
Abstract
Background and Objective
Daridorexant is a dual orexin receptor antagonist for the treatment of insomnia. In two phase III, 12-week studies in patients with insomnia disorder, daridorexant improved sleep and daytime functioning while maintaining a favorable safety profile. The objective of this 40-week extension study was to assess the long-term safety and tolerability of daridorexant.
Methods
Adults with insomnia disorder who completed the 12-week studies were invited to enroll in this double-blind extension study. Patients originally randomised to daridorexant (10 mg/25 mg/50 mg) remained on their respective treatments; patients randomised to placebo were re-randomised to daridorexant 25 mg or placebo. The 40-week treatment period was followed by a 7-day placebo run-out. The primary objective was to assess safety/tolerability. Exploratory objectives were to evaluate the efficacy of daridorexant on sleep (self-reported total sleep time) and daytime functioning (Insomnia Daytime Symptoms and Impacts Questionnaire).
Results
In total, 804 patients were enrolled in the study, of whom 801 received at least one dose of the study treatment and 550 patients (68.4%) completed the study. Overall incidence of treatment-emergent adverse events was similar across groups (35–40%). Daridorexant did not induce next-morning sleepiness and no withdrawal-related symptoms or rebound were observed after treatment discontinuation. Improvements in sleep and daytime functioning were maintained through to the end of the study and were most pronounced with daridorexant 50 mg. Daridorexant 50 mg, compared with placebo, increased self-reported total sleep time by a least-squares mean of 20.4 (95% confidence interval [CI] 4.2, 36.5), 15.8 (95% CI − 0.8, 32.5) and 17.8 (95% CI − 0.4, 35.9) minutes and decreased (i.e., improved) Insomnia Daytime Symptoms and Impacts Questionnaire total scores by a least-squares mean of − 9.3 (95% CI − 15.1, − 3.6), − 9.5 (95% CI − 15.4, − 3.5) and − 9.1 (95% CI − 15.6, − 2.7), at weeks 12, 24 and 36 of the extension study, respectively.
Conclusions
Treatment with daridorexant, for up to 12 months, was generally safe and well tolerated. Exploratory efficacy analyses suggest that the sustained improvements in sleep and daytime functioning with daridorexant 50 mg support its use for long-term treatment of insomnia disorder, without concerns of new safety signals.
Clinical Trial Registration
ClinicalTrials.gov (NCT03679884) [first posted: 21 September, 2018], https://clinicaltrials.gov/ct2/show/NCT03679884.
Plain Language Summary
Insomnia disorder is the long-term inability to fall asleep or stay asleep with a significant impact on daily life. Left inadequately treated, this disorder may increase the risk of other health problems. For patients with insomnia disorder who require a sleep medication, many drugs are not recommended for long-term use and there is an unmet need for one that can be used safely and effectively over the long term. Daridorexant is a new insomnia treatment that was approved for adults following positive results in two 12-week clinical studies. Both studies showed that, in patients with insomnia disorder, daridorexant improved night-time sleep and patients’ ability to function during the day, while avoiding major safety concerns. Patients who completed these two studies could continue into a 40-week extension study enabling the safety and tolerability of daridorexant to be investigated for up to 1 year. Treatment remained double blind for the entire 1-year period. The extension study showed that daridorexant, at all doses studied (10 mg, 25 mg, 50 mg), continued to be generally safe and well tolerated. Patients showed no signs of tolerance, physical dependence, rebound nor any excessive daytime sleepiness. Exploratory efficacy analyses suggest that improved night-time and daytime symptoms of insomnia were sustained, in particular with the highest approved dose, 50 mg, and there were no signs that the benefits of the drug were wearing off at the end of the 1 year. These results support the use of daridorexant 50 mg for the long-term treatment of insomnia disorder in adults.
Similar content being viewed by others
In patients with insomnia disorder, daridorexant administered for up to 1 year was generally safe with no signs of tolerance, physical dependance or rebound. |
Exploratory efficacy endpoints show daridorexant sustained improvements in night-time sleep variables and daytime functioning. |
The effects of daridorexant were most favorable with the highest approved dose of 50 mg, without concern for new safety signals. |
1 Introduction
Some 50 years ago, sleep alterations were at the forefront of emerging biological psychiatry as specific changes in sleep architecture were recognised as biological markers of, for example, depressive illnesses [1]. In the International Classification of Diseases, Tenth Revision, difficulties to initiate or maintain sleep were operationalised as symptoms of mental disorders leaving just a small group of patients with primary insomnia to be treated with specific sleep-related therapy [2, 3]. Over the past 30 years, pivotal functions of sleep have been described including its influence on brain maturation, neuronal plasticity, memory consolidation, learning processes, metabolic coordination and immune system functioning [4]. Thus, good-quality sleep of sufficient length is a basic physiological necessity, essential for well-being, performance and health.
In 2019, the World Health Organization passed the International Classification of Diseases, 11th Revision, including Chapter 7 on Sleep-Wake Disorders. Insomnia disorder was operationalised to be diagnosed whenever disturbances to initiate or maintain sleep in conjunction with day-time impairment are present [5], aligning the International Classification of Diseases, 11th Revision with the definition adapted in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition published in 2013 [6]. Epidemiologic studies consistently report ~10% of the general population experience insomnia or nonrestorative sleep [7]. Thus, insomnia disorder seems to be one of the most prevalent and chronic medical disorders. Insomnia disorder is associated with a substantial burden of disease [8]. The daytime impairment can negatively influence quality of life in several ways, including decreased workplace productivity, and an increased risk for injuries and accidents [9,10,11,12,13,14]. For many patients, this sleep-wake disorder becomes a persistent condition and beyond the short-term effects of not getting enough good sleep, undertreated or untreated insomnia poses some serious long-term health consequences, including increasing the risk of hypertension, diabetes mellitus, depression, neurodegenerative disorders and cancer [15,16,17].
The ideal treatment for insomnia disorder should be one that is safe and effective, in the short and long term. It should improve not only the quantitative measures such as total sleep time, wake-time after sleep onset and sleep-onset latency, but also sleep quality, to improve daytime performance and general health [18, 19]. Several drugs are available for the treatment of insomnia, with GABA-A modulators (benzodiazepines and Z-drugs) being the most commonly prescribed, and generally considered effective for short-term treatment [20]. However, many of these medications do not, for example, provide meaningful effects on sleep maintenance or address the daytime functioning impairments [21]. Concerns regarding cognitive and motor impairment, falls, dependence and high levels of somnolence further limit their use and guidelines strongly caution against, or discourage, their long-term use [2, 11, 20,21,22]. This creates a critical unmet need for patients who require a safe and effective long-term treatment.
Dual orexin receptor antagonists (DORAs) have in the last decade emerged as a novel treatment option for insomnia disorder. They act specifically on the brain’s orexin system that regulates the sleep-wake cycle, without causing broad depression of the central nervous system. The orexin A and B neuropeptides bind to the G-protein-coupled orexin-1 and orexin-2 receptors to promote wakefulness [23, 24]. Normally, the orexin neurons exert their highest activity during the day as wakefulness is promoted and are virtually silent at night [25, 26]. Unusual activity of the orexin system during night-time (or sleep time) could lead to abnormal hyperarousal and insomnia [27, 28]. Dual orexin receptor antagonists suppress the excessive wakefulness during sleep time by selectively targeting and blocking the binding of orexin neuropeptides to the two receptors [29]. This mechanism of action avoids the more widespread inhibition of neuronal pathways and side effects (e.g. next-morning residual sleepiness, falls, motor incoordination, tolerance and physical dependence) that are intrinsic to GABA-A modulators and often limit their use [30,31,32,33,34,35].
The DORA daridorexant, approved for the treatment of insomnia in adults aged ≥ 18 years [14], has a pharmacokinetic (PK) and pharmacodynamic profile with a rapid absorption enabling a fast onset, and a rapid elimination enabling night-time sleep maintenance whilst avoiding next-morning sleepiness [36,37,38]. Beneficial effects of daridorexant were shown in two pivotal 12-week trials in adult patients with insomnia disorder [39]. Daridorexant significantly improved sleep onset and sleep maintenance with the highest dose (daridorexant 50 mg) being the most efficacious. Progressive improvements in daytime functioning (sleepiness, alert/cognition and mood) were also observed, assessed using the validated patient-reported outcomes instrument, the Insomnia Daytime Symptoms and Impacts Questionnaire (IDSIQ) [40], with no evidence of next-morning sleepiness. Patients who completed the 12-week studies were eligible to enroll in a 40-week extension study. Here, we report the long-term safety, tolerability and efficacy findings for daily daridorexant on self-reported sleep quality and daytime functioning.
2 Methods
2.1 Study Design
The extension study was a phase III, international, randomised, double-blind, parallel-group, placebo-controlled study (ClinicalTrials.gov NCT03679884) designed to evaluate the long-term safety and efficacy of daridorexant in patients with insomnia disorder who had completed one of the two pivotal 12-week phase III trials (Trial 1: NCT03545191; Trial 2: NCT03575104). Protocol details for the 12-week studies have been reported [39].
The extension study consisted of a 40-week treatment period followed by a 30-day safety follow-up period, which included a 7-day single-blind placebo run-out period (Fig. S1 of the Electronic Supplementary Material [ESM]). Patients who completed the 12-week studies [39], and had been randomised to oral daridorexant (10 mg, 25 mg or 50 mg) continued their same double-blind treatment for an additional 40 weeks (52 weeks total treatment). Patients originally randomised to 12 weeks of placebo were re-randomised (1:1) to receive double-blind daridorexant 25 mg (‘ex-placebo/daridorexant 25 mg’) or placebo for 40 weeks. Randomisation was stratified by age (< 65 and ≥ 65 years) and treatment was allocated using an interactive response technology system. The randomisation list remained confidential until after the database lock. Patients, investigators and study/sponsor personnel remained blinded to all treatment assignments for the duration of the study. Please refer to Table S1 in the ESM for the full list of investigators.
The study adhered to the Declaration of Helsinki, International Conference on Harmonization-Good Clinical Practice and local regulations. Protocols were approved by an institutional review board or ethics committee. All participants provided written informed consent. The study was monitored by an independent data-monitoring committee and an independent safety board (ISB) [see Table S2 and S3 in the ESM].
2.2 Study Participants
Inclusion criteria for the extension study were completion of the 12-week double-blind treatment plus the placebo run-out of the pivotal trials. Eligibility criteria for the 12-week studies have been described [39]. Briefly, this included adult patients (aged ≥ 18 years), with a diagnosis of insomnia disorder (Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition), an Insomnia Severity Index score ≥ 15 and a self-reported history of disturbed sleep (≥ 30 min to fall asleep, ≥ 30 min awake during sleep time, self-reported total sleep time [sTST] ≤ 6.5 h) for ≥ 3 nights per week for ≥ 3 months. During the placebo run-in period, the following polysomnography criteria were required to be met: latency to persistent sleep ≥ 20 min, wake after sleep onset ≥ 30 min and mean total sleep time < 7 h. Patients with a history of sleep-related breathing disorders, other sleep disorders, suicide ideation/attempt, self-reported excessive daytime napping, acute/unstable psychiatric conditions or alcohol/drug abuse were excluded [39]. Patients with unstable medical conditions, significant medical disorders or acute illnesses, clinically relevant electrocardiographic findings, suicidality, hematology or biochemistry test results reported during the 12-week trials that could affect the patient’s safety or interfere with assessments were excluded from entering the extension study.
2.3 Primary Outcomes: Safety and Tolerability
The primary objective of this extension study was to assess the long-term safety and tolerability of daridorexant. Measures included treatment-emergent adverse events (TEAEs), including serious TEAEs, that started/worsened on or after the double-blind treatment start date of the extension study up to 30 days after the double-blind treatment end date, as well as TEAEs leading to premature discontinuation of double-blind treatment, and adverse events of special interest (AESIs) after adjudication by an ISB. Adverse events of special interest were defined as narcolepsy-like symptoms related to excessive daytime sleepiness, cataplexy, complex sleep behaviour events including hallucinations and sleep paralysis, and suicide/self-injury. Adverse events were coded using the Medical Dictionary for Regulatory Activities Preferred Terms.
Change from the baseline in the Epworth Sleepiness Scale (ESS) total score (range 0–24; higher scores indicate greater daytime sleepiness) was assessed at weeks 14, 27, 40 and 41 (run-out). Change from the baseline in the visual analogue scale-assessed morning sleepiness score (range 0–100, higher score indicates a better outcome) was assessed as an average of daily entries over 1 week, every 4 weeks. Vital signs and electrocardiographic parameters were monitored throughout the study and a change in body weight was assessed at week 40.
Withdrawal effects upon treatment discontinuation were assessed based on changes from the last assessment on double-blind treatment (week 40) to the end of the placebo run-out (week 41) in the Benzodiazepine Withdrawal Symptom Questionnaire (BWSQ) total score and the occurrence of relevant adverse events during the placebo run-out. Rebound insomnia was assessed based on a change in sTST from the baseline to the average over the 1-week placebo run-out period.
2.4 Exploratory Outcomes: Night-Time Sleep and Daytime Functioning
Exploratory objectives were to evaluate the long-term efficacy of daridorexant on sleep and daytime functioning. Analyses focused on the self-reported variables that were included as secondary endpoints in the 12-week studies, and for which a type I error was controlled in these studies [39]. The endpoints were changes from the baseline in sTST, recorded by the patient in the sleep diary questionnaire, and in IDSIQ scores. The IDSIQ is a validated instrument developed in accordance with US Food and Drug Administration guidance for assessing patient-reported outcomes [40]. The IDSIQ contains 14 different questions assessing daytime functioning in patients with insomnia disorder; the questions are grouped into three domains each representing the main daytime symptoms and impacts of insomnia on alert/cognition (six questions), sleepiness (four questions) and mood (four questions), and patients rate each question on a scale of 0–10. The sleep diary questionnaire and IDSIQ were completed daily for 7 days every 4 weeks and sTST and IDSIQ scores were the mean of the entries each week. Treatment exposure was measured as the duration of double-blind treatment (days) in the extension study.
2.5 Statistical Analyses
No formal sample size calculation was performed; the extension study was open to all patients who completed the 12-week trials. Baseline was defined as the last observed measurement before or on the first day of double-blind treatment in the 12-week study (termed “pivotal 12-week study baseline” for daridorexant 10-mg, 25-mg, 50-mg and placebo groups) or the extension study (termed “extension study baseline” for ex-placebo/daridorexant 25-mg and placebo groups).
Safety endpoints were summarised descriptively and analysed using the safety set (all patients who received one or more doses of the study treatment) except those evaluating the placebo run-out period, withdrawal symptoms or rebound insomnia, which were analysed using the treatment withdrawal set (all patients in the safety set who received one or more doses of the single-blind placebo treatment in the run-out).
Exploratory efficacy outcomes were analysed using the full analysis set (all patients randomised to study treatment). Changes from the baseline were analysed using a mixed model for repeated measures and a restricted maximum likelihood approach. The model included terms for age group as per assigned strata (< 65; ≥ 65 years), treatment, visit (week 12, 24 and 36 of extension study), and interaction of treatment by visit, and baseline by visit. Results are reported as least-squares mean with 95% confidence interval (CI) for change from the baseline and difference to placebo; these are provided as a measure of the strength of the findings and should be viewed as descriptive only. Post-hoc analyses of the change from the pivotal 12-week study baseline for the efficacy endpoints were performed according to which 12-week study patients entered from (Trial 1 and 2); any p-values presented are descriptive only. For the post-hoc analyses, a similar mixed model for repeated measures and a restricted maximum likelihood approach were used but with the respective treatment in Trial 1 or 2 and visit (month 1, 3, 6, 9 and 12). Missing data were handled by a mixed model for repeated measures and were not imputed.
3 Results
3.1 Study Participants
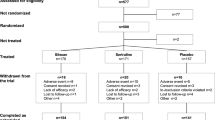
The study was conducted between October 2018 and February 2021 at 94 sites in 14 countries (Belgium, Bulgaria, Canada, Denmark, Finland, France, Germany, Hungary, Poland, South Korea, Spain, Sweden, Switzerland, USA). Overall, 1684 patients completed the 12-week double-blind treatment and placebo run-out of the pivotal trials (Trial 1: N = 847; Trial 2: N = 837). Of these, 804 (47.7%) entered the extension study and 801 received at least one dose of the study treatment (daridorexant 10 mg, n = 142; daridorexant 25 mg, n = 268; daridorexant 50 mg, n = 137; ex-placebo/daridorexant 25 mg, n = 126; placebo, n = 128) (Fig. 1). No information was collected on reasons for non-participation. Overall, 550 patients (68.4%) completed 40 weeks of double-blind treatment. Treatment discontinuations were more frequent in the placebo group than the daridorexant groups with the most common reason being a lack of efficacy (23% vs 8–11%, placebo vs daridorexant groups, Fig. 1). Mean (standard deviation) treatment exposure during the extension study double-blind treatment period was slightly higher in the daridorexant treatment groups (227 [91.6], 231 [85.3], 222 [93.2] and 232 [88.6] days in the 10-mg, 25-mg, 50-mg and ex-placebo/daridorexant 25-mg groups, respectively) versus placebo (210 [97.2] days).

Patient disposition. Patients could withdraw from the study (and hence double-blind [DB] treatment) at any time. However, patients who prematurely discontinued DB treatment were not considered withdrawn from the study and could remain in the extension study and continue with all planned study procedures until the end of the study (apart from the placebo run-out). Therefore, the reason for a patient prematurely discontinuing treatment and for discontinuing the study could be different. SB single-blind
Demographic and baseline characteristics were overall balanced between treatment groups (Table 1) and similar to the intention-to-treat population (N = 1854) in the 12-week trials [39]. Overall mean age was 58 years (range 19–85 years), with 41.7% aged ≥ 65 years. The majority of patients were female (71.5%) and White (89.8%). At the start of the extension study, comorbidities and concomitant therapies were reported in 72.1% and 64.8% of patients, respectively; both were balanced across treatment groups (Table 1).
3.2 Safety
All results presented are during the extension study. The overall incidence and severity of TEAEs (up to 30 days after the end of the double-blind treatment) were similar across groups (35–40%) (Table 2). Most TEAEs occurring during the 40-week double-blind period were mild/moderate in severity (91.2%). The most commonly reported TEAE during double-blind treatment in all groups was nasopharyngitis. All other TEAEs, including falls, headache and somnolence were reported in < 3% of patients, with dizziness and fatigue in < 2% of patients in any group. Treatment-emergent adverse events leading to discontinuation of double-blind treatment were reported only for single patients in any given group (Table S4 in the ESM).
The incidence of serious TEAEs was < 5.5% in all groups (Table 2). Two serious TEAEs assessed as related to study medication by the investigator were reported: orthostatic intolerance (daridorexant 25 mg) and depression/suicidal ideation (placebo) [Table S5 in the ESM]. Two deaths, both cardiovascular related, were reported during the study (daridorexant 10 mg, n = 1; daridorexant 25 mg, n = 1) and assessed by the investigator as not related to study treatment.
Independent safety board-adjudicated AESIs were infrequent and reported in three patients, all considered by the investigator as drug related and not requiring treatment (Table 2). One non-serious event related to excessive daytime sleepiness was reported in the daridorexant 25-mg group. This AESI had onset on day 44 (of extension study) and was resolved on day 93. During this period, the patient experienced several separate episodes of short duration (fell asleep or napped during daytime less than or once per week). The patient discontinued the study on day 98 because of this AESI. A second non-serious TEAE related to hallucinations/sleep paralysis was reported in the daridorexant 50-mg group; this patient experienced mild events of abnormal dreams (no impression of fear or being in a nightmare) on days 1, 2 and 4 of the extension study, awaking 1–1.5 h after taking the study drug. The AESI resolved thereafter. A third serious AESI of suicidal ideation was reported in the placebo group. No complex sleep behavior nor AESI related to cataplexy was reported in any patient.
Accidental overdose (patients who unintentionally took or were uncertain of whether they had taken an extra tablet) was the most frequently reported TEAE pertaining to drug abuse potential and was reported in 15 patients receiving daridorexant (10 mg, n = 4 [2.8%]; 25 mg, n = 3 [1.1%]; 50 mg, n = 4 [2.9%]; ex-placebo/daridorexant 25 mg, n = 4 [3.2%]) [Table S6 in the ESM]. All cases were asymptomatic, mild and non-serious. Other TEAEs pertaining to drug abuse were reported for only one or two patients in any one treatment group and no TEAEs denoting euphoria were reported.
Fall was the most frequently reported TEAE pertaining to accidents and injuries, reported for 14 patients across treatment groups (10 mg, n = 2 [1.4%]; 25 mg, n = 6 [2.2%]; 50 mg, n = 3 [2.2%]; ex-placebo/daridorexant 25 mg, n = 1 [0.8%]; placebo, n = 2 [1.6%] (Table S7 in the ESM). None was serious nor occurred during the night and, in all cases, external contributing factors (e.g. stumbling, slippery floor) were reported. No evidence of any central nervous system depressant effects, such as somnolence or impaired attention, was reported at the time of any fall. The incidence of falls was low in patients aged < 65 years (1.1%) and ≥ 65 years (2.7%).
Patients taking daridorexant did not show excessive daytime sleepiness relative to placebo. At all timepoints, and in all groups, compared with baseline, mean visual analogue scale scores for morning sleepiness were numerically higher (Table S8 in the ESM) and mean ESS scores numerically lower (i.e. both improved) [Table S9 in the ESM]. There were no clinically significant findings for any changes in hematology or clinical chemistry parameters, vital signs, body weight or electrocardiographic parameters.
3.3 Withdrawal
There was no evidence of any withdrawal-related symptoms upon cessation of the study treatment (Table S10 in the ESM). Mean BWSQ total scores were low and similar across treatment groups (Table S11 in the ESM). Changes from the last assessment on the double-blind treatment to the placebo run-out period were minor with no relevant differences between daridorexant and placebo groups. No patient had a BWSQ score > 20 at the end of the run-out. The number of patients with at least one symptom scored as severe on BWSQ was low in all groups, and no dose dependency was observed.
3.4 Rebound Insomnia
No signal suggestive of any rebound effect after treatment discontinuation was observed with daridorexant versus baseline (Table S10 in the ESM). During the placebo run-out, mean sTST was numerically higher than baseline (i.e. improved) in all daridorexant and placebo groups (Table S12 in the ESM).
3.5 Efficacy
Improvements from the baseline in sTST and IDSIQ scores observed in the 12-week studies [39] were maintained through to the end of the extension study. Least-square mean changes from the pivotal 12-week study baseline in sTST and IDSIQ scores at week 12, 24 and 36 of the extension study were numerically greater for all daridorexant doses versus placebo and most pronounced with daridorexant 50 mg (Table S13 in the ESM). In the 50-mg group, least-squares mean increases in sTST from the 12-week study baseline versus placebo were 20.4 min (95% CI 4.2, 36.5; p = 0.014) at week 12, 15.8 min (95% CI − 0.8, 32.5; p = 0.063) at week 24 and 17.8 minutes (95% CI − 0.4, 35.9; p = 0.055) at week 36. For the IDSIQ total score, least-squares mean reductions from baseline versus placebo in the 50-mg group were − 9.3 (95% CI − 15.1, − 3.6; p = 0.0015), −9.5 (95% CI − 15.4, − 3.5; p = 0.0019) and − 9.1 (95% CI − 15.6, − 2.7; p = 0.0058) at weeks 12, 24 and 36, respectively. Similar findings were observed for IDSIQ sleepiness, alert/cognition and mood domain scores with all p values < 0.05 at weeks 12, 24 and 36 in the daridorexant 50-mg group.
For patients who entered the extension study from Trial 1, the increases in sTST and reductions in IDSIQ total scores and domain scores from baseline were consistently larger with daridorexant 50 mg versus 25 mg and placebo throughout the extension study (Fig. 2 and Fig. S2 and Table S14 in the ESM). For patients who entered from Trial 2, the treatment effect on sTST was maintained for both daridorexant doses (10 and 25 mg) and placebo (Fig. S3 and Table S15 in the ESM). For the IDSIQ total score, a large treatment effect was maintained throughout the extension study for daridorexant 50 mg from Trial 1 (Fig. 2) and daridorexant 25 mg from Trial 2 (Fig. S3 in the ESM) and changes were consistently larger than with placebo. Similar findings were observed for the individual IDSIQ domain scores. For patients who received placebo during the 12-week studies and were re-randomised to daridorexant 25 mg in the extension study, mean changes from the extension study baseline indicated numerical improvements in sTST and IDSIQ total scores and domain scores compared with patients randomised to continue placebo (Table S16 in the ESM).

Mean changes from baselinea in self-reported total sleep time (sTST, minutes) and Insomnia Daytime Symptoms and Impacts Questionnaire (IDSIQ) total score over time from Trial 1. Mean of observed a sTST values and b IDSIQ total scores, at study timepoints in patients who entered the extension study from Trial 1 and received daridorexant 25 mg, daridorexant 50 mg or placebo. Two-sided p values shown are versus placebo, calculated using the linear-effects model for repeated measures. p values are for descriptive purposes only. Grey bars indicate the 7-day placebo run-out periods. Left panel shows the change from baseline over time in the pivotal 12-week study; right panel shows the change over time in the 40-week extension study. Figures do not include the placebo patients who were re-randomised to receive daridorexant 25 mg (ex-placebo/daridorexant 25 mg). aPivotal 12-week study baseline. RO run-out
4 Discussion
This is the first long-term study to investigate, in parallel, the safety, the night-time efficacy and the impact on daytime functioning of an insomnia medication in a placebo-controlled setting. Over a total of 52 weeks of nightly treatment (including the pivotal 12-week trials), daridorexant in patients with insomnia disorder demonstrated a favorable long-term safety and tolerability profile consistent with the 12-week study findings [39]. No new safety signals were reported and there was no evidence of physical dependence, tolerance or rebound. In exploratory efficacy analyses, daridorexant 50 mg showed the most favorable improvements that were sustained from the baseline in patient-reported total sleep time and daytime functioning compared with placebo. There were no signs that the benefits were wearing off at the end of the study.
In patients who received 52 weeks of active treatment, daridorexant was generally safe and well tolerated without any residual sleepiness the next morning, or other safety concerns at any dose studied. In fact, consistent with observations from the 12-week studies [39], daridorexant improved next morning sleepiness (visual analogue scale). Moreover, the incidences of falls, somnolence, fatigue and dizziness were low in all groups. As in the 12-week studies, no adverse events of cataplexy or other narcolepsy-related symptoms were reported during the extension study. The abuse of sleep medications is a major concern to both prescribing physicians and patients [41]. Here, the incidence of abuse-associated adverse events was low and, following abrupt cessation of 52-week treatment, there was no evidence of withdrawal symptoms, or rebound insomnia, suggesting low potential for abuse of daridorexant. This is consistent with all clinical data to date in which there have been no reports of misuse, abuse or diversion of daridorexant [39, 42,43,44], and with preclinical data that show daridorexant does not bind to any known abuse-associated central nervous system targets [45].
Daridorexant provided sustained efficacy in increasing sTST and decreasing all IDSIQ scores, versus placebo. Many patients require long-term treatment for insomnia disorder and thus this sustained efficacy over time, and lack of any evidence of attenuation of efficacy over time, is noteworthy. For patients who received placebo during the 12-week studies and were subsequently randomised to daridorexant 25 mg for 40 weeks, the mean changes in sTST and IDSIQ scores indicated clear improvements, suggesting that daridorexant adds benefit even in patients who previously improved on placebo in the pivotal trials.
The sustained improvement in all aspects of daytime functioning, observed with daridorexant 50 mg, addresses a crucial medical need for patients with insomnia disorder. The chronology of the improvement of daytime functioning is also of interest. Evidently, the improvement in night-time function arises before the plateau of improvements in daytime functioning during the first 3 months of treatment. It could be speculated that an accumulation of nights with increased quality and quantity of sleep may be required to observe this improved daytime functioning.
Despite the chronicity of insomnia disorder, there is a lack of long-term clinical trials and as such, most sleep medications are not approved for use beyond 1 month. For benzodiazepines, to our knowledge, the longest study is an 8-week trial of temazepam [46]. For Z-drugs, eszopiclone has been studied up to 12 months and shown to maintain efficacy without development of tolerance [47, 48]. Nevertheless, because of a lack of evidence and remaining safety concerns, sedative hypnotics remain recommended only for short-term use [2, 11, 22]. With regard to DORAs, 12-month studies have been performed for suvorexant [49] and lemborexant [50]; however, these studies have some limitations. For suvorexant, no long-term data on the approved 10-mg dose are available [49]. For lemborexant (5 or 10 mg), only the first 6 months of the study were placebo controlled and daytime functioning was not assessed using a validated instrument such as the IDSIQ.
A strength of the current study is the collection of self-reported patient relevant endpoints for up to 1 year, including the prospective daily measurements of daytime functioning. A further strength is that treatment remained double blind for the entire 1 year without any reports of any code breaks or indications of patient unblinding. Given this study design, the study completion rate (68.4%) was relatively high; often such long-term extension studies with a continued placebo arm do not provide the same incentives to patients as shorter term studies. This may be indicative of the continued effectiveness and safety of daridorexant. However, it does likely contribute to the proportion of patients who decided not to participate in the extension study. Reasons for non-participation were not collected; such information may have been insightful in helping to understand the reasons for patients who do not wish to take sleep medications in the months following acute remediation of their insomnia. Non-participation may however be partly explained by a potential reluctance to continue long-term treatment with a sleep medication (given general recommendations for short-term use only) plus the possibility of remaining on placebo for a further 40 weeks. Those who felt a benefit while receiving placebo in the 12-week studies may have been more likely to continue into the extension study. Further, there may be attrition bias during the extension study owing to more patients in the placebo group prematurely discontinuing treatment than in the daridorexant groups. Further, efficacy endpoints, which should be interpreted as exploratory, are limited to those that were key endpoints in the two 12-week trials and considered of most relevance. Other efficacy endpoints will be considered for separate publication. There are also the limitations associated with the 12-week studies [39], including that most patients were White and represented a population with moderate and severe insomnia (Insomnia Severity Index score ≥ 15), and perhaps more severe than is generally seen in practice; this study population may not be fully representative of the wider patient population, and patient numbers may not have been big enough to detect rare adverse events. It should also be acknowledged that, in the absence of head-to-head trials comparing daridorexant to other DORAs, any inferences about comparative efficacy or safety are limited.
As with any long-term treatment, many patients will take other medications concomitantly for a variety of conditions and thus consideration should be given to the PK and pharmacodynamic drug–drug interactions with daridorexant. No PK analyses were performed from this study but within the clinical pharmacology program of daridorexant, the pharmacokinetics/pharmacodynamics of daridorexant has been evaluated in healthy young and older adults [37, 38]. Specific safety aspects, including the modulating effects of intrinsic [51] and extrinsic factors [52,53,54], human abuse potential [44] and effects on driving performance [55] and safety in special populations (subjects with respiratory disease [56, 57] and hepatic or renal impairment [58, 59]) have also been evaluated. To summarise, daridorexant is quickly absorbed and cleared from the plasma and the distinct pharmacodynamic effects observed are in accordance with the pharmacokinetics, i.e. for a certain dose, the extent and duration of effects are explained by its PK profile, also demonstrated by the dose–response relationship for sleep parameters observed in phase III [39].
5 Conclusions
Results from this study indicate that daridorexant, at all doses studied, administered for up to 1 year is generally safe and well tolerated in patients with insomnia disorder. Exploratory analyses suggest that the sustained efficacy in improving night-time and daytime symptoms of insomnia, with no evidence of tolerance or dependency, supports the use of daridorexant 50 mg for the long-term treatment of insomnia disorder in adults.
References
Kupfer DJ, Foster FG. Interval between onset of sleep and rapid-eye-movement sleep as an indicator of depression. Lancet. 1972;2:684–6. https://doi.org/10.1016/s0140-6736(72)92090-9.
Riemann D, Baglioni C, Bassetti C, Bjorvatn B, Dolenc Groselj L, Ellis JG, et al. European guideline for the diagnosis and treatment of insomnia. J Sleep Res. 2017;26(6):675–700. https://doi.org/10.1111/jsr.12594.
Ferini-Strambi L, Auer R, Bjorvatn B, Castronovo V, Franco O, Gabutti L, et al. Insomnia disorder: clinical and research challenges for the 21st century. Eur J Neurol. 2021;28(7):2156–67. https://doi.org/10.1111/ene.14784.
Niethard N, Born J. Back to baseline: sleep recalibrates synapses. Nat Neurosci. 2019;22(2):149–51. https://doi.org/10.1038/s41593-018-0327-6.
World Health Organization. International statistical classification of diseases and related health problem. 11th ed. 2022. https://icd.who.int/browse11/l-m/en. Accessed 25 Nov 2022.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington: American Psychiatric Association; 2013.
Ohayon MM. Epidemiology of insomnia: what we know and what we still need to learn. Sleep Med Rev. 2002;6(2):97–111. https://doi.org/10.1053/smrv.2002.0186.
Roth T, Coulouvrat C, Hajak G, Lakoma MD, Sampson NA, Shahly V, et al. Prevalence and perceived health associated with insomnia based on DSM-IV-TR; international statistical classification of diseases and related health problems, tenth revision; and research diagnostic criteria/international classification of sleep disorders, second edition criteria: results from the America Insomnia Survey. Biol Psychiatry. 2011;69(6):592–600. https://doi.org/10.1016/j.biopsych.2010.10.023.
Ustinov Y, Lichstein KL, Wal GS, Taylor DJ, Riedel BW, Bush AJ. Association between report of insomnia and daytime functioning. Sleep Med. 2010;11(1):65–8. https://doi.org/10.1016/j.sleep.2009.07.009.
Fullerton DS. The economic impact of insomnia in managed care: a clearer picture emerges. Am J Manag Care. 2006;12(8 Suppl.):S246–52.
Sateia MJ, Buysse DJ, Krystal AD, Neubauer DN, Heald JL. Clinical practice guideline for the pharmacologic treatment of chronic insomnia in adults: an American Academy of Sleep Medicine Clinical Practice Guideline. J Clin Sleep Med. 2017;13(2):307–49. https://doi.org/10.5664/jcsm.6470.
Carey TJ, Moul DE, Pilkonis P, Germain A, Buysse DJ. Focusing on the experience of insomnia. Behav Sleep Med. 2005;3(2):73–86. https://doi.org/10.1207/s15402010bsm0302_2.
Laugsand LE, Strand LB, Vatten LJ, Janszky I, Bjørngaard JH. Insomnia symptoms and risk for unintentional fatal injuries: the HUNT Study. Sleep. 2014;37(11):1777–86. https://doi.org/10.5665/sleep.4170.
Markham A. Daridorexant: first approval. Drugs. 2022;82(5):601–7. https://doi.org/10.1007/s40265-022-01699-y.
Kyle SD, Morgan K, Espie CA. Insomnia and health-related quality of life. Sleep Med Rev. 2010;14(1):69–82. https://doi.org/10.1016/j.smrv.2009.07.004.
Wilson S, Anderson K, Baldwin D, Dijk D-J, Espie A, Espie C, et al. British Association for Psychopharmacology consensus statement on evidence-based treatment of insomnia, parasomnias and circadian rhythm disorders: an update. J Psychopharmacol. 2019;33:026988111985534. https://doi.org/10.1177/0269881119855343.
Hertenstein E, Feige B, Gmeiner T, Kienzler C, Spiegelhalder K, Johann A, et al. Insomnia as a predictor of mental disorders: a systematic review and meta-analysis. Sleep Med Rev. 2019;43:96–105. https://doi.org/10.1016/j.smrv.2018.10.006.
Schutte-Rodin S, Broch L, Buysse D, Dorsey C, Sateia M. Clinical guideline for the evaluation and management of chronic insomnia in adults. J Clin Sleep Med. 2008;4(5):487–504.
Kunz D. Rethinking the use of hypnotics for treatment of insomnia in the elderly. Expert Opin Pharmacother. 2021;22(8):953–7. https://doi.org/10.1080/14656566.2021.1900116.
Krystal AD, Prather AA, Ashbrook LH. The assessment and management of insomnia: an update. World Psychiatry. 2019;18(3):337–52. https://doi.org/10.1002/wps.20674.
Neubauer DN, Pandi-Perumal SR, Spence DW, Buttoo K, Monti JM. Pharmacotherapy of insomnia. J Cent Nerv Syst Dis. 2018. https://doi.org/10.1177/1179573518770672.
Qaseem A, Kansagara D, Forciea MA, Cooke M, Denberg TD. Management of chronic insomnia disorder in adults: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2016;165(2):125–33. https://doi.org/10.7326/m15-2175.
Kantor S, Mochizuki T, Janisiewicz AM, Clark E, Nishino S, Scammell TE. Orexin neurons are necessary for the circadian control of REM sleep. Sleep. 2009;32(9):1127–34. https://doi.org/10.1093/sleep/32.9.1127.
Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437(7063):1257–63. https://doi.org/10.1038/nature04284.
Scammell TE, Winrow CJ. Orexin receptors: pharmacology and therapeutic opportunities. Annu Rev Pharmacol Toxicol. 2011;51:243–66. https://doi.org/10.1146/annurev-pharmtox-010510-100528.
Gotter AL, Winrow CJ, Brunner J, Garson SL, Fox SV, Binns J, et al. The duration of sleep promoting efficacy by dual orexin receptor antagonists is dependent upon receptor occupancy threshold. BMC Neurosci. 2013;14:90. https://doi.org/10.1186/1471-2202-14-90.
Grafe LA, Bhatnagar S. Orexins and stress. Front Neuroendocrinol. 2018;51:132–45. https://doi.org/10.1016/j.yfrne.2018.06.003.
Levenson JC, Kay DB, Buysse DJ. The pathophysiology of insomnia. Chest. 2015;147(4):1179–92. https://doi.org/10.1378/chest.14-1617.
Janto K, Prichard JR, Pusalavidyasagar S. An update on dual orexin receptor antagonists and their potential role in insomnia therapeutics. J Clin Sleep Med. 2018;14(08):1399–408. https://doi.org/10.5664/jcsm.7282.
Griffin CE 3rd, Kaye AM, Bueno FR, Kaye AD. Benzodiazepine pharmacology and central nervous system-mediated effects. Ochsner J. 2013;13(2):214–23.
Wesensten NJ, Balkin TJ, Belenky GL. Effects of daytime administration of zolpidem and triazolam on performance. Aviat Space Environ Med. 1996;67(2):115–20.
Frey DJ, Ortega JD, Wiseman C, Farley CT, Wright KP Jr. Influence of zolpidem and sleep inertia on balance and cognition during nighttime awakening: a randomized placebo-controlled trial. J Am Geriatr Soc. 2011;59(1):73–81. https://doi.org/10.1111/j.1532-5415.2010.03229.x.
Wang PS, Bohn RL, Glynn RJ, Mogun H, Avorn J. Zolpidem use and hip fractures in older people. J Am Geriatr Soc. 2001;49(12):1685–90. https://doi.org/10.1111/j.1532-5415.2001.49280.x.
Weaver MF. Prescription sedative misuse and abuse. Yale J Biol Med. 2015;88(3):247–56.
Asnis GM, Thomas M, Henderson MA. Pharmacotherapy treatment options for insomnia: a primer for clinicians. Int J Mol Sci. 2015. https://doi.org/10.3390/ijms17010050.
Boss C, Gatfield J, Brotschi C, Heidmann B, Sifferlen T, von Raumer M, et al. The quest for the best dual orexin receptor antagonist (daridorexant) for the treatment of insomnia disorders. ChemMedChem. 2020;15:2286–305. https://doi.org/10.1002/cmdc.202000453.
Muehlan C, Boehler M, Brooks S, Zuiker R, van Gerven J, Dingemanse J. Clinical pharmacology of the dual orexin receptor antagonist ACT-541468 in elderly subjects: exploration of pharmacokinetics, pharmacodynamics and tolerability following single-dose morning and repeated-dose evening administration. J Psychopharmacol. 2020;34(3):326–35. https://doi.org/10.1177/0269881119882854.
Muehlan C, Brooks S, Zuiker R, van Gerven J, Dingemanse J. Multiple-dose clinical pharmacology of ACT-541468, a novel dual orexin receptor antagonist, following repeated-dose morning and evening administration. Eur Neuropsychopharmacol. 2019;29(7):847–57. https://doi.org/10.1016/j.euroneuro.2019.05.009.
Mignot E, Mayleben D, Fietze I, Leger D, Zammit G, Bassetti CLA, et al. Safety and efficacy of daridorexant in patients with insomnia disorder: results from two multicentre, randomised, double-blind, placebo-controlled, phase 3 trials. Lancet Neurol. 2022;21(2):125–39. https://doi.org/10.1016/S1474-4422(21)00436-1.
Hudgens S, Phillips-Beyer A, Newton L, Seboek Kinter D, Benes H. Development and validation of the Insomnia Daytime Symptoms and Impacts Questionnaire (IDSIQ). Patient. 2020;14:249–68. https://doi.org/10.1007/s40271-020-00474-z.
Griffiths R, Johnson M. Relative abuse liability of hypnotic drugs: a conceptual framework and algorithm for differentiating among compounds. J Clin Psychiatry. 2005;66(Suppl. 9):31–41.
Dauvilliers Y, Zammit G, Fietze I, Mayleben D, Seboek Kinter D, Pain S, et al. Daridorexant, a new dual orexin receptor antagonist to treat insomnia disorder. Ann Neurol. 2020;87(3):347–56. https://doi.org/10.1002/ana.25680.
Zammit G, Dauvilliers Y, Pain S, Sebok Kinter D, Mansour Y, Kunz D. Daridorexant, a new dual orexin receptor antagonist, in elderly subjects with insomnia disorder. Neurology. 2020;94:e2222–32. https://doi.org/10.1212/wnl.0000000000009475.
Ufer M, Kelsh D, Schoedel KA, Dingemanse J. Abuse potential assessment of the new dual orexin receptor antagonist daridorexant in recreational sedative drug users as compared to suvorexant and zolpidem. Sleep. 2021. https://doi.org/10.1093/sleep/zsab224.
Ufer M, Steiner MA, Post A, Dingemanse J, Toeroek M, Giusepponi M, et al. W150. Assessment of the abuse potential of daridorexant, a new dual orexin receptor antagonist for the treatment of insomnia disorder: data from preclinical and clinical studies. Neuropsychopharmacology. 2020;45(1):354. https://doi.org/10.1038/s41386-020-00892-5.
Morin CM, Colecchi C, Stone J, Sood R, Brink D. Behavioral and pharmacological therapies for late-life insomnia: a randomized controlled trial. JAMA. 1999;281(11):991–9. https://doi.org/10.1001/jama.281.11.991.
Krystal AD, Walsh JK, Laska E, Caron J, Amato DA, Wessel TC, et al. Sustained efficacy of eszopiclone over 6 months of nightly treatment: results of a randomized, double-blind, placebo-controlled study in adults with chronic insomnia. Sleep. 2003;26(7):793–9. https://doi.org/10.1093/sleep/26.7.793.
Roth T, Walsh JK, Krystal A, Wessel T, Roehrs TA. An evaluation of the efficacy and safety of eszopiclone over 12 months in patients with chronic primary insomnia. Sleep Med. 2005;6(6):487–95. https://doi.org/10.1016/j.sleep.2005.06.004.
Michelson D, Snyder E, Paradis E, Chengan-Liu M, Snavely DB, Hutzelmann J, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(5):461–71. https://doi.org/10.1016/s1474-4422(14)70053-5.
Yardley J, Kärppä M, Inoue Y, Pinner K, Perdomo C, Ishikawa K, et al. Long-term effectiveness and safety of lemborexant in adults with insomnia disorder: results from a phase 3 randomized clinical trial. Sleep Med. 2021;80:333–42. https://doi.org/10.1016/j.sleep.2021.01.048.
Krause A, Lott D, Brussee JM, Muehlan C, Dingemanse J. Population pharmacokinetic modeling of daridorexant, a novel dual orexin receptor antagonist. CPT Pharmacomet Syst Pharmacol. 2022. https://doi.org/10.1002/psp4.12877.
Boof ML, Alatrach A, Ufer M, Dingemanse J. Interaction potential of the dual orexin receptor antagonist ACT-541468 with CYP3A4 and food: results from two interaction studies. Eur J Clin Pharmacol. 2019;75(2):195–205. https://doi.org/10.1007/s00228-018-2559-5.
Berger B, Brooks S, Zuiker R, Richard M, Muehlan C, Dingemanse J. Pharmacological interactions between the dual orexin receptor antagonist daridorexant and ethanol in a double-blind, randomized, placebo-controlled, double-dummy, four-way crossover phase I study in healthy subjects. CNS Drugs. 2020;34(12):1253–66. https://doi.org/10.1007/s40263-020-00768-8.
Gehin M, Wierdak J, Sabattini G, Sidharta PN, Dingemanse J. Effect of gastric pH and of a moderate CYP3A4 inducer on the pharmacokinetics of daridorexant, a dual orexin receptor antagonist. Br J Clin Pharmacol. 2022;88(2):810–9. https://doi.org/10.1111/bcp.15029.
Muehlan C, Brooks S, Vaillant C, Meinel M, Jacobs GE, Zuiker RG, et al. Driving performance after bedtime administration of daridorexant, assessed in a sensitive simulator. Clin Pharmacol Ther. 2022;111(6):1334–42. https://doi.org/10.1002/cpt.2592.
Boof M-L, Dingemanse J, Lederer K, Fietze I, Ufer M. Effect of the new dual orexin receptor antagonist daridorexant on nighttime respiratory function and sleep in patients with mild and moderate obstructive sleep apnea. Sleep. 2021;44(6):zsaa275. https://doi.org/10.1093/sleep/zsaa275.
Boof ML, Dingemanse J, Brunke M, Esselmann A, Heymer P, Kestermann O, et al. Effect of the novel dual orexin receptor antagonist daridorexant on night-time respiratory function and sleep in patients with moderate chronic obstructive pulmonary disease. J Sleep Res. 2021;30(4): e13248. https://doi.org/10.1111/jsr.13248.
Berger B, Dingemanse J, Sabattini G, Delahaye S, Duthaler U, Muehlan C, et al. Effect of liver cirrhosis on the pharmacokinetics, metabolism, and tolerability of daridorexant, a novel dual orexin receptor antagonist. Clin Pharmacokinet. 2021;60(10):1349–60. https://doi.org/10.1007/s40262-021-01028-8.
Berger B, Muehlan C, Klein G, Dingemanse J. Pharmacokinetics of daridorexant, a dual orexin receptor antagonist, are not affected by renal impairment. Clin Transl Sci. 2021;14(6):2132–8. https://doi.org/10.1111/cts.13079.
Acknowledgements
The authors thank all study investigators, IDMC and ISB members and participants, study staff and nursing teams for their participation. The authors thank Jean-Paul Clozel (Idorsia Pharmaceuticals) for provision of pertinent comments and critical review of the manuscript. Medical writing support (writing the draft, revising the manuscript and journal styling) was provided by Jessica Beake, Ph.D. (Beake Medicom Ltd), funded by Idorsia Pharmaceuticals Ltd, and figures were created by Nicolas Weber (Idorsia Pharmaceuticals). Data from this study were presented at World Sleep 2022, in abstract, oral and poster form: Kunz D, et al., Long-term safety and efficacy of daridorexant in patients with insomnia disorder [oral presentation]; Dauvilliers Y., et al. Efficacy of long-term treatment with daridorexant in patients with insomnia disorder on sleep and daytime functioning: a post-hoc analysis [poster #109].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by Idorsia Pharmaceuticals Ltd, Allschwil, Switzerland. Idorsia Idorsia Pharmaceuticals Ltd funded the manuscript writing support (manuscript preparation) and the open access fee.
Conflict of interest
DK has participated in advisory boards from Idorsia; DGB has participated in advisory boards from Idorsia, Roche and received a research grant from MSD; GP has participated in advisory boards from Takeda, Bioprojet, Jazz, Fidia and Idorsia; HB has participated in advisory boards from UCB, Jazz and Idorsia; ST has nothing to disclose; YD participated in advisory boards from UCB, Takeda, Bioprojet, Jazz, Orexia, Avadel and Idorsia; DSK, PC, MR and MS-S are employees of Idorsia Pharmaceuticals Ltd. The IDSIQ was developed by Buysse DJ, Thompson W, Scott J and Franzen PL. Germain A, Hall M, Moul, DE, Nofzinger , EA and Kupfer DJ of the University of Pittsburgh and as amended by Idorsia Pharmaceuticals Ltd. IDSIQ© 2020, University of Pittsburg. All rights reserved. IDSIQ-14 derivative created 2020 by Idorsia Pharmaceuticals Ltd under license and distributed by Idorsia Pharmaceuticals Ltd under license.
Ethics approval
The study protocol was approved by the appropriate institutional review boards or independent ethics committees and the study was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Consent to participate
All patients provided written informed consent.
Consent for publication
Not applicable.
Availability of data and material
The study sponsor will receive requests for individual participant data that underlie the results reported in this article, after deidentification, from researchers who provide a methodologically sound proposal. Please direct any requests to medicalinformationus@idorsia.com.
Code availability
Not applicable.
Authors’ contributions
Dalma Seboek Kinter was involved in the study conception and design. Dalma Seboek Kinter, Magdalene Rausch, Dieter Kunz, Yves Dauvilliers, Heike Benes, Diego García-Borreguero, Giuselle Plazzi and Stephen Thein were all involved in data acquisition. Mouna Sassi-Sayadi was involved in the data analysis, validation and visualisation. All authors were involved in interpretation of the data. Dieter Kunz, Yves Dauvilliers, Heike Benes, Diego García-Borreguero, Giuseppe Plazzi and Stephen Thein were trial investigators or participated in the conduct of the study. All authors had full access to the data, were involved with review and editing of the manuscript for important intellectual content, approved the final version of the manuscript and had final responsibility for the decision to submit for publication and agreed to be accountable for the work.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kunz, D., Dauvilliers, Y., Benes, H. et al. Long-Term Safety and Tolerability of Daridorexant in Patients with Insomnia Disorder. CNS Drugs 37, 93–106 (2023). https://doi.org/10.1007/s40263-022-00980-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-022-00980-8