
Abstract
Nonalcoholic fatty liver disease (NAFLD) is one of the most common chronic diseases of the liver, even in pediatrics. NAFLD ranges from simple fat accumulation (steatosis) to inflammation and fibrosis [nonalcoholic steatohepatitis (NASH)]. The progression of disease is a critical aspect, since the evolution of fatty liver to fibrotic stages may be the prelude to cirrhosis and hepatocellular carcinoma. Accumulating evidence has shown that the connection between the gut and the liver is crucial among the factors involved in liver disease progression. In patients with NAFLD, small bowel bacterial overgrowth and an increased intestinal permeability are present. In such conditions, the hepatobiliary system is inevitably exposed to a high level of bacterial products and is thus able to activate the innate immune system and initiate the cascade of proinflammatory signal transduction leading to liver inflammation and fibrosis. This review will provide an overview of the current knowledge on the pathophysiology of the cross-talk between the gut and the liver in the pathogenesis of NAFLD and its progression to NASH, and will discusses potential therapeutic means to manipulate the intestinal microbiota in order to modulate liver disease development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is one of the most common chronic diseases of the liver, even in pediatrics. It nowadays reaches a prevalence of up to 40 % in the general population and up to 13 % in children [1]. These prevalence data reflect the parallel increase of the risk factors of this disease, such as obesity and type 2 diabetes mellitus.
NAFLD ranges from simple fat accumulation (steatosis) to inflammation and fibrosis [nonalcoholic steatohepatitis (NASH)]. The progression of NAFLD to the more severe form, NASH, is linked to various genetic and environmental factors [2•]. The progression of disease is a crucial aspect, since the evolution of fatty liver to fibrotic stages may be the prelude to cirrhosis and hepatocellular carcinoma (HCC) [3], even if in children only one case of steatosis-dependent HCC has been reported in the absence of a fibrotic background [4]. Plenty of research has focused on the interaction between the liver and gut, since the so-called “gut–liver axis” appears to play a major role among the factors leading to disease progression [5, 6•].
Normally the intestinal epithelium allows the absorption of nutrients while also functioning as a barrier, which prevents antigens and pathogens from entering the mucosal tissues and potentially causing disease. The intestinal tract is inhabited by 1014 microbes [7], and it has become evident that they are involved in molecular cross-talk with the intestinal epithelium and affect the intestinal barrier function [8]. An important component of the intestinal barrier is the intercellular junctional complex, crucial for the maintenance of barrier integrity. Tight junctions (TJs) constitute a multifunctional complex that forms a seal between adjacent epithelial cells near the apical surface [9]. TJs seal the paracellular space between epithelial cells, thus preventing paracellular diffusion of microorganisms and other antigens across the epithelium. They are not a static barrier, but highly dynamic structures that are constantly being remodeled because of interactions with external stimuli, such as food residues and pathogenic and commensal bacteria. In this way, TJs can regulate the entry of nutrients, ions and water, but restrict pathogen entry and maintain the barrier function of the epithelium.
NAFLD is associated with increased intestinal permeability (IP), since TJs appear to go through a disruption process making the intestine in these liver patients “leaky.” In in vitro and animal models of NAFLD, increased IP and alterations of gut microbiota have been shown to augment the exposure of the liver to gut-derived bacterial products. Increased serum levels of these products [for example lipopolysaccharides (LPS)] may cause low-grade endotoxemia, especially in the portal system and in the liver. Endotoxemia stimulates innate immune receptors, which activate signaling pathways involved in liver inflammation and fibrogenesis [10•].
In this setting, it is not surprising that probiotics reduce hepatic injury and inflammation in ob/ob mice [11], suggesting that modulation of the gut microbiota by probiotics may represent a feasible approach for the prevention or treatment of NASH.
The purpose of this review is to provide an overview of the current knowledge on the pathophysiology of the cross-talk between the gut and the liver in the pathogenesis of NAFLD and its progression to NASH, trying to open new treatment horizons in this field.
Role of Intestinal Permeability in the Gut–Liver Cross-Talk
It is now well known that in case of small bowel bacterial overgrowth (SBBO) and increased IP, the hepatobiliary system is inevitably exposed to a high level of these bacterial products and is thus able to activate the receptors on cell surfaces and to initiate the cascade of proinflammatory signal transduction leading to liver inflammation and fibrosis. In patients with NAFLD and correspondents in animal models, both alterations are demonstrated.
In a case-control study, Wigg et al. [12] showed that patients with NASH had a higher prevalence of SBBO, as well as blood levels of tumor necrosis factor alpha (TNF-α), compared to controls. Another case-control study by Miele et al. [13] has given particularly significant results. This study was able to confirm not only that NAFLD is associated with SBBO, but also that it is associated with an increased IP because of a process of destruction of the TJs of the intestinal wall, which in turn is associated with the severity of hepatic steatosis. Miele et al. [13] studied 35 consecutive patients with biopsy-proven NAFLD, 27 with untreated celiac disease (as a model of IP), and 24 healthy volunteers. They assessed the presence of SBBO by glucose breath testing, IP by means of the urinary excretion of 51Cr-ethylene diamine tetraacetate (51Cr-EDTA) test, and the integrity of TJs within the gut by immunohistochemical analysis of zona occludens-1 (ZO-1) expression in duodenal biopsy specimens. Patients with NAFLD had significantly increased gut permeability (compared with healthy subjects; p < 0.001) and a higher prevalence of SBBO, although both were lower than in the untreated celiac patients. In patients with NAFLD, both IP and the prevalence of SBBO were correlated with the severity of steatosis, but not with the presence of NASH. Our group has recently conducted a case-control study on a pediatric population [14], investigating the prevalence of altered IP in children with biopsy-proven NAFLD and studying its potential association with the stage of liver disease. LPS blood levels were also evaluated. IP in children was measured using the lactulose-mannitol bowel permeability test [15]. In line with previous findings, IP was significantly higher in children with NAFLD compared to controls (p < 0.05). Within the NAFLD group, IP was increased in children with NASH compared to those with steatosis only (p < 0.05) and was correlated with the presence and stage of portal inflammation (p = 0.02), fibrosis (p = 0.0002) and ballooning of hepatocytes (p = 0.003). Moreover, circulating LPS levels were higher in children with steatohepatitis (p < 0.05). IP and LPS blood levels increased in patient with NAFLD and were correlated with the severity of disease. Therefore, qualitative or quantitative changes in the intestinal bacterial flora and in their toxic products may lead to disruption of the intestinal barrier, bacterial translocation across the gut mucosa and the development of portal endotoxemia.
Why a Link Between the Liver and the Gut?
The liver is a very peculiar organ, since it receives blood from both the portal vein and the hepatic artery. The blood via the portal vein, draining the mesenteric veins, contains not only products derived from food digestion, but also from the bacteria that physiologically colonize the gut. In fact, it has been suggested that the portal vein serves as a “super highway” from the intestine to the liver [16••].
The portal system carries about 70–75 % of the total hepatic blood flow and releases bacterial products such as the LPS, bacterial DNA and peptidoglycan into the liver. All these molecules belong to distinct classes of endogenous signals, called pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), that trigger the host immune response by the activation of Toll-like receptors (TLRs) [17]. The protective role of TLRs against numerous microbes is mostly achieved by activating some of the dominant signaling cascades such as the nuclear factor kappa B (NFκB) transcriptional pathways, which are referred to as proinflammatory since they promote immune cell recruitment [17].
It has been demonstrated that in NASH patients, even low blood levels of microbial products—endotoxemia—might activate TLR pathways in the liver. In fact, TLRs are a class of receptors that recognize structurally conserved microbe-derived molecules and are expressed on Kupffer cells, biliary epithelial cells, hepatocytes, hepatic stellate cells (HSC), epithelial cells and dendritic cells of the liver [6•]. They are part of the mucosal immune system and have the major role in detecting and clearing most food-borne pathogens as well as preventing harmful bacteria from harming beneficial bacteria and host tissues.
While immune cell recruitment exerts a containment action against pathogens, it can also result in host tissue damage. It is well known that cytokines that mediate the inflammatory responses such as TNF-α also have a variety of effects on differentiation pathways; thus, there is a potential for microbial products to have broad effects on host phenotype. The latter seem to happen in the development and progression of fatty liver disease. In fact, some studies have shown that patients with NAFLD have an increased blood level of LPS, especially if affected by NASH or initial stages of liver fibrosis [6•, 18]. Moreover, levels of bacterial endotoxins seem correlated to the levels of proinflammatory cytokines, such TNFα. Yang et al. [19] were the first researchers to observe that LPS is able to promote the evolution of NAFLD in NASH in animal NAFLD models through the activation of TNFα. Wigg et al. [12] also confirmed these findings in humans in a case-control study: patients with NASH had higher a prevalence of SBBO as well as higher blood levels of TNFα compared to controls. This demonstrates the cross-talk between the gut and liver in NAFLD.
The Cross-Talk Between the Liver and Gut in NAFLD and Its Related Fibrosis
TLRs are present on the cells of innate immunity and are able to recognize highly conserved motifs in PAMPs and DAMPs. TLRs, acting as immune sensors of PAMPs and DAMPs, initiate an adaptive immune response and a signaling cascade leading to activation of proinflammatory genes. The most studied PAMP is LPS, a component of the gram-negative bacterial cell membrane, the active component of endotoxin. The latter binds to LPS-binding protein, which subsequently binds to CD14.
The LPS–LPS-binding protein (LBP)–CD14 complex activates TLR-4, present on Kupffer cells, triggering an essential inflammatory cascade. Because of the unique “blood link” between the liver and the gut described above, the liver is constantly exposed to TLR ligands. Normal livers do not show signs of inflammation because of low expression of TLR-4 and its adaptor molecules in the liver and the ability to modulate TLR-4 signaling by liver tolerance. But under pathologic conditions such as the increased IP that is seen in NAFLD, altered TLR-4/LPS signaling plays an important role in the pathogenesis and progression of chronic fatty liver disease.
Of all the 13 TLRs known, TLR-2, TLR-4, its coreceptor CD14 and TLR-9 have been well studied in NAFLD. TLR4 specifically is the receptor of LPS. Szabo and coworkers investigated the role of TLR-2 and TLR-4 polymorphism on liver damage and on cytokine induction in a methionine–choline-deficient (MCD) diet-induced model of NASH [20]. They found that in TLR-2−/− mice there is an increase in liver injury associated with NASH, which may suggest a protective role for TLR-2−/− mediated signals in liver injury [20]. Similarly, Rivera et al. [21] found that hepatocellular damage was notably more severe and the TNF-α level was more elevated in TLR-2−/− mice. Possibly the TLR-2 deficiency exacerbates NASH by altering the signaling via the TLR-4 pathway, whereas its presence may play a protective role against the induction of NASH.
Chronic liver injury leads to the development of hepatic fibrosis because of an inflammatory trigger and the consequent increased accumulation of extracellular matrix in the liver [22]. It is now clear that if one hand is made by the Kupffer cells, which are those initiating fibrogenesis by secreting proinflammatory and profibrogenic cytokines, the HSCs are the predominant source of extracellular matrix production in the fibrotic liver [23].
Noteworthily, abundant data demonstrate that LPS is elevated in experimental models of hepatic fibrosis [24–26] and in patients with cirrhosis [27–29]. It is believed that alterations of the intestinal microbiota and a failure of the intestinal mucosal barrier cause increases in bacterial translocation and LPS levels, especially in later stages of hepatic fibrosis and cirrhosis [30–35]. Recent studies on mice models have demonstrated the crucial role for the LPS–TLR4 pathway in hepatic fibrogenesis. In fact, TLR4 is expressed on two key mediators of hepatic fibrogenesis: Kupffer cells and HSC [2•, 36, 37]; although Kupffer cells express the highest levels of TLR4 in the liver and are considered a prime target of LPS, TLR4 expressed on quiescent and activated HSC is the main mediator of fibrosis. It has been demonstrated that TLR4-chimeric mice display an important reduction in hepatic fibrogenesis [24]. LPS directly targets HSC in vivo to upregulate chemokines and attract Kupffer cells. At the same time, TLR4 activation induces a downregulation of the TGFβ pseudoreceptor Bambi on HSC. These two mechanisms work hand in hand to promote the activation of HSC by Kupffer cell-released TGFβ and subsequently hepatic fibrosis. In fact, HSCs [38] have a TLR4-mediated NF-κB activation in response to a fairly low concentrations of LPS. NF-κB is reported to be the predominant target through which TLR4 ligands promote fibrosis in the liver [39].
The crucial role of LPS is also supported by the finding that LBP-deficient as well as gut-sterilized mice also have a marked reduction of hepatic fibrosis [24, 40]. In addition to LPS, endogenous TLR ligands such as the two most known DAMPs, hyaluronan and high-mobility group box 1 (HMGB1), are also elevated in murine fibrogenesis [24]. Interestingly, a recent experimental study demonstrated that HMGB1 is able to induce proliferation and activation of HSC and to trigger their proinflammatory and profibrogenic phenotype by increasing the expression of monocyte chemoattractant protein-1 (MCP-1), enhancing the effect of TGFβ [41•].
Also the TLR9 recognizes fragments of bacterial DNA in intestinal derivation and is able to induce the production of proinflammatory cytokines; in fact TLR9 appears to be involved in the evolution of nonalcoholic fibrotic liver disease through the production of IL1β by the Kupffer cells. Indeed knockout mice for TLR9 have a reduced degree of steatohepatitis and fibrosis [6•].
Modulating the intestinal microflora and consequent endotoxemia levels through the use of probiotics and selective intestinal decontamination may open new ways to avoid NAFLD complications with hepatic fibrosis, cirrhosis and HCC.
Effects of Intestinal Microbiota Modulation on NASH
Probiotics are live commensal microorganisms that can modulate the intestinal microbiota. A first systematic review in 2007 [42] underlined the absence of randomized controlled trials on the administration of probiotics in patients with NAFLD. However, to date, several studies have been performed in both animal models and in humans, some of which are randomized controlled trials (see Table 1), showing encouraging results for the use of these formulations.
Changes in the intestinal microbiota through the use of probiotics seem to be able to vary NAFLD progression, limiting the progression of fibrogenesis in several animal models of NAFLD [43, 44]. Performing interventions on intestinal microbiota using probiotics has been demonstrated to modulate the expression of nuclear receptors, correcting insulin resistance in the liver and the adipose tissues and protecting against the development of NASH and progression to fibrosis.
VSL#3 is the most largely studied probiotic in the field. In particular, Velayudham et al. demonstrated VSL#3, a mixture of eight probiotic strains (Streptococcus salivarius subsp. Thermophilus, Bifidobacterium [B. breve, B. infantis, B. longum], Lactobacillus acidophilus, L. plantarum, L. casei, and L. delbrueckii subsp. Bulgaricus), exerted a protective action against fibrogenesis, but not on hepatic inflammation, in a MCD diet-induced mouse model of NAFLD [43]. In mousee models of genetic dyslipidemia (Apo-E-deficient mice) failing to develop NASH-like lesions on a standard diet, it has been shown that destrane sulfate sodium-induced intestinal inflammation and the consequently increased IP triggered the transition of steatosis to NASH and that these disorders were efficiently prevented by a therapeutic intervention with VSL#3 [45]. Similar results were also obtained in animal models of high-fat-diet-induced liver disease that was attenuated by VSL#3 treatment [46].
More recently, the MIYAIRI 588 strain of C. butyricum has been reported to induce a decrease in hepatic fibrous deposition in rat models of choline-deficient/l-amino acid-defined (CDAA)-diet-induced NAFLD [43].
In intestinal tissues in a rodent model of colitis, it was demonstrated that probiotics can modulate IP and correct the inflammation-driven metabolic dysfunction [46]. Therefore, it is conceivable that in theory probiotic administration might reset the “leaky gut” of NAFLD patients, offering an interesting approach to counteracting liver damage in NAFLD even though no well-designed clinical trials with effective results are available yet [47].
Conclusions
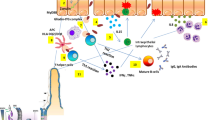
The gut and liver are anatomically strictly linked to one another. Alterations in the gut barrier through a TJ disruption process whose mechanisms are not entirely clear break the established balance between the two organs. Consequent to destruction of the IP integrity, harmful molecules are released in the portal vein blood and act against the liver to activate inflammation and fibrogenesis. TLRs appear to play a crucial role at this point, since their signaling cascade induces the production and release of proinflammatory signals in Kupffer cells and the expression of a profibrogenic pattern in HSCs. The host inflammatory response of Kupffer cells and HSC activation via TLRs therefore develop into a vicious cycle that, in a dangerous liaison between the gut and liver, causes low-grade hepatic inflammation and damage, inducing the shift from simple steatosis to a more aggressive NASH coupled with fibrosis (Fig. 1).

The role of the gut in the pathogenesis of NASH
As the diet-dependent alteration of the composition of the intestinal microbiota is at the root of this process, therapeutic approaches, including probiotic-based treatments, seem able to restore the physiological microbioma. Interventions on the microflora using probiotics appear to be promising against NAFLD-related tissue damage. Despite early experimental studies, no clinical evidence to support the use of any such probiotics is available. Hence, further studies are needed to understand whether probiotics may realistically open a new page in the treatment of NAFLD-related damage or at least may represent a concrete support to increase the effectiveness of already proven targeted drugs.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Giorgio V, Prono F, Graziano F, Nobili V. Pediatric non alcoholic fatty liver disease: old and new concepts on development, progression, metabolic insight and potential treatment targets. BMC Pediatr. 2013;25(13):40. doi:10.1186/1471-2431-13-40.
• Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol. 2013. doi:10.1038/nrgastro.2013.149. This paper goes through the pathogenesis of NAFLD and NASH, which is the result of a complex interplay between host and environmental factors. Recently uncovered aspects of the genetic, biochemical, immunological and molecular events are widely discussed here.
Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, Benson JT, Enders FB, Angulo P. The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut. 2009;58:1538–44.
Nobili V, Alisi A, Grimaldi C, Liccardo D, Francalanci P, Monti L, Castellano A, de Ville de Goyet J. Non-alcoholic fatty liver disease and hepatocellular carcinoma in a 7-year-old obese boy: coincidence or comorbidity? Pediatr Obes. 2013;. doi:10.1111/j.2047-6310.2013.00209.x.
Miele L, Marrone G, Lauritano C, Cefalo C, Gasbarrini A, Day C, Grieco A. Gut–liver axis and microbiota in NAFLD: insight pathophysiology for novel therapeutic target. Curr Pharm Des. 2013;19:5314–24.
• Frasinariu OE, Ceccarelli S, Alisi A, Moraru E, Nobili V. Gut–liver axis and fibrosis in nonalcoholic fatty liver disease: an input for novel therapies. Dig Liver Dis. 2013;45:543–51. doi:10.1016/j.dld.2012.11.010. In this review it is clearly and easily explained how the link between the gut and the liver seems to play a major role among the factor leading to NAFLD progession, and widely clarifies the role of the innate immunity in this disease.
Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–48.
Wells JM, Rossi O, Meijerink M, van Baarlen P. Microbes and Health Sackler Colloquium: epithelial crosstalk at the microbiota–mucosal interface. Proc Natl Acad Sci USA. 2010;. doi:10.1073/pnas.1000092107.
Ulluwishewa D, Anderson RC, McNabb WC, Moughan PJ, Wells JM, Roy NC. Regulation of tight junction permeability by intestinal bacteria and dietary components. J Nutr. 2011;141(5):769–76. doi:10.3945/jn.110.135657.
• Roh YS, Seki E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J Gastroenterol Hepatol. 2013;28(Suppl 1):38–42. doi:10.1111/jgh.12019. Toll-like receptors are the most important component of the innate immunity that are involved in the so-called gut–liver axis. It is here explained how they act in the interplay between the gut and the liver.
Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, Desimone C, Song XY, Diehl AM. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37:343–50.
Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48(2):206–11.
Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Mascianà R, Forgione A, Gabrieli ML, Perotti G, Vecchio FM, Rapaccini G, Gasbarrini G, Day CP, Grieco A. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–87. doi:10.1002/hep.22848.
Giorgio V, Miele L, Principessa L, Ferretti F, Villa MP, Negro V, Grieco A, Alisi A, Nobili V. Intestinal permeability is increased in children with non-alcoholic fatty liver disease, and correlates with liver disease severity. Dig Liver Dis. 2014;. doi:10.1016/j.dld.2014.02.010.
Dastych M, Dastych M Jr, Novotná H, Cíhalová J. Lactulose/mannitol test and specificity, sensitivity, and area under curve of intestinal permeability parameters in patients with liver cirrhosis and Crohn’s disease. Dig Dis Sci. 2008;53:2789–92. doi:10.1007/s10620-007-0184-8.
•• Chassaing B, Etienne-Mesmin L, Gewirtz AT. Microbiota-liver axis in hepatic disease. Hepatology. 2014;59(1):328–39. doi:10.1002/hep.26494. Accumulating evidence indicates that the gut microbiota plays a key role in chronic inflammatory disease of the liver. This is an extensive review on the potential therapeutic means to manipulate the microbiota so as to prevent and/or treat liver disease.
Yang L, Seki E. Toll-like receptors in liver fibrosis: cellular crosstalk and mechanisms. Front Physiol. 2012;22(3):138.
Mehal WZ. The Gordian Knot of dysbiosis, obesity and NAFLD. Nat Rev Gastroenterol Hepatol. 2013;10(11):637–44. doi:10.1038/nrgastro.2013.146.
Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA. 1997;94(6):2557–62.
Szabo G, Velayudham A, Romics L Jr, Mandrekar P. Modulation of non-alcoholic steatohepatitis by pattern recognition receptors in mice: the role of Toll-like receptors 2 and 4. Alcohol Clin Exp Res. 2005;29(Suppl 11):140S–5S.
Rivera CA, Gaskin L, Allman M, Pang J, Brady K, Adegboyega P, Pruitt K. Toll-like receptor-2 deficiency enhances non-alcoholic steatohepatitis. BMC Gastroenterol. 2010;10:52.
Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol. 2013;3(4):1473–92. doi:10.1002/cphy.c120035.
Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–18.
Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGFbeta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–32.
Nolan JP, Leibowitz AI. Endotoxin and the liver. III. Modification of acute carbon tetrachloride injury by polymyxin b—an antiendotoxin. Gastroenterology. 1978;75:445–9.
Grinko I, Geerts A, Wisse E. Experimental biliary fibrosis correlates with increased numbers of fat-storing and Kupffer cells, and portal endotoxemia. J Hepatol. 1995;23:449–58.
Fukui H, Brauner B, Bode JC, Bode C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J Hepatol. 1991;12:162–9.
Chan CC, Hwang SJ, Lee FY, Wang SS, Chang FY, Li CP, Chu CJ, Lu RH, Lee SD. Prognostic value of plasma endotoxin levels in patients with cirrhosis. Scand J Gastroenterol. 1997;32:942–6.
Lin RS, Lee FY, Lee SD, Tsai YT, Lin HC, Lu RH, Hsu WC, Huang CC, Wang SS, Lo KJ. Endotoxemia in patients with chronic liver diseases: relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J Hepatol. 1995;22:165–72.
Wiest R, Garcia-Tsao G. Bacterial translocation (BT) in cirrhosis. Hepatology. 2005;41:422–33.
Chesta J, Defilippi C. Abnormalities in proximal small bowel motility in patients with cirrhosis. Hepatology. 1993;17:828–32.
Guarner C, Runyon BA, Young S, Heck M, Sheikh MY. Intestinal bacterial overgrowth and bacterial translocation in cirrhotic rats with ascites. J Hepatol. 1997;26:1372–8.
Ramachandran A, Prabhu R, Thomas S, Reddy JB, Pulimood A, Balasubramanian KA. Intestinal mucosal alterations in experimental cirrhosis in the rat: role of oxygen free radicals. Hepatology. 2002;35:622–9.
Rajkovic IA, Williams R. Abnormalities of neutrophil phagocytosis, intracellular killing and metabolic activity in alcoholic cirrhosis and hepatitis. Hepatology. 1986;6:252–62.
Garcia-Tsao G, Lee FY, Barden GE, Cartun R, West AB. Bacterial translocation to mesenteric lymph nodes is increased in cirrhotic rats with ascites. Gastroenterology. 1995;108:1835–41.
Su GL, Klein RD, Aminlari A, Zhang HY, Steinstraesser L, Alarcon WH, Remick DG, Wang SC. Kupffer cell activation by lipopolysaccharide in rats: role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology. 2000;31:932–6.
Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37:1043–55.
Schromm AB, Lien E, Henneke P, Chow JC, Yoshimura A, Heine H, Latz E, Monks BG, Schwartz DA, Miyake K, Golenbock DT. Molecular genetic analysis of an endotoxin nonresponder mutant cell line: a point mutation in a conserved region of MD-2 abolishes endotoxininduced signaling. J Exp Med. 2001;194:79–88.
Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, Lee JO. Crystal structure of the TLR4–MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–17.
Isayama F, Hines IN, Kremer M, Milton RJ, Byrd CL, Perry AW, McKim SE, Parsons C, Rippe RA, Wheeler MD. LPS signaling enhances hepatic fibrogenesis caused by experimental cholestasis in mice. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1318–28.
• Zhang Z, Lin C, Peng L, Ouyang Y, Cao Y, Wang J, Friedman SL, Guo J. High mobility group box 1 activates Toll like receptor 4 signaling in hepatic stellate cells. Life Sci 2012;91:207–12. This paper is on an experimental model that explains how Toll-like receptors interact with hepatic stellate cells to enhance their inflammatory phenotype, finally triggering the development of liver fibrosis.
Lirussi F, Mastropasqua E, Orando S, Orlando R. Probiotics for non-alcoholic fatty liver disease and/or steatohepatitis. Cochrane Database Syst Rev. 2007;24:(1):CD005165.
Velayudham A, Dolganiuc A, Ellis M, Petrasek J, Kodys K, Mandrekar P, Szabo G. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology. 2009;49(3):989–97.
Endo H, Niioka M, Kobayashi N, Tanaka M, Watanabe T. Butyrate-producing probiotics reduce nonalcoholic fatty liver disease progression in rats: new insight into the probiotics for the gut–liver axis. PLoS One. 2013;8(5):e63388.
Mencarelli A, Cipriani S, Renga B, Bruno A, D’Amore C, Distrutti E, Fiorucci S. VSL#3 resets insulin signaling and protects against NASH and atherosclerosis in a model of genetic dyslipidemia and intestinal inflammation. PLoS One. 2012;7(9):e45425. doi:10.1371/journal.pone.0045425.
Mencarelli A, Distrutti E, Renga B, D’Amore C, Cipriani S, Palladino G, Donini A, Ricci P, Fiorucci S. Probiotics modulate intestinal expression of nuclear receptor and provide counter-regulatory signals to inflammation-driven adipose tissue activation. PLoS One. 2011;6(7):e22978. doi:10.1371/journal.pone.0022978.
Younossi ZM, Reyes MJ, Mishra A, Mehta R, Henry L. Systematic review with meta-analysis: non-alcoholic steatohepatitis—a case for personalised treatment based on pathogenic targets. Aliment Pharmacol Ther. 2014;39(1):3–14.
Vajro P, Mandato C, Licenziati MR, Franzese A, Vitale DF, Lenta S, Caropreso M, Vallone G, Meli R. Effects of Lactobacillus rhamnosus strain GG in pediatric obesity-related liver disease. J Pediatr Gastroenterol Nutr. 2011;52(6):740–3. doi:10.1097/MPG.0b013e31821f9b85.
Aller R, De Luis DA, Izaola O, Conde R, Gonzalez Sagrado M, Primo D, De La Fuente B, Gonzalez J. Effect of a probiotic on liver aminotransferases in nonalcoholic fatty liver disease patients: a double blind randomized clinical trial. Eur Rev Med Pharmacol Sci. 2011;15(9):1090–5.
Malaguarnera M, Vacante M, Antic T, Giordano M, Chisari G, Acquaviva R, Mastrojeni S, Malaguarnera G, Mistretta A, Li Volti G, Galvano F. Bifidobacterium longum with fructo-oligosaccharides in patients with non alcoholic steatohepatitis. Dig Dis Sci. 2012;57(2):545–53. doi:10.1007/s10620-011-1887-4.
Wong VW, Won GL, Chim AM, Chu WC, Yeung DK, Li KC, Chan HL. Treatment of nonalcoholic steatohepatitis with probiotics. A proof-of-concept study. Ann Hepatol. 2013;12(2):256–62.
Loguercio C, Federico A, Tuccillo C, Terracciano F, D’Auria MV, De Simone C, Del Vecchio Blanco C. Beneficial effects of a probiotic VSL#3 on parameters of liver dysfunction in chronic liver diseases. J Clin Gastroenterol. 2005;39(6):540–3.
Alisi A, Bedogni G, Baviera G, Giorgio V, Porro E, Paris C, Giammaria P, Reali L, Anania F, Nobili V. Randomised clinical trial: the beneficial effects of VSL#3 in obese children with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2014. doi:10.1111/apt.12758.
Disclosure
Valentina Giorgio, Anna Alisi, Hoshemand Mohammad Kazem, Stefano Monti and Valerio Nobili declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Giorgio, V., Alisi, A., Kazem, H.M. et al. NASH and the Cross-Talk Between the Gut and Liver. Curr Pediatr Rep 2, 211–217 (2014). https://doi.org/10.1007/s40124-014-0047-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40124-014-0047-7