrhea (25 %) [80].
4.5 Other Antivirals
4.5.1 Ribavirin
In the past, ribavirin was frequently used in combination with peginterferon-α, but it is now used as part of DAA regimens. Anemia was frequently reported in trials where cirrhotic patients were treated with ribavirin, varying from 2 to 40 % dependent on the combination treatment [63, 66, 71, 81, 82].
Patients with severe and moderate renal impairment who were treated with peginterferon-α-2a and a daily dose of ribavirin 400 or 600 mg, respectively, frequently needed dosage reductions (71 and 54 %). Dosage reductions were required in 22 and 23 % of ESRD patients treated with a daily dose of ribavirin 200 mg and subjects with normal renal function, respectively. ESRD patients had a safety profile comparable with that of subjects with normal renal function [56, 59].
5 Discussion
5.1 Influence of Hepatic Impairment on Drug Pharmacokinetics
Liver cirrhosis is the main complication of chronic HCV infection. Cirrhosis changes the liver architecture into one with nodules causing reduction in hepatic blood flow, portal systemic shunting, capiliarization of sinusoids, and loss in number and function of hepatocytes. The liver is the main metabolizing organ, and therefore these changes have a profound influence on metabolism and elimination of drugs [6].
Cirrhosis curtails the metabolizing capacity of the liver due to decreased levels of CYP enzymes. Various CYP enzymes are affected (e.g., CYP3A4, CY2A6, CYP2C9), but the susceptibility depends of the type and severity of the liver disease. For example, CYP1A2 and CYP2C19 are sensitive to liver disease, whereas CYP2E1 is less susceptible. These changes in CYP enzymes may cause increased drug concentrations of enzyme substrates. This explains the increased exposure of grazoprevir and paritaprevir in cirrhotic patients, because these drugs are metabolized by CYP3A4. Likewise, the increased sofosbuvir concentration or increased t ½ of ledipasvir might be related to the reduced capacity of the liver to metabolize drugs by enzymes other than CYP enzymes, e.g., uridine diphosphate-glucuronosyltransferase (UGT) conjugation is affected in cirrhotic livers. In addition, efflux transporters may be upregulated, while uptake transporters may be downregulated. These alterations are not yet fully understood [83]. However, they provide an explanation of increased simeprevir and grazoprevir concentrations, because OATP uptake transporters may be downregulated, causing decreased uptake from the circulation into hepatocytes, resulting in increased plasma concentrations.
Reduced hepatic metabolism affects the first-pass effect. This pre-systemic metabolism is responsible for metabolizing orally administered drugs prior to entering the systemic circulation. Decreased pre-systemic metabolism results in elevated bioavailability, as seen with paritaprevir and grazoprevir. In addition, portal shunting affects hepatic blood flow. Blood bypasses the liver, leading to an increased systemic drug concentration as a result of decreased hepatic metabolism.
In cirrhosis, the liver produces less drug-binding proteins (albumin, α1-acid-glycoprotein). Fewer proteins are available, and thus the unbound fraction of drugs may be elevated. Only this unbound fraction of drugs is available for uptake in the tissues and therefore is responsible for the pharmacological effect of a drug. The increased unbound fraction might even cause toxicity despite the total exposure being decreased. This was seen in ombitasvir: the unbound fraction increased twofold in patients with severe hepatic impairment but the total AUC was decreased [52]. On the other hand, the AUC of total daclatasvir was decreased, but the unbound fraction of daclatasvir remained unchanged in patients with cirrhosis due to increased clearance of free daclatasvir. This means that no differences were found in the active concentration of daclatasvir and therefore there was no need for dose modifications [19].
Finally, the ribavirin C max was increased in patient with cirrhosis compared with controls. Ribavirin is renally cleared and extensively metabolized (site unknown). These changes in C max may be caused by alterations in, for example, hepatic drug transporter activity; however, these findings could also be caused by the higher inter-individual variability of ribavirin. Since other pharmacokinetic parameters were not affected, no dose adjustments are necessary when using ribavirin in cirrhotic patients.
Figure 2 summarizes the physiological alterations during cirrhosis that influence drug concentrations and Table 1 shows the recommended doses or contraindications for CP-A/B/C patients.

Overview of the pathophysiological changes in patients with liver cirrhosis that influence drug metabolism and therefore the pharmacokinetics of drugs. CYP cytochrome P450, UGT uridine diphosphate-glucuronosyltransferase, ↓ indicates decrease, ↑ indicates increase
5.2 Influence of Renal Impairment on Drug Pharmacokinetics
The prevalence of HCV in dialysis patients in Europe and the USA varies from 3 to 20 %. In 2002, 8 % of the dialyzed patients in the USA were infected with HCV [84]. HCV is both a cause and a consequence of renal impairment: first, patients on dialysis have an increased infection risk due to medical procedures and, secondly, HCV causes pathological changes to the kidneys [84, 85].
Renal dysfunction influences the renal clearance of drugs. Glomerular filtration, tubular secretion, and tubular reabsorption are responsible for renal clearance of drugs. The clearance may be altered due to damage to glomeruli or by altered activity of drug transporters in tubular cells. In general, renal impairment results in increased drug concentrations of renally cleared drugs.
Patients with ESRD are often dependent on dialysis. An important factor that affects the clearance of drugs is the molecular weight of the drug in relation to the pore size. Other parameters influencing drug clearance during hemodialysis are protein binding, V d, water solubility, and plasma clearance. Characteristics of the dialyzer are also important for the pharmacokinetics of drugs, e.g., the flow of the blood and dialysate, and the concentration gradient. As described in Sect. 3, most HCV drugs are highly bound to plasma proteins, which explains why these drugs are not removed by dialysis (Table 4) as only unbound drugs can be removed by dialysis. Additionally, most drugs are metabolized by the liver, and therefore the contribution of hemodialysis to the clearance of drugs is relatively low [86].
In general, DAAs are hepatically cleared, meaning no dose modifications are necessary in patients with renal impairment. However, sofosbuvir and ribavirin, which are primary renally eliminated, are exceptions to this.
Exposure to GS-331007 increased with a decreasing GFR, but—as explained earlier—dose modification may not always be advisable. Sofosbuvir is removed during dialysis and thus it is recommended that it is administered after dialysis [23].
Ribavirin is mainly situated in the erythrocyte and not effectively cleared from the body during hemodialysis [59]. This causes increased plasma concentrations, which are related to (severe) anemia. However, while higher ribavirin plasma concentrations are related to anemia, they are also associated with improved SVR. Plasma concentrations of ribavirin can explain toxicity or give information regarding whether the ribavirin exposure is sufficient. In other words: is the patient treated with the right dose [62, 87–89]? Due to toxicity, dose modifications are needed in ribavirin-treated patients who have a GFR ≤50 mL/min and in ESRD patients.
A second consideration is that non-renal clearance is affected in patients with renal disease. It should be noted that this phenomenon is mostly studied in patients with ESRD. CYP enzymes, UGT enzymes, and drug transporters have altered activity in patients with renal insufficiency, but protein-binding may also decrease. One hypothesis is that the uremic toxins cause these alterations in drug transporters and enzymes; dialysis then removes these toxins, which improves CYP3A- and transporter-related clearance [90]. These alterations in hepatic function may affect hepatically cleared drugs such as daclatasvir, grazoprevir, and elbasvir. Daclatasvir exposure (bound and unbound) was elevated in patients with decreasing renal function [20]. Similarly, grazoprevir and elbasvir exposure increased significantly in patients with a GFR <30 mL/min [32, 50].
In conclusion, it is hard to predict what the influence of renal function on the exposure of drugs is. This should be taken into account when interpreting the dose recommendations in Table 4.
5.3 Studies in Patients with Renal and Hepatic Impairment During Clinical Development
The effect of renal or hepatic impairment on the pharmacokinetics of HCV drugs is often studied in single-dose studies in HCV-negative patients with renal insufficiency or liver cirrhosis. These studies give an idea of the pharmacokinetics in these patients groups, but the influence of HCV is missing. The virus itself may also influence drug metabolism, as inflammation and infection are known to affect CYP activity in the liver [91, 92].
Therefore, studies in HCV-infected patients with renal or hepatic impairment are performed after licensing and post-marketing surveillance takes place (e.g., collecting AEs). These findings might then be used to change the prescribing information. For example, the labels of both simeprevir and paritaprevir/ritonavir plus ombitasvir with or without dasabuvir were recently changed due to information that became available after licensing. In our opinion, these studies should be part of the pre-registration process, since HCV patients suffer from these conditions and therefore they will be treated with the novel DAAs. In comparison, sofosbuvir is contraindicated for patients with a GFR <30 mL/min because no safety studies have been performed and data are missing. This might exclude patients unnecessarily from treatment.
6 Conclusion
This review described the pharmacokinetics, efficacy, and safety of HCV drugs in patients with renal and hepatic dysfunction. All of the available drugs for the treatment of HCV can be used in patients with CP-A and in those with a GFR ≥30 mL/min. Some drugs are contraindicated in patients with advanced liver disease (CP-B or CP-C), and sofosbuvir plus ledipasvir or daclatasvir are the best options for this group. Patients with a GFR <30 mL/min can be treated with grazoprevir plus elbasvir or paritaprevir/ritonavir and ombitasvir with or without dasabuvir. Sofosbuvir is an important part of HCV therapy, and therefore data on its use in renally impaired patients is essential information; however, data on sofosbuvir are still pending. Lastly, it would be helpful if more pharmacokinetics, efficacy, and safety data became available for the treatment of patients with advance liver disease or severe renal impairment. These patients might benefit the most from therapy, possibly preventing the need for liver transplantation.
References
Singal AK, Guturu P, Hmoud B, Kuo YF, Salameh H, Wiesner RH. Evolving frequency and outcomes of liver transplantation based on etiology of liver disease. Transplantation. 2013;95(5):755–60. doi:10.1097/TP.0b013e31827afb3a.
Thomas DL. Global control of hepatitis C: where challenge meets opportunity. Nat Med. 2013;19(7):850–8. doi:10.1038/nm.3184.
Gower E, Estes C, Blach S, Razavi-Shearer K, Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol. 2014;61(1 Suppl.):S45–57. doi:10.1016/j.jhep.2014.07.027.
WHO. Hepatitis C fact sheet. 2014. http://www.who.int/mediacentre/factsheets/fs164/en/. Accessed 15 May 2014.
Highleyman L. Hepatitis C: the changing picture. 2009. http://www.thebody.com/content/art53550.html. Accessed 14 Jan 2016.
Verbeeck R. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64(12):1147–61. doi:10.1007/s00228-008-0553-z.
Fukazawa K, Lee HT. Updates on hepato-renal syndrome. J Anesth Clin Res. 2013;4(9):352. doi:10.4172/2155-6148.1000352.
Sun H, Frassetto L, Benet LZ. Effects of renal failure on drug transport and metabolism. Pharmacol Ther. 2006;109(1–2):1–11. doi:10.1016/j.pharmthera.2005.05.010.
EASL. EASL recommendations on treatment of hepatitis C 2015. 2015. http://www.easl.eu/research/our-contributions/clinical-practice-guidelines/detail/recommendations-on-treatment-of-hepatitis-c-2015/report/1. Accessed 8 June 2015.
AASLD. HCV guidance: recommendations for testing, managing, and treating hepatitis C. 2015. http://www.hcvguidelines.org/. Accessed 27 Jan 2016.
EMA. Olysio product information. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002777/WC500167867.pdf. Accessed 29 Sept 2015.
Ouwerkerk-Mahadevan S, Simion A, Spittaels K, Beumont-Mauviel M. Pharmacokinetics of simeprevir (TMC435) in volunteers with moderate or severe hepatic impairment [abstract no. 887]. J Hepatol. 2013;58(Suppl. 1):S229. doi:10.1016/S0168-8278(13)60889-2.
Sekar V, Simion A, Peeters M, Spittaels K, Lawitz E, Marbury TC, et al. Pharmacokinetics of TMC435 in subjects with moderate hepatic impairment [abstract no. 472]. J Hepatol. 2011;54(Suppl. 1):S193. doi:10.1016/S0168-8278(11)60474-1.
Eley T, He B, Chang I, Colston E, Child M, Bedford W, et al. The effect of hepatic impairment on the pharmacokinetics of asunaprevir, an HCV NS3 protease inhibitor. Antivir Ther. 2015;20(1):29–37. doi:10.3851/imp2773.
Eley T, Garimella T, Li W, Bertz RJ. Asunaprevir: a review of preclinical and clinical pharmacokinetics and drug-drug interactions. Clin Pharmacokinet. 2015;54(12):1205–22. doi:10.1007/s40262-015-0299-6.
Garimella T, He B, Luo WL, Colston E, Zhu LM, Kandoussi H, et al. Asunaprevir pharmacokinetics and safety in subjects with impaired renal function [abstract no. 463]. 64th annual meeting of the american association for the study of liver diseases, 1–3 Nov 2013, Washington, DC.
Adamczyk R, Sims K, Hesney M, Wind-Rotolo M, Stonier M, Penn R, et al. The effect of renal impairment on multiple-dose pharmacokinetics of the fixed-dose combination of daclatasvir/asunaprevir/beclabuvir. J Hepatol. 2015;62(Suppl. 2):S628. doi:10.1016/S0168-8278(15)30993-4.
EMA. Daklinza product information. 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003768/WC500172848.pdf. Accessed 29 Sept 2015.
Bifano M, Sevinsky H, Persson A, Chung E, Wind-Rotolo M, Hwang C, et al. Single-dose pharmacokinetics of daclatasvir (DCV; BMS-790052) in subjects with hepatic impairment compared with healthy subjects [abstract no. 1004A]. 62nd annual meeting of the American association for the study of liver diseases, 6–9 Nov 2011, San Fransisco.
Garimella T, Wang R, Luo WL, Hwang C, Sherman D, Kandoussi H, et al. Single-dose pharmacokinetics and safety of daclatasvir in subjects with renal function impairment. Antivir Ther. 2015;5:535–43. doi:10.3851/imp2941.
EMA. Harvoni product information. 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003850/WC500177995.pdf. Accessed 29 Sept 2015.
FDA. Sovaldi prescribing information. 2013. http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/204671s000lbl.pdf. Accessed 25 Jan 2016.
EMA. Sovaldi product information. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002798/WC500160597.pdf. Accessed 29 Sep 2015.
Lawitz E, Rodriguez-Torres M, Cornpropst M, Denning JM, Clemons D, McNair L, et al. The effect of hepatic impairment on the safety, pharmacokinetics, and antiviral activity of PSI-7977 in hepatitis C infected subjects treated for seven days [abstract no. 1130]. J Hepatol. 2012;56(Suppl. 2):S445–6. doi:10.1016/S0168-8278(12)61142-8.
Cornpropst MT, Dennning JM, Clemons D, Marbury TC, Alcorn H, Smith WB, et al. The effect of renal impairment and end stage renal disease on the single-dose pharmacokinetics of GS-7977 [abstract no. 1101]. J Hepatol. 2012;56(Suppl. 2):S433. doi:10.1016/S0168-8278(12)61113-1.
Desnoyer A. Sofosbuvir in hemodialysis: 400mg daily of only the day of hemodialysis? [abstract no. 19]. 16th international workshop of clincial pharmacology of HIV and hepatitis therapy, 26 May 2015, Washington, DC.
Gane EJ, Robson RA, Bonacine M, Maliakkal B, Kirby B, Liu LJ, et al. Safety, antiviral efficacy, and pharmacokinetics of sofosbuvir in patients with severe renal impairment [abstract no. 966]. 65th annual meeting of the American association for the study of liver diseases, 7 Nov 2014, Boston, MA.
Liu L, Sajwani K, Kirby B, Denning J, Stamm LM, Brainard D, et al. Safety and efficacy of treatment with daily sofosbuvir 400mg + ribavirin 200mg for 24 weeks in genotype 1 and 3 HCV-infected patients with severe renal impairment. 66th annual meeting of the American association for the study of liver diseases, 13–17 Nov 2015, San Fransisco.
Gilead Sciences. Sofosbuvir plus ribavirin, or ledipasvir/sofosbuvir in adults With HCV infection and renal insufficiency [ClinicalTrials.gov Identifier: NCT01958281]. US National institues of Health, ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01958281. Accessed 14 Mar 2016.
FDA. Clinical Pharmacology and Biopharmaceutics review(s): sovaldi (sofosbuvir). 2013. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204671Orig1s000ClinPharmR.pdf. Accessed 04 Mar 2016.
Saxena V, Koraishy FM, Sise M, Lim JK, Chung RT, Liapakis A, et al. Safety and efficacy of sofosbuvir-containing regimines in hepatitis C infected patients with reduces renal function: real-world experience from HCV-TARGET [abstract no. LP08]. J Hepatol. 2015;62(Suppl. 2):S267. doi:10.1016/S0168-8278(15)30162-8.
Yeh WW, Caro L, Guo Z, Feng HP, Davis H, Kozisek M, et al. Pharmacokinetics of co-administered HCV protease inhibitor grazoprevir (MK-5172) and NS5A inhibitor elbasvir (MK-8742) in volunteers with end-stage renal disease on hemodialysis or sever renal impairment not on hemodialysis [poster no. 1940]. 65th annual meeting of the American association for the study of liver diseases, 7 Nov 2014, Boston, MA.
Nazario HE, Ndungu M, Modi AA. Sofosbuvir and simeprevir in hepatitis C genotype 1- patients with end-stage renal disease on hemodialysis or GFR <30mL/min. Liver Int. 2015. doi:10.1111/liv.13025.
German P, Mathias A, Yang JC, McNair L, Shen G, Vimal M, et al. Pharmacokinetics of ledipasvir, an HCV-specific NS5A inhibitor, in HCV-uninfect subjects with moderate or severe hepatic impairment [abstract no. 1952]. 64th annual meeting of the American association for the study of liver diseases, 1–3 Nov 2013, Washington, DC.
Mogalian E, Mathias A, Yang J, Moorehead L, Hernandez J, Lasseter K, et al. The pharmacokinetics of ledipasvir, an HCV-specific NS5A inhibitor, in HCV-uninfected subjects with severe renal impairment [abstract no. 1952]. 65th annual meeting of the American association for the study of liver diseases, 7–11 Nov 2014, Boston, MA.
Foster GR, Afdhal N, Roberts SK, Brau N, Gane EJ, Pianko S, et al. Sofosbuvir and velpatasvir for HCV genotype 2 and 3 infection. N Engl J Med. 2015;373(27):2608–17. doi:10.1056/NEJMoa1512612.
Feld JJ, Jacobson IM, Hezode C, Asselah T, Ruane PJ, Gruener N, et al. Sofosbuvir and velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. N Engl J Med. 2015;373(27):2599–607. doi:10.1056/NEJMoa1512610.
Mogalian E, German P, Yang CY, Moorehead L, Brainard D, McNally J, et al. Evaluation of transporter and cytochrome P450-mediated drug-drug interactions between pan-genotypic HCV NS5A inhibitor GS-5816 and phenotypic probe drugs [abstract O_07]. 15th international workshop on clinical pharmacology of HIV & hepatitis therapy, 19–21 May 2014, Washington, DC.
Mogalian E, German P, Kearney BP, Yang CY, Brainard D, McNally J, et al. Use of multiple probes to assess transporter- and cytochrome P450-mediated drug-drug interaction potential of the pangenotypic hcv NS5A inhibitor velpatasvir. Clin Pharmacokinet. 2015. doi:10.1007/s40262-015-0334-7.
Mogalian E, Mathias A, Brainard D, McNally J, Moorehead L, Hernandez M, et al. The pharmacokinetics of GS-5816, a pan-genotype HCV NS5A inhibitor, in HCV-uninfected subject with moderate and severe hepatic impairment [abstract no. P742]. J Hepatol. 2014;60(Suppl. 1):S317. doi:10.1016/S0168-8278(14)60903-X.
Mogalian E, Mathias A, Brainard D, Shen G, McNally J, Sajwani K, et al. The pharmacokinetics of GS-5816, a pangenotypic HCV-specific NS5A inhibitor, in HCV-uninfected subjects with severe renal impairment [abstract no. P0712]. J Hepatol. 2015;62(Suppl. 2):S590–1. doi:10.1016/S0168-8278(15)30915-6.
Gilead Sciences. Safety, tolerability, pharmacokinetics, and antiviral activity of GS-9857 in subjects with chronic hepatitis C virus infection. US National Institues of Health, ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02185794. Accessed 14 Mar 2016.
Yeh WW, Fraser IP, Reitmann C, Caro L, Mitselos A, Denef JF, et al. Pharmacokinetic interaction Of HCV protease inhibitor MK-5172 and ritonavir in healthy subjects [abstract no. 52]. HEPDART, 8–12 Dec 2013, Hawaii.
Gentile I, Buonomo AR, Borgia F, Zappulo E, Castaldo G, Borgia G. MK-5172: a second-generation protease inhibitor for the treatment of hepatitis C virus infection. Expert Opin Inv Drug. 2014;23(5):719–28. doi:10.1517/13543784.2014.902049.
Petry A, Brainard DM, Anderson MS, Mitselos A, Laethem T, Heriman I, et al. Safety, tolerability, and pharmacokinetics after single and multiple doses of MK-5172, a novel HCV NS3/4a protease inhibitor with potent activity against known resistance mutants, in healthy subjects [poster]. 61st annual meeting of the American association for the study of liver diseases, 29 Oct–2 Nov 2010, Boston, MA.
Caro L, Anderson MS, Du L, Palcza J, Han L, van Dyck K, et al. Pharmacokinetics and pharmacokinetic/pharmacodynamic relationship for MK-5172, a novel hepatitis C virus (HCV) NS3/4A protease inhibitor, in genotype 1 and genotype 3 HCV-infected patients [abstract no. 13716]. 62nd annual meeting of the American association for the study of liver diseases, 6–9 Nov 2011, San Francisco.
Marshall WL, Yeh WW, Bethel-Brown C, Stypinski D, Auger P, Brandquist C, et al. No evidence of pharmacokinetic drug-drug interaction in healthy subjects between coadministered grazoprevir (MK-5172)/elbasvir (MK-8742) and sofosbuvir [abstract no. P0910]. J Hepatol. 2015;62(Suppl. 2):S686. doi:10.1016/S0168-8278(15)31113-2.
Roth D, Nelson DR, Bruchfeld A, Liapakis A, Silva M, Monsour H Jr, et al. Grazoprevir plus elbasvir in treatment-naive and treatment-experienced patients with hepatitis C virus genotype 1 infection and stage 4–5 chronic kidney disease (the C-SURFER study): a combination phase 3 study. Lancet. 2015;386(10003):1537–45. doi:10.1016/S0140-6736(15)00349-9.
Jacobson IM, Poordad F, Firpi-Morell R, Everson GT, Verna EC, Bhanja S, et al. Efficacy and safety of grazoprevir and elbasvir in hepatitis C genotype 1-infected patients with child–pugh class B cirrhosis (C-salt part A) [abstract no. O008]. J Hepatol. 2015;62(Suppl. 2):S193–4. doi:10.1016/S0168-8278(15)30015-5.
FDA. Zepatier prescribing information. 2016. https://www.merck.com/product/usa/pi_circulars/z/zepatier/zepatier_pi.pdf. Accessed 4 Mar 2016.
EMA. Viekirax product information. 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003839/WC500183997.pdf. Accessed 29 Sept 2015.
Khatri A, Menon R, Marbury TC, Lawitz E, Podsadecki TJ, Mullally V, et al. Pharmacokinetics and safety of coadministered paritaprevir plus ritonavir (paritaprevir/r), ombitasvir, and dasabuvir in hepatic impairment. J Hepatol. 2015;63(4):805–12. doi:10.1016/j.jhep.2015.05.029.
EMA. Exviera product information. 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003837/WC500182233.pdf. Accessed 29 Sept 2015.
Abbvie. Abbvie update U.S. Labels for viekira pak and technivie. 2015. http://abbvie.mediaroom.com/press_releases?item=122646. Accessed 23 Mar 2016.
FDA. FDA drug safety communication: FDA warns of serious liver injury risk with hepatitis C treatments viekira pak and technivie. 2015. http://www.fda.gov/Drugs/DrugSafety/ucm468634.htm. Accessed 27 Oct 2015.
FDA. Copegus prescribing information. 2011. http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021511s023lbl.pdf. Accessed 30 Sept 2015.
EMA. Rebetol product information. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000246/WC500048210.pdf. Accessed 29 Sept 2015.
Glue P, Schenker S, Gupta S, Clement RP, Zambas D, Salfi M. The single dose pharmacokinetics of ribavirin in subjects with chronic liver disease. Br J Clin Pharmacol. 2000;49(5):417–21. doi:10.1046/j.1365-2125.2000.00186.x.
Brennan BJ, Wang K, Blotner S, Magnusson MO, Wilkins JJ, Martin P, et al. Safety, tolerability, and pharmacokinetics of ribavirin in hepatitis C virus-infected patients with various degrees of renal impairment. Antimicrob Agents Chemother. 2013;57(12):6097–105. doi:10.1128/aac.00608-13.
Gupta SK, Kantesaria B, Glue P. Exploring the influence of renal dysfunction on the pharmacokinetics of ribavirin after oral and intravenous dosing. Drug Discov Ther. 2014;8(2):89-95.
de Kanter CT, Koning L, Berden FA, Wasmuth JC, Grintjes-Huisman KJ, Becker B, et al. The ARRIBA concept: adequate resorption of ribavirin. Antivir Ther. 2015;20(5):515–20. doi:10.3851/imp2935.
van Vlerken LG, de Kanter CT, Boland GJ, van Loon AM, van Soest H, Koek GH, et al. Measuring ribavirin concentrations during the earliest stages of antiviral therapy for hepatitis C: potential relevance for treatment outcome. Ther Drug Monit. 2013;35(4):546–51. doi:10.1097/FTD.0b013e31828a9fbf.
Aqel B, Pungpapong S, Leise M, Werner KT, Chervenak AE, Watt KD, et al. Multicenter experience using simeprevir and sofosbuvir with or without ribavirin to treat hepatitis C genotype 1 in patients with cirrhosis. Hepatology. 2015;62(4):1004–12. doi:10.1002/hep.27937.
Saxena V, Nyberg L, Pauly M, Dasgupta A, Nyberg A, Piasecki B, et al. Safety and efficacy of simeprevir/sofosbuvir in hepatitis C infected patients with compensated and decompensated cirrhosis. Hepatology. 2015;62(3):715–25. doi:10.1002/hep.27922.
FDA. Olysio prescribing Information. 2013. http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/205123s001lbl.pdf. Accessed 13 Jan 2016.
Kalyan Ram B, Frank C, Adam P, Cynthia L, Maria H, Lennox J, et al. Safety, efficacy and tolerability of half-dose sofosbuvir plus simeprevir in treatment of hepatitis C in patients with end stage renal disease. J Hepatol. 2015;63(3):715-25. doi:10.1016/j.jhep.2015.06.004.
Czul F, Peyton A, O’Brien CB, Jeffers L, Martin P, Bhamidimarri KR. Safety, efficacy and tolerability in the treatment of patients with chronic hepatitis C and severe renal impairment [poster. no 2256]. 66th annual meeting of the American association for the study of liver diseases, 7–13 Nov 2015, San Fransisco.
Muir AJ, Poordad F, Lalezari J, Everson G, Dore GJ, Herring R, et al. Daclatasvir in combination with asunaprevir and beclabuvir for hepatitis C virus genotype 1 infection with compensated cirrhosis. JAMA. 2015;313(17):1736–44. doi:10.1001/jama.2015.3868.
Kao JH, Heo J, Yoffe B, Sievert W, Jacobson I, Bessone F, et al. Efficacy and safety of daclatasvir in combination with asunaprevir in cirrhotic and non-cirrhotic patients with HCV genotype 1b: results of the HALLMARK-DUAL study [abstract no. P1300]. J Hepatol. 2015;60(Suppl. 1):S527–8. doi:10.1016/S0168-8278(14)61467-7.
Suda G, Kudo M, Nagasaka A, Furuya K, Yamamoto Y, Kobayashi T, et al. Efficacy and safety of daclatasvir and asunaprevir combination therapy in chronic hemodialysis patients with chronic hepatitis C. J Gastroenterol. 2016. doi:10.1007/s00535-016-1162-8.
Poordad F, Schiff ER, Vierling JM, Landis C, Fontana RJ, Yang R, et al. Daclatasvir with sofosbuvir and ribavirin for HCV infection with advanced cirrhosis or post-liver transplant recurrence. Hepatology. 2016. doi:10.1002/hep.28446.
Leroy V, Angus P, Bronowicki JP, Dore G, Hezode C, Pianko S, et al. All-oral treatment with daclatasvir (DV) plus sofosbuvir (SOF) plus ribavirin (RBV) for 12 or 16 weeks in HCV genotype (GT) 3-infected patients with advanced fibrosis of cirrhosis: the ally-3+ phase 3 study [abstract no. lb-3]. 66th annual meeting of the American association for the study of liver diseases, 13–17 Nov 2015, San Fransisco.
Ho SB, Byrne S, Moon S, Zhu Y, Seyedkazemi S, Rossaro L, et al. Twelve weeks of sofosbuvir plus ribavirin effective for treatment of genotype 2 HCV in difficult to treat U.S. veterans with cirrhosis - results of the valor-HCV study [abstract no. 1041]. 66th annual meeting of the American association for the study of liver diseases, 13–17 Nov 2015, San Fransisco.
Kayali Z, Ashouri B. Prospectieve stuy for the efficacy of sofosbuvir and simeprevir +/- ribavirin in hepatitis C genotype 1 and 4 compensated cirrhotic patients. Single center study and real life expierence [abstract no. 1100]. 66th annual meeting of the American association for the study of liver diseases, 13–17 Nov 2015, San Fransisco.
Charlton M, Everson GT, Flamm SL, Kumar P, Landis C, Brown RS Jr, et al. Ledipasvir and sofosbuvir plus ribavirin for treatment of HCV infection in patients with advanced liver disease. Gastroenterology. 2015;149(9):649–59. doi:10.1053/j.gastro.2015.05.010.
Bourliere M, Bronowicki JP, de Ledinghen V, Hezode C, Zoulim F, Mathurin P, et al. Ledipasvir-sofosbuvir with or without ribavirin to treat patients with HCV genotype 1 infection and cirrhosis non-responsive to previous protease-inhibitor therapy: a randomised, double-blind, phase 2 trial (SIRIUS). Lancet Infect Dis. 2015;15(4):397–404. doi:10.1016/s1473-3099(15)70050-2.
Curry MP, O’Leary JG, Bzowej N, Muir AJ, Korenblat KM, Fenkel JM, et al. Sofosbuvir and velpatasvir for HCV in patients with decompensated cirrhosis. N Engl J Med. 2015;373(27):2618–28. doi:10.1056/NEJMoa1512614.
Lawitz E, Gane E, Pearlman B, Tam E, Ghesquiere W, Guyader D, et al. Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY): a randomised, open-label phase 2 trial. Lancet. 2015;385(9973):1075-86.
Poordad F, Hezode C, Trinh R, Kowdley KV, Zeuzem S, Agarwal K, et al. ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med. 2014;370(21):1973–82. doi:10.1056/NEJMoa1402869.
Pockros PJ, Reddy KR, Mantry PS, Cohen E, Bennett M, Sulkowski MS, et al. Safety of ombitasvir/paritaprevir/ritonavir plus dasabuvir for treating HCV GT1 infection in patients with severe renal impairment or end-stage renal disease: the RUBY-1 study [abstract no. L01]. J Hepatol. 2015;62(Suppl. 2):S257. doi:10.1016/S0168-8278(15)30147-1.
Hezode C, Fontaine H, Dorival C, Zoulim F, Larrey D, Canva V, et al. Effectiveness of telaprevir or boceprevir in treatment-experienced patients with HCV genotype 1 infection and cirrhosis. Gastroenterology. 2014;147(1):132–42.e4. doi:10.1053/j.gastro.2014.03.051.
Buti M, Agarwal K, Horsmans Y, Sievert W, Janczewska E, Zeuzem S, et al. Telaprevir twice daily is noninferior to telaprevir every 8 hours for patients with chronic hepatitis C. Gastroenterology. 2014;146(3):744–53.e3. doi:10.1053/j.gastro.2013.11.047.
Zollner G, Fickert P, Silbert D, Fuchsbichler A, Marschall H-U, Zatloukal K, et al. Adaptive changes in hepatobiliary transporter expression in primary biliary cirrhosis. J Hepatol. 2003;38(6):717–27. doi:10.1016/S0168-8278(03)00096-5.
Aguirre Valadez J, Garcia Juarez I, Rincon Pedrero R, Torre A. Management of chronic hepatitis C virus infection in patients with end-stage renal disease: a review. Ter Clin Risk Manag. 2015;11:329–38. doi:10.2147/tcrm.s74282.
Dalrymple LS, Koepsell T, Sampson J, Louie T, Dominitz JA, Young B, et al. Hepatitis C virus infection and the prevalence of renal insufficiency. Clin J Am Soc Nephrol. 2007;2(4):715–21. doi:10.2215/cjn.00470107.
Johnson CA, Simmons WD. Dialysis of drugs. Nephrology Pharmacy Associates. http://www.just.edu.jo/DIC/Manuals/Dialysis%20of%20Drugs.pdf. Accessed 23 Mar 2016.
Morello J, Rodriguez-Novoa S, Jimenez-Nacher I, Soriano V. Usefulness of monitoring ribavirin plasma concentrations to improve treatment response in patients with chronic hepatitis C. J Antimicrob Chemother. 2008;62(6):1174–80. doi:10.1093/jac/dkn421.
Larrat S, Stanke-Labesque F, Plages A, Zarski JP, Bessard G, Souvignet C. Ribavirin quantification in combination treatment of chronic hepatitis C. Antimicrob Agents Chemother. 2003;47(1):124–9.
de Kanter CTMM. Introducing clinical pharmacology to hepatitis C therapy. Nijmegen: Radboud University Medical Center; 2015.
Verbeeck RK, Musuamba FT. Pharmacokinetics and dosage adjustment in patients with renal dysfunction. Eur J Clin Pharmacol. 2009;65(8):757–73. doi:10.1007/s00228-009-0678-8.
Aitken AE, Richardson TA, Morgan ET. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu Rev Pharmacol. 2006;46123–49. doi:10.1146/annurev.pharmtox.46.120604.141059.
Morgan ET, Goralski KB, Piquette-Miller M, Renton KW, Robertson GR, Chaluvadi MR, et al. Regulation of drug-metabolizing enzymes and transporters in infection, inflammation, and cancer. Drug Metab Dispos. 2008;36(2):205–16. doi:10.1124/dmd.107.018747.
Lawitz E, Gane E, Pearlman B, Tam E, Ghesquiere W, Guyader D, et al. Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY): a randomised, open-label phase 2 trial. Lancet. 2015;385(9973):1075–86. doi:10.1016/s0140-6736(14)61795-5.
Lawitz E, Poordad F, Gutierrez JA, Evans B, Hwang P, Howe A, et al. C-SWIFT: grazoprevir (MK-5172) + elbasvir (MK-8472) + sofosbuvir in treatment-naïve patients with hepatitis C virus genotype 1 infection, with and without cirrhosis, for durations of 4, 6 or 8 weeks (interim results) [abstract]. 65th annual meeting of the America association for the study of liver diseases, 7–11 Nov 2014, Boston, MA.
Bruchfeld A, Lindahl K, Reichard O, Carlsson T, Schvarcz R. Pegylated interferon and ribavirin treatment for hepatitis C in haemodialysis patients. J Hepatol. 2006;13(5):316–21. doi:10.1111/j.1365-2893.2005.00680.x.
Qaqish RB, Pilot-Matias T, Yu Y, Redman R, Mobashery N, Asselah T, et al. Efficacy and safety of ombitasvir/paritaprevir/ritonavir co-administrated with ribavirin in adults with genotype 4 chronic hepatitis C infedction and cirrhosis (AGATE-1) [abstract no. 714]. 66th annual meeting of the American association for the study of liver diseases, 16–17 Nov 2015, San Fransisco.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were used for the preparation of this review.
Conflict of interest
Elise J. Smolders and Clara T. M. M. de Kanter declare that they have no conflicts of interest that are directly relevant to the content of this review. Bart van Hoek has been a member of advisory boards of Abbvie, BMS, Falk, Janssen, and MSD and has participated recently in clinical trials for Abbvie, BMS, Gilead, Janssen, and Merck. Johannes E. Arends has been a member of advisory boards of Abbvie, BMS, Gilead, Janssen, Merck, and Viiv and has received sponsorship/research Grants from Abbvie and BMS. Johannes P. H. Drenth has been a member of advisory boards of Abbvie, BMS, Gilead, Janssen, and Merck and has received sponsorship/research Grants from Abbvie and Janssen. David M. Burger has been a member of advisory boards of Abbvie, BMS, Gilead, Janssen, and Merck and has received sponsorship/research grants from BMS, Janssen, Merck, and Viiv. However, these conflicts of interest did not influence the preparation of this review.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Smolders, E.J., de Kanter, C.T.M.M., van Hoek, B. et al. Pharmacokinetics, Efficacy, and Safety of Hepatitis C Virus Drugs in Patients with Liver and/or Renal Impairment. Drug Saf 39, 589–611 (2016). https://doi.org/10.1007/s40264-016-0420-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-016-0420-2