
Abstract
Background and objective
Within the UNIVERSAL project (RIA2019PD-2882) we aim to develop a paediatric dolutegravir (DTG)/emtricitabine (FTC or F)/tenofovir alafenamide (TAF) fixed-dose combination. To inform dosing of this study, we undertook a relative bioavailability (RBA) study in healthy volunteers to investigate a potential pharmacokinetic effect when paediatric formulations of DTG and F/TAF are taken together.
Methods
Participants received all of the following treatments as paediatric formulations in randomised order: a single dose of 180/22.5 mg F/TAF; a single dose of 30 mg DTG; a single dose of 180/22.5 mg F/TAF plus 30 mg DTG. Blood concentrations of DTG, FTC, TAF, and tenofovir (TFV) were measured over 48 h post-dose. If the 90% confidence intervals (CIs) of the geometric least squares mean (GLSM) ratios of area under the curve (AUC) and maximum concentration (Cmax) of each compound were within 0.70–1.43, we considered this as no clinically relevant PK interaction.
Results
A total of 15 healthy volunteers were included. We did not observe a clinically relevant PK interaction between the paediatric DTG and F/TAF formulations for the compounds DTG, FTC, and TFV. For TAF, the lower boundaries of the 90% CIs of the GLSM ratios of the AUC0–∞ and Cmax fell outside our acceptance criteria of 0.70–1.43.
Conclusions
Although TAF AUC and Cmax 90% CIs fell outside the pre-defined criteria (0.62–1.11 and 0.65–1.01, respectively), no consistent effect on TAF PK was observed, likely due to high inter-subject variability. Moreover, there are several reasons to rely on TFV exposure as being more clinically relevant than TAF exposure. Therefore, we found no clinically relevant interactions in this study.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
This is a relative bioavailability study in healthy volunteers, conducted in the Netherlands. |
We investigated if there is a potential pharmacokinetic interaction when paediatric formulations of dolutegravir (DTG) and emtricitabine (FTC)/tenofovir alafenamide fumarate (TAF) were administered together. |
The data of this study will inform on the dose ratio as well as dose selection for a paediatric DTG/FTC/TAF fixed-dose combination to be developed in the EDCTP-funded UNIVERSAL project (RIA2019PD-2882). |
1 Introduction
Worldwide, approximately 1.5 million children younger than 15 years are estimated to be living with HIV (1). These children need lifelong treatment with antiretroviral therapy (ART) to prevent HIV-related morbidity and mortality. However, only 57% of the children living with HIV had access to ART at the end of 2022, whereas that percentage among adults is much higher: 77% (1). Moreover, over a third of those children receiving ART in 2021 were treated with suboptimal regimens and formulations, which contributes to worse treatment outcomes (1).
According to the most recent World Health Organization guidelines (2), the preferred treatment for children living with HIV is a combination of dolutegravir (DTG) with two nucleoside reverse transcriptase inhibitors (NRTIs). If these three medicines could be combined in one paediatric formulation, this would allow easy administration and simplified procurement by programmes in resource-limited settings. Furthermore, fixed-dose combination (FDC) tablets will lead to a minimal pill burden, which will ensure better adherence to the therapy and hence improved treatment outcomes. However, the development of an ideal combined pill containing different HIV medicines for children remains challenging, considering the preferred specific combinations, strengths, and formulations of HIV medicines that vary by age and weight.
Within the EDCTP2-funded UNIVERSAL project (RIA2019PD-2882), complementary antiretroviral FDCs for infants and children will be developed. One of the products to be developed is a dispersible FDC tablet of dolutegravir/emtricitabine/tenofovir alafenamide fumarate (DTG/FTC/TAF). In adults, tenofovir disoproxil fumarate (TDF) together with FTC is one of the preferred NRTI backbones (3). However, TDF is generally not recommended in young children due to potential bone and renal toxicity in growing children. TAF, as a pro-drug of tenofovir (TFV), showed lower plasma exposure to TFV compared to TDF, and therefore these adverse effects are substantially reduced. The treatment combination of DTG with TAF and FTC has recently shown excellent antiviral activity in children in Africa who used it as a second-line treatment (4). Moreover, a dispersible formulation of this combination will ensure that this can also be administered to the younger children who are not able to swallow a tablet. This paediatric-friendly FDC will simplify treatment for children living with HIV in low- and middle- income countries.
Prior to the development of this FDC, the pharmacokinetics (PK) of DTG, FTC, and TAF need to be considered when combined. Earlier drug–drug interaction studies showed no clinically relevant effect of emtricitabine/tenofovir alafenamide fumarate (F/TAF) on DTG exposure, but 17% higher TAF and 25% higher TFV concentrations in combination with DTG were observed (5, 6). These studies were, however, performed with oral solid-dose formulations of DTG and F/TAF not suitable for younger children. The PK data on the combination of DTG dispersible tablets (DT) and F/TAF paediatric tablets for oral suspension (TOS) are currently lacking. DTG DT had 1.6 times higher area under the curve (AUC) than adult film-coated DTG tablets (7). Paediatric formulations contain excipients different to those in the adult tablets, which could potentially play a role in the absorption phase when combining these compounds in one formulation.
In this study, we undertook a relative bioavailability (RBA) study in healthy volunteers to investigate a potential PK effect when paediatric DTG DT (30-mg dose) and F/TAF TOS (180/22.5-mg dose) formulations are taken together. These data will support the UNIVERSAL project in selecting doses for the paediatric DTG/FTC/TAF FDC.
2 Methods
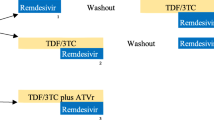
This single-dose, open-label, three-period, randomised, cross-over trial in healthy adult volunteers was conducted from November 2022 to March 2023 at the Radboud university medical Center, Nijmegen, the Netherlands. To investigate the RBA of DTG, TAF, and FTC when taken as paediatric TOS formulations simultaneously, the PK of single doses of DTG DT and F/TAF TOS were compared to a co-administration of DTG DT and F/TAF TOS dispersed together. Since TAF is a pro-drug of TFV, both compounds were measured. We used Gilead’s F/TAF 60/7.5-mg TOS (clinical batch) and ViiV Healthcare’s TIVICAY DTG 5-mg DT. Due to bioassay sensitivity, we chose to use three tablets of F/TAF 60/7.5 mg (180/22.5 mg), which also provides a similar dose to the adult product (200/25 mg). A dose of 30 mg DT DTG, given as six tablets of 5 mg, was chosen because of its bioequivalence to the adult 50-mg film-coated tablet of DTG (7).
The protocol was approved by our national ethics committee as well as the local ethics committee MREC Oost-Nederland. Data were collected using Castor EDC (Castor Electronic Data Capture CB, Amsterdam, the Netherlands). The sample size of this three-period, crossover study was calculated using a mixed linear model with fixed factors period and treatment. A total sample size of 13 participants was considered sufficient for a power of 80% to show a 30% difference for AUC and maximum concentration (Cmax) considering an intrasubject variability for TAF AUC has a coefficient of variation percentage (CV%) of ~ 35%. The goal was to include a total number of 16 participants to account for dropouts.
Healthy volunteers were randomised to one of the following treatment sequences: ABC, ACB, BCA, BAC, CAB, or CBA. The treatment regimens were as follows: reference treatment A—a single dose of 180/22.5 mg (given as 3 × 60/7.5 mg) F/TAF TOS; reference treatment B—a single dose of 30 mg (given as 6 × 5 mg) DTG DT; and test treatment C—a single dose of 180/22.5 mg F/TAF TOS co-administered with a single dose of 30 mg DTG DT. Reference treatments A and B were given as a dispersed suspension and prepared according to the pharmaceutical companies' instructions. In the test treatment, F/TAF TOS were dispersed first, then DTG DT were added to provide a co-dispersed suspension. Every treatment period was followed by a washout period of 7 days.
All participants had an overnight fast before the treatments were administered on the PK days. From 4 h after dosing until release from confinement, consumption of available beverages was allowed as desired and meals were standardised.
3 Blood Sampling
For every participant, blood samples were taken over a period of 48 h during all three treatment periods to construct the PK curves of DTG, FTC, TAF, and TFV. Blood samples were collected at the following time points: t = 0 (pre-dose), 0.17, 0.33, 0.5, 0.75, 1.0, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 24, and 48 h post dose. All samples were centrifuged within 1.5 h after collection. To ensure the stability of TAF, the samples for measuring TAF were acidified with acetic acid. After centrifugation plasma samples were stored at −80 °C at the research clinical centre, until transport to the laboratory of the Department of Pharmacy at the Radboud University Medical Center (Nijmegen, The Netherlands).
4 Analysis
The primary objective of this study was to investigate the RBA of paediatric formulations for DTG, FTC, and TAF when taken simultaneously, from which the potential drug–drug interaction between DTG DT and F/TAF TOS could be assessed. The main PK parameters to be evaluated with respect to the exposure of DTG, FTC, TAF, and TFV were the AUC and Cmax.
Concentrations of DTG, FTC, TAF, and TFV in plasma were analysed using a validated liquid chromatography with tandem mass spectrometry (LC-MS/MS) method at the laboratory of the Pharmacy of the Radboud University Medical Center. The assays were externally validated through the International Interlaboratory Quality Control Program for Measurement of Antiretroviral Drugs in Plasma and the Clinical Pharmacology Quality Assurance (CPQA) Program (8). The lower limits of quantification in these assays were as follows: DTG 0.05 mg/L, FTC 0.015 mg/L, TAF 1.0 ng/mL, and TFV 1.0 ng/mL.
PK parameters were determined by means of non-compartmental analysis using Phoenix 64 WinNonlin (version 8.4). For DTG, FTC, and TAF, the AUC0–∞ was determined using the linear up/log down trapezoidal method. For the exposure of TFV, the AUClast instead of the AUC0–∞ was determined because of the long half-life of TFV after TAF ingestion and subsequent over-extrapolation of the AUC. Additionally, the Cmax, elimination half-life (T½), and time to maximum plasma concentration (Tmax) were determined for each individual curve.
Geometric least squares mean (GLSM) ratios and 90% confidence intervals (CIs) of the PK parameters of the test treatment (DTG DT and F/TAF TOS combined) to both reference treatments (DTG DT or F/TAF TOS separately) were determined using the method described in the European Medicines Agency (EMA) guidance on investigations of bioequivalence (9). An analysis of variance (ANOVA) was performed on log-transformed data with fixed effects: treatment, period, sequence, and subject within sequence (bioequivalence module in WinNonlin/Phoenix version 8.4). To determine whether there is a potential, clinically relevant PK interaction, the following pre-defined acceptance criteria were used: if after a single dose, the 90% CIs of the GLSM ratios of AUC and Cmax of each compound are within the range 0.70–1.43, there is no clinically relevant PK interaction. The 0.70 and 1.43 boundaries were selected based on the dose–exposure relationship for the antiretroviral drugs studied, which indicated that a 30% change in exposure was not clinically relevant. In addition, these no-effect boundaries have been previously used in the interaction tables of the substance summary of product characteristics (SmPCs) to inform dosing of adults and children, where a difference less than 30% did not lead to any dose adjustments for clinical practice. In addition, the safety and tolerability of the treatments in healthy participants were evaluated. All adverse events reported spontaneously by the participant or observed by the investigator, or his staff were recorded. For each adverse event, the following information was recorded and formally reported: start and stop date and time, severity, relationship to trial medication, action taken, and outcome.
5 Results
Sixteen healthy volunteers (seven female, nine male) were enrolled in this study. One female discontinued before study medication was taken, because of recently started antibiotic treatment. Two dropped out after the first period of the study because of personal considerations that were unrelated to the study. The latter two received treatment B (30 mg DT DTG) only; the corresponding PK curves of DTG were included in the analysis. The median age of the 15 included participants was 27.0 (interquartile range [IQR] 21.0–31.0) years. The median body mass index was 25.1 (IQR 21.6–26.0) kg/m2. Most of the participants were Caucasian (86.7%), with only one black and one Asian participant. All participants provided written informed consent for this study.
Figure 1 shows the median concentration–time profiles of all compounds for each treatment. Table 1 shows the geometric means and coefficient of variation for PK parameters of the test treatment (C) and the reference treatments (A and B) and the GLSM ratios and the corresponding 90% CIs of the test treatments versus the reference treatment. Test treatment C (DTG and F/TAF combined) met the pre-specified acceptance criteria when compared to the reference treatments (DTG and F/TAF separately) for DTG, TFV, and FTC components. The DTG GLSM ratios (90% CI) were 1.02 (0.94–1.10) for AUC0–∞ and 1.16 (1.10–1.23) for Cmax. The TFV values were 0.84 (0.70–1.01) for AUC0–last and 1.06 (0.92–1.23) for Cmax. The FTC values were 0.97 (0.92–1.02) for AUC0–∞ and 1.05 (0.94–1.17) for Cmax. All 90% CIs were within the range of 0.70–1.43 except for TAF; the lower boundaries of the 90% CIs of the GLSM ratios for AUC0–∞ and Cmax were 0.62 and 0.65, respectively. Figure 2 shows the individual ratios of the test treatment versus the reference treatments for all compounds.

Median plasma concentration–time curves of a DTG after single-dose 30 mg DTG (treatment B) or single-dose 30 mg DTG + 280/22.5 mg F/TAF (treatment C); b TAF after single-dose 280/22.5 mg F/TAF (treatment A) or single-dose 30 mg DTG + 280/22.5 mg F/TAF (treatment C); c TFV after single-dose 280/22.5 mg F/TAF (treatment A) or single-dose 30 mg DTG + 280/22.5 mg F/TAF (treatment C); d FTC after single-dose 280/22.5 mg F/TAF (treatment A) or single-dose 30 mg DTG + 280/22.5 mg F/TAF (treatment C). DTG dolutegravir, F/TAF FTC/TAF, FTC emtricitabine, TAF tenofovir alafenamide fumarate, TFV tenofovir

Individual AUC and Cmax ratios of a DTG, b TAF, c TFV, and d FTC. The grey areas indicate the pre-defined acceptance criteria for 90% CI (0.70–1.43). The dotted line in the middle indicates a GLSM ratio of 1, meaning that there is no change in exposure or Cmax between the test treatment C and the reference treatment A or B. AUC area under the curve, CI confidence interval, Cmax maximum concentration, DTG dolutegravir, FTC emtricitabine, GLSM geometric least squares mean, TAF tenofovir alafenamide fumarate, TFV tenofovir
No serious adverse events were observed, and none of the drop-outs in the study were related to adverse events. In total, 19 adverse events of mild to moderate grade were observed and reported during the study. Four adverse events were judged not to be related to study medication. Three adverse events were judged to be possibly related to the study medication. The others were unlikely related to the study medication. Table 2 shows the number, grade, and relation to study medication of these adverse events.
6 Discussion
In this study, we did not observe a clinically relevant PK interaction between DTG DT and F/TAF TOS for the compounds DTG, FTC, and TFV. For TAF, the lower boundaries of the 90% CIs of the GLSM ratios of the AUC0–∞ and Cmax fell outside our acceptance criteria of 0.70–1.43.
For DTG, we found a GLSM ratio of 1.16 (90% CI 1.10–1.23) for Cmax, with all but one individual ratio above 1. This may indicate that there is a non-significant increase in the absorption of DTG when combined with F/TAF TOS. As there was no impact on AUC0–∞, and the Tmax was reached earlier (0.75 h in the test treatment versus 1 h in the reference treatment), this effect may occur in the absorption phase only. A significant higher Cmax of DTG has been observed and reported when the adult film-coated formulation of DTG was given with F/TAF, suggesting that this minor effect may be related to co-administration, but the actual mechanism for this potential effect is currently unknown (5, 10). Given that the safety data for DTG has been reassuring (11-13), and we expect not to reach the Cmax of 5.4 mg/L found in adults taking DTG 50 mg twice daily and which has been demonstrated to be safe (12), we see no cause for concern. Finally, we found comparable AUC0–∞ and Cmax values for DTG in our study after a single dose of 30-mg DT compared to other single-dose studies with the 50-mg adult DTG product, which is bioequivalent to the dose we gave (14, 15).
For FTC, there were no differences in Cmax and AUC0–∞ GLSM ratios compared to previous studies (5, 6, 16). In addition, the individual values are equally distributed; hence no trend was observed in higher or lower exposure in either of the treatments.
For TAF, we could not rule out a potentially relevant PK interaction between DTG DT and F/TAF TOS based on AUC0–∞ and Cmax GLSM ratios on 90% CIs. However, when evaluating the individual ratios of TAF in the test treatment versus the reference treatment, no consistent trend was seen in TAF exposure in either of the treatments: 50% of the participants in this study had a ratio below 1 and 50% had a ratio above 1. In contrast to our study, higher TAF exposure was reported when given with DTG as adult formulations (5, 6). Furthermore, as TAF is a pro-drug of TFV, and since TFV exposures were not impacted, the clinical impact was doubtful. It has been shown that higher concentrations of intracellular TFV diphosphate (TFV-DP) are found after administration of TAF compared to administration of TDF, with much lower TFV concentrations (6). TAF exhibits greater stability in the intestine and plasma compared to TDF; as a consequence, TAF has improved intracellular accumulation of TFV-DP in HIV target cells with lower circulating levels of TFV. Therefore, we expect that the lower exposure of TAF observed in our study is negligible in terms of clinical relevance. In addition, the AUC0–∞ levels observed within this study, were still within ranges that were found to be safe and effective in adults, indicating that the observed changes are not clinically relevant (17). We suggest that our results mainly reflect the high intersubject variation which has been reported for TAF. Previous studies measuring TAF (25-mg dose) administered to adults in a fasting state found PK results in the same order of magnitude as we found with a 22.5-mg TAF dose (6, 18), which reinforces confidence in the study results.
In conclusion, we did not find any clinically relevant PK interaction between DTG DT and F/TAF TOS for DTG, TFV, and FTC when co-administered, compared to products administered separately. Although TAF AUC0–∞ and Cmax 90% CIs fell outside the pre-defined criteria, no consistent effect on TAF PK was observed, likely due to high intersubject variability. Moreover, there are several reasons to rely on TFV exposure as being more clinically relevant than TAF exposure. Therefore, we assume that we found no clinically relevant interactions in this study. These data will inform on the dose ratio as well as dose selection for a paediatric DTG/FTC/TAF FDC to be developed in the UNIVERSAL project.
Change history
02 May 2024
A Correction to this paper has been published: https://doi.org/10.1007/s40262-024-01378-z
References
UNAIDS. Global HIV & AIDS statistics fact sheet 2023 2023 [Available from: https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf. Accessed 10 nov 2023
Organisation WH. Consolidated guidelines on HIV prevention, testing, treatment, service delivery and monitoring. 2021. Accessed 10 nov 2023
DHHS. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV 2023 [Available from: https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/adult-adolescent-arv/guidelines-adult-adolescent-arv.pdf. Accessed 10 nov 2023
Musiime VAJS, Bwakura Dangarembizi M, Chabala C, Doerholt K, Dukakia N, Kityo Mutuluza C, Lugemwa A, Lungu J, Makumbi S, Mcllleron H, Mujuru HA, Mulenga V, Musoro G, Mwamabazi M, Nambi E, Nazzinda R, Ndebele W, Nduna B, Shakeshaft C, Siziba G, Tafeni S, Turkova A, Walker AS, Gibb DM. Editor Increasing second-line antiretroviral therapy options for children with HIV in Africa: week-96 efficacy and safety results of the CHAPAS-4 randomised trial IAS 2023; 2023. Accessed 10 nov 2023
Begley R, Das M, Zhong L, Ling J, Kearney BP, Custodio JM. Pharmacokinetics of tenofovir alafenamide when coadministered with other HIV antiretrovirals. J Acquir Immune Defic Syndr. 2018;78(4):465–72. Accessed 10 nov 2023
FDA. Descovy: Clinical Pharmacology Biopharmaceutics Review 2016 [Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208215Orig1_toc.cfm. Accessed 10 nov 2023
EMA. Tivicay; Summary of Product Characteristics 2022 [updated May 21, 2019. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002753/WC500160680.pdf. Accessed 10 nov 2023
Clinical Pharmacology Quality Assurance Program [Available from: http://www.buffalo.edu/tprc/section-2/CPQA.html. Accessed 10 nov 2023
EMA. Guideline on the investigation of bioequivalence 2010 [Available from: http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf. Accessed 10 nov 2023
FDA. Clinical Pharmacology Review Abbendum F/TAF or FTC/TAF 2015 [Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208215Orig1s000ClinPharmR.pdf. Accessed 10 nov 2023
Min S, Sloan L, DeJesus E, Hawkins T, McCurdy L, Song I, et al. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of dolutegravir as 10-day monotherapy in HIV-1-infected adults. AIDS. 2011;25(14):1737–45. Accessed 10 nov 2023
Eron JJ, Clotet B, Durant J, Katlama C, Kumar P, Lazzarin A, et al. Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING Study. J Infect Dis. 2013;207(5):740–8. Accessed 10 nov 2023
ViiV Healthcare. A Pilot Study Assessing the Integrase Inhibitor GSK1349572 in HIV-infected Persons With Virus Resistant to Raltegravir. Study Results ClinicalTrials.gov2013 [Available from: https://clinicaltrials.gov/ct2/show/results/NCT00950859?term=dolutegravir&recrs=e&cond=Hiv&outc=AUC&age=1&draw=4&rank=30. Accessed 10 nov 2023
Song IH, Borland J, Savina PM, Chen S, Patel P, Wajima T, et al. Pharmacokinetics of single-dose dolutegravir in HIV-seronegative subjects with moderate hepatic impairment compared to healthy matched controls. Clin Pharmacol Drug Dev. 2013;2(4):342–8. Accessed 10 nov 2023
Song I, Borland J, Arya N, Wynne B, Piscitelli S. Pharmacokinetics of dolutegravir when administered with mineral supplements in healthy adult subjects. J Clin Pharmacol. 2014;XX(XX):1–7. Accessed 10 nov 2023
FDA. Multi-Discipline review Biktarvy [updated 18 March 2018 cited 20 May 2021 Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210251Orig1s000MultidisciplineR.pdf. Accessed 10 nov 2023
Ruane PJ, DeJesus E, Berger D, Markowitz M, Bredeek UF, Callebaut C, et al. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of tenofovir alafenamide as 10-day monotherapy in HIV-1-positive adults. J Acquir Immune Def Syndr. 2013;63(4):449–55. Accessed 10 nov 2023
EMA. Product Information: Biktary 2020 [Available from: https://www.ema.europa.eu/en/documents/product-information/biktarvy-epar-product-information_en.pdf. Accessed 10 nov 2023
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Trial medication (F/TAF TOS) was provided, and the trial funded, by Gilead Sciences, Inc. This study was conducted as an addendum to the EDCTP2-funded UNIVERSAL project (RIA2019PD-2882).
Conflict of Interest/Competing Interests
AC and DB have received research grants (paid to the institution) from ViiV Healthcare, Gilead Sciences, and Merck. All other authors have no conflicts of interest to disclose.
Ethics Approval
Yes.
Consent to Participate
Yes.
Consent for Publication
Yes.
Availability of Data and Material
The data that support the findings of this study are available on request from the corresponding author.
Code Availability
Not applicable.
Authors’ Contributions
LB and AC have written the protocol. LB and AK arranged all study preparations, including recruiting participants. LW was the study physician and did the screening of the participants and had overall medical responsibility. LB and AK processed the samples during the study and were the main contacts during the study days. LB and AK performed the analysis of the study. LB, AK, AC, DB, and RL took part in all substantive discussions. LB and AK wrote the manuscript. All co-authors reviewed the manuscript and provided substantial input that led to the final manuscript.
Additional information
The original online version of this article was revised to correct Table 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bevers, L.A.H., Kamphuis, A.E.M., van der Wekken-Pas, L.C.W. et al. Relative Bioavailability of Dolutegravir (DTG) and Emtricitabine/Tenofovir Alafenamide Fumarate (F/TAF) Administered as Paediatric Tablet Formulations in Healthy Volunteers. Clin Pharmacokinet (2024). https://doi.org/10.1007/s40262-024-01365-4
Accepted:
Published:
DOI: https://doi.org/10.1007/s40262-024-01365-4