
Abstract
Lipoprotein(a) (Lp(a)) is a highly proatherogenic lipid fraction that is genetically determined and minimally responsive to lifestyle or behavior changes. Mendelian randomization studies have suggested a causal link between elevated Lp(a) and heart disease, stroke, and aortic stenosis. There is substantial inter-ethnic variation in Lp(a) levels, with persons of African descent having the highest median values. Monitoring of Lp(a) has historically been limited by lack of standardization of assays. With the advent of novel therapeutic modalities to lower Lp(a) levels including proprotein convertase subtilisin/kexin 9 (PCSK9) inhibitors and targeted antisense oligonucleotides, it is increasingly important to screen patients who have family or personal history of atherosclerotic cardiovascular disease for elevations in Lp(a). Further study is needed to establish a causal relationship between elevated Lp(a) and cardiovascular disease across diverse ethnic populations.
Similar content being viewed by others
Lp(a) is a highly atherogenic lipid fraction with numerous studies showing a strong association between elevated Lp(a) and coronary artery disease stroke and aortic stenosis. |
The expression of Lp(a) is largely determined by autosomal dominant inheritance with behavioral factors having minimal influence over serum levels. |
Persons of African and South Asian descent have significantly higher mean Lp(a) levels than persons of European Latin American or East Asian descent. |
Novel therapies including PCSK-9 inhibitors and antisense oligonucleotides significantly reduce Lp(a) levels independently of LDL reductions. |
Further studies on the role of Lp(a) reduction in primary cardiovascular disease prevention are needed. |
Introduction
What is Lipoprotein(a)?
Lipoprotein(a) [Lp(a)] is a low-density lipoprotein(LDL)-like lipid fraction consisting of two subunits: a single apolipoprotein a [Apo(a)] molecule, which is a lipid-rich fraction produced by the liver; and apolipoprotein B-100 (ApoB100), which has an LDL receptor and is also a constituent of very low density lipoprotein(VLDL), intermediate density lipoprotein(IDL), and LDL [1]. Apo(a) consists of 11 different kringle domains, which are subdomains of the protein with a structure that resembles the folded Danish pastry of the same name. While most kringle domains are expressed exactly once in the final Apo(a) quaternary structure, the primary constituent by mass is the kringle IV type 2 (KIV-2) domain, which can be repeated in the Apo(a) molecule from fewer than 13 times to more than 50 times [2]. The expression profile of the KIV-2 domain is predominantly determined genetically in an autosomal dominant fashion, and there is an inverse relationship between the number of KIV-2 repeats and the concentration of Lp(a) in serum [3].
Considerable overlap exists between the structure of the apo(a) molecule and those of plasminogen and tissue plasminogen activator. The kringle IV type 10 has a lysine binding site similar to plasminogen but the cleavage site has a mutation that prevents proteolysis. Lp(a) thus can compete with plasminogen for the binding site on fibrin but since it lacks the proteolytic subdomain it functionally inhibits fibrinolysis [4].
Lp(a) has also been identified as an acute-phase reactant after tissue injury. Lp(a) was positively correlated with IL-6, C-reactive protein, and alpha-1 antitrypsin after acute myocardial infarcts [5, 6]. Apo(a) may also act as an immune suppressor by decreasing recruitment of neutrophils [7].
Lp(a) is a preferred carrier for oxidized phospholipids (OxPL), which are demonstrated to be immunogenic, proinflammatory, and proatherogenic [8]. In a prospective cohort study of 1400 healthy adults followed for 15 years, highest tertile OxPL was associated with hazard ratios of 2.4 for cardiovascular disease (CVD) and 3.6 for stroke compared to the lowest tertile [9]. Lp(a) is believed to exert part of its proatherogenic effects via its carriage of OxPL. Lp(a) is also believed to increase the propensity to form coronary calcification. Korean men and women in the highest Lp(a) quartile were found to have odds ratios of 1.2 and 1.6, respectively, for coronary artery calcium score greater than 0, even after adjusting for LDL and metabolic syndrome [10].
While the expression of Lp(a) is largely determined by genetic inheritance, behavioral factors such as diet and exercise can affect Lp(a) levels to a modest degree. For example, in a study of 37 healthy women fed two low-fat diets, one low in vegetable content and the other high in vegetable content, they experienced a 7% and 9% increase in Lp(a), respectively [11]. Sustained physical exercise can increase Lp(a) levels by 10% for moderate exercise and up to over 100% for high-endurance athletes [12,13,14,15]. The rise in Lp(a) during exercise is likely as an acute-phase reactant in response to soft tissue damage in the musculature and vasculature during repeated movements. Two studies found no significant relationship between exercise and Lp(a) but did note a non-significant trend of higher mean Lp(a) in the active group [16, 17]. Many studies, however, find no relationship between exercise and Lp(a) levels, and the benefits of exercise on cardiovascular health are well established [18,19,20,21,22,23,24,25].
This article reviews the up-to-date literature related to Lp(a) and its role in cardiovascular disease, ethnic disparities in the levels and pathogenicity of this lipid fraction, clinical monitoring guidelines, and treatment modalities (Table 1). This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Associations Between Lp(a) and Cardiovascular Disease
Lp(a) is believed to be a highly atherogenic lipid fraction with numerous studies showing a strong association between elevated Lp(a) and coronary artery disease, stroke, and aortic stenosis, which will be explored in the following section.
Studies on Lp(a) have leveraged longitudinal data the Copenhagen City Heart Study (CCHS), the Copenhagen General Population Study (CGPS), and the Copenhagen Ischemic Heart Disease Study (CIDHS). All studies contained genetic as well as outcomes data. These studies employ Mendelian randomization, an epidemiological method that uses the independent assortment of risk alleles as a randomization event to estimate the association between a genetic profile and a clinical outcome of interest [26]. Mendelian randomization studies are not able to determine a causal relationship between a gene and a clinical outcome in the same way that randomized controlled trials can, but they can be stronger than normal observational studies at determining causation if the risk allele that is studied is not confounded by linkage disequilibrium or population stratification [27]. In the aforementioned Danish studies, the population in question was ethnically homogenous (i.e., no population stratification effects) and LPA, the gene under study, is not known to be in linkage disequilibrium with any other genes, though this possibility can rarely be excluded.
Coronary Artery Disease
In the Lipid Research Clinics Coronary Primary Prevention Trial, Lp(a) was demonstrated prospectively to be 21% higher in white men who went on to develop coronary heart disease over the 7–10-year follow-up period [28]. The elevation in Lp(a) remained significant after controlling for age, body mass index, cigarette smoking, blood pressure, LDL cholesterol level, and HDL cholesterol level, but not after controlling for treatment with cholestyramine vs. placebo.
One study using the CCHS, CGPS, and CIDHS populations found a hazard ratio of 1.2 for all-cause mortality and 1.5 for cardiovascular mortality in subjects with greater than the 96th percentile of Lp(a) serum concentration compared to those with less than the 10th percentile of Lp(a) [29].
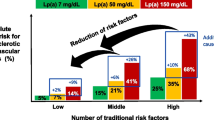
The CCHS, CGPS, and CIDHS study populations were also analyzed to evaluate the impact of secondary prevention in subjects with a history of CAD. Major adverse cardiovascular events (MACE) incidence ratios were proportional to the Lp(a) concentration. The researchers estimated that a 20% reduction in MACE could be achieved by lowering Lp(a) by 50 mg/dl for 5 years and a 40% reduction in MACE achieved by lowering Lp(a) my 99 mg/dl for 5 years [30].
In a case control Mendelian randomization study of Lp(a) in 143,000 Icelanders, several deleterious effects were demonstrated. A 50 nmol/l increase in Lp(a) concentration was associated with a 0.22–0.27-year reduction in life expectancy. The same 50 nmol/l Lp(a) increase was associated with odds ratios of 1.16 for CAD, 1.27 for coronary artery bypass grafting, 1.18 for MI, 1.16 for peripheral arterial disease, and 1.05 for heart failure [31].
Elevations in Lp(a) are associated with unfavorable outcomes after percutaneous intervention (PCI). As an acute-phase reactant, Lp(a) has been demonstrated to increase rapidly after PCI before resolving to baseline in 1–2 days [32]. In patients who have comorbid chronic kidney disease or diabetes, Lp(a) was independently associated with worse outcomes after PCI [33, 34].
The role of Lp(a) in secondary prevention of CVD remains unclear. A combination study of over 25,000 patients demonstrated an odds ratio of 1.40 for cardiovascular events in patients with CVD and the highest quantile of Lp(a), a relationship that remained significant when patients had LDL > 130 mg/dl but not when below this threshold [35].
Stroke
In the CCHS, CGPS, and CIDHS populations, subjects with greater than 96th percentile of Lp(a) concentration (> 93 mg/dl or 199 nmol/l) had an HR of 1.6 for ischemic stroke when compared to subjects with less than the median Lp(a) concentration (< 10 mg/dl or 18 nmol/l) with the highest risks in patients above the age of 70 who smoked or had hypertension [36]. There was a graded relationship between Lp(a) concentration and MI risk, with an HR ranging from 1.2 at the 66th percentile to 2.6 at greater than the 95th percentile [37]. In both the CCHS and CGPS, the number of kringle IV type 2 (KIV2) repeats was inversely correlated with Lp(a) levels. In the CCHS, the HR for MI was 1.5, 1.3, and 1.1 in the first, second, and third quartiles of KIV2 compared to the fourth quartile, and similar results were seen in the CGPS and CIDHS [37]. These results suggest a causal relationship between Lp(a) elevation and increased CVD risk in a Caucasian population.
The Icelandic study mentioned in the previous section noted an HR of 1.03 for stroke for a 50 nmol/l increase in Lp(a). These associations were similar for the inverse of number of KIV2 repeats but lost significance when controlling for Lp(a) concentration, suggesting the greater role of absolute concentration and not particle size in pathogenicity [31].
The 2011 American Heart Association/American Stroke Association guidelines do not recommend screening for Lp(a) for primary prevention of strokes unless the patient has unexplained early cardiovascular events in first-degree relatives or high Lp(a) is known to be present in first-degree relatives [38]. The 2014 guidelines do not mention screening for Lp(a), although they do recommend considering niacin in the treatment of Lp(a) elevations, a treatment which has since fallen out of favor, as discussed below [39].
Aortic Stenosis
In two prospective cohort studies evaluating the role of Lp(a) in primary prevention of aortic stenosis, researchers estimated that lowering Lp(a) among the top quintile of the population could prevent one in seven cases of aortic stenosis, while targeting the top 10% could prevent one in 12 aortic stenosis cases [40].
The Cohorts for Heart Aging Research in Genomic Epidemiology (CHARGE) consortium performed a genome-wide association study to identify genes associated with the presence of aortic calcification identified on CT scanning and found that the LPA gene was associated with aortic valve calcification, incident aortic stenosis, and aortic valve replacement [41]. In the Icelandic Mendelian randomization study, a 50 nmol/l increase in Lp(a) was associated with a hazard ratio of 1.17 for aortic stenosis [31].
In patients with aortic stenosis, patients in the top tertile of Lp(a) had increased valve calcification at baseline and experienced faster progression of valvular calcium scores compared with lower tertiles [42].
Racial Differences in Lp(a) Levels and Pathogenicity
Persons of African and South Asian descent have higher mean Lp(a) levels [43,44,45]. In African Americans, Lp(a) displays a bell-shaped frequency distribution around the median and mean [46]. By contrast, the distribution of Lp(a) concentrations in Caucasians is skewed, with the mean value exceeding the median due to a small number of individuals with very high Lp(a) levels [3, 47]. In the Charleston Heart Study cohort, elderly African-American men demonstrated a more skewed distribution of Lp(a) concentration frequencies, resembling that of the white cohort participants [48]. Thus it may be the case that Lp(a) decreases as African Americans age or that those with elevated Lp(a) are less likely to survive to the 7th, 8th, or 9th generation.
Even at young ages, African-American children have Lp(a) levels that are 1.7 times that of white children [49]. While white children with parental history of MI were more likely to have Lp(a) greater than 25 mg/dl compared to those without parental MI, African-American children did not display a similar pattern, suggesting a different risk profile associated with elevated Lp(a) in African Americans [49]. Indeed, the Multiethnic Study of Atherosclerosis demonstrated a significant relationship between Lp(a) and carotid atherosclerosis for whites, but was equivocal for Hispanics and not significant for African Americans [50].
African Americans have been demonstrated to have favorable total cholesterol and triglyceride profiles compared to Caucasians despite Lp(a) levels that were 2.2 times greater than levels seen in Caucasians [45]. When comparing overweight and obese prediabetic African-American and Caucasian women, African-American women were found to have higher HDL and ApoA1 and lower VLDL but they also had higher CVD mortality than their Caucasian counterparts [51].
The contribution of the apo(a) allele frequencies to Lp(a) levels varies by ethnicity. Ghanaians with the same apo(a) allele polymorphs as Germans still had up to a three-fold increased Lp(a) serum concentration than their German counterparts, and the difference in expression was especially pronounced in patients who had high molecular weight polymorphs [52]. The Coronary Artery Risk Development in Young Adults (CARDIA) trial, a prospective longitudinal epidemiological study of African-American and Caucasian subjects, demonstrated that at very high molecular weights of apo(a), both groups had low Lp(a) levels. By contrast, there was significant difference in Lp(a) in African Americans and Caucasians who had intermediate size isoforms, with African Americans expressing a wider distribution of Lp(a) serum concentrations than Caucasians [53]. The SNP rs9457951 was strongly associated with variation in Lp(a) level in African Americans in the Jackson Heart Study and was confirmed to be influential in the Dallas Heart Study. Of note, this difference was not demonstrated to have an association with increased cardiovascular mortality [54].
In a post hoc analysis of the African Americans and Caucasians in the Atherosclerosis Risk in Communities (ARIC) study, it was demonstrated that African Americans had a wider interindividual distribution of Lp(a) concentrations compared to Caucasians. For African Americans, a race-specific 1-SD–greater log-transformed Lp(a) was associated with a hazard ratio of 1.13 for incident CVD, 1.11 for incident CHD, and 1.21 for ischemic strokes. For Caucasians, the respective hazard ratios were 1.09, 1.10, and 1.07, with ischemic strokes not reaching statistical significance [55].
A retrospective cohort study in East and Southeast Asian populations residing in California found that Indian Americans had higher Lp(a) levels than non-Hispanic whites (36 vs. 29 nmol/l) and that Chinese Americans had the lowest average Lp(a) level (22 nmol/l). There was a trend toward greater burden of ischemic heart disease in subjects with the highest quintile of Lp(a) compared to the lowest quintile, but this relationship was not significant [43].
Monitoring Lp(a) in Clinical Practice
Unlike HDL, LDL, and triglycerides, Lp(a) is not currently routinely measured in primary care settings. The American College of Cardiology/American Heart Association blood cholesterol guidelines from 2018 consider Lp(a) ≥ 50 mg/dl or ≥ 125 nmol/l to be a risk-enhancing factor and recommend measuring Lp(a) in patients who have a family history of premature atherosclerotic cardiovascular disease (ASCVD) or a personal history of ASCVD not explained by major risk factors [56]. It is recommended to screen Lp(a) levels in women with a history of hypercholesterolemia, but there was minimal improvement in cardiovascular mortality and morbidity risk prediction in adult women with total cholesterol less than 220 mg/dl [57].
One of the main barriers to studying and treating Lp(a) is that measurement is not standardized. Results may vary depending on the specific assay used due to the high degree of polymorphism of Apo(a). Using a monoclonal antibody against the highly variable KIV-2 epitope yields an overestimation of molar concentration for larger Apo(a) molecules and underestimates molar concentration in subjects with smaller Apo(a) phenotypes [58]. A Lp(a) standardization group supported by the World Health Organization and the International Federation of Clinical Chemistry was established to evaluate the extant assays and make recommendations regarding clinical assessment of Lp(a). The group measured Lp(a) serum concentrations using six different assays, providing Lp(a) in mg/dl (Denka Seiken, Abbott Quantia, Beckman, Diasys 21FS, and Siemens N Latex) or in nmol/l (Roche TinaQuant, Diasys 21 FS). There was significant variation between assays at the higher and lower ends of Lp(a) concentration as well as significant intra-assay variability, suggesting that more work needs to be done to standardize Lp(a) assessment across varying labs [59].
Given that Apo(a) isoforms have highly variable molecular weights depending on the number of KIV-2 repeats, it has been suggested that clinicians discontinue using mg/dl units in favor of nmol/l [60]. This would have the added benefit of encouraging further standardization of assays and would align with prevailing evidence that molar concentration is more predictive than volume measurements.
Therapeutics for Lp(a) Reduction
As noted, until recently, there were no drug candidates to target Lp(a) reduction. Statins have not been demonstrated to lower Lp(a) levels. On the contrary, administration of atorvastatin and pravastatin into hepatocytes increased LPA mRNA by 1.5-fold in addition to increasing synthesis of apo(a) [61] (Table 2).
Plasma lipid apheresis is demonstrated to be quite effective at reducing Lp(a), but it also reduces a broader array of lipids and lipoproteins [62, 63]. A German cohort study demonstrated that plasma lipid apheresis reduced MACE by 78% in the year after initiation in assessing patients with combined elevated triglycerides, LDL, and Lp(a) [64]. The German cohort recommends lipid apheresis in patients who have isolated Lp(a) elevations greater than 60 mg/dl absent other lipid derangements. In the UK, Lp(a) can be considered as an additional risk factor to be considered in the initiation of lipid apheresis to treat elevated LDL-C [62]. Lipid apheresis is limited in its practicability due to the significant side-effect profile of fatigue, hypotension, and neurohormonal dysregulation, and thus is only recommended in the treatment of familial hypercholesterolemia in the US, Japan, Australia, and Spain [62].
Niacin has long been known to reduce Lp(a) levels in addition to increasing HDL and reducing LDL and triglycerides. The effect of niacin on cardiovascular outcomes has been called into question after large randomized controlled trials failed to show any benefit. The Heart protection Study 2-Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE) trial was a multicenter randomized controlled trial of statin therapy plus either 2 g of extended-release niacin plus 40 mg laropiprant versus placebo in patients with vascular disease. While patients in the treatment arm had higher HDL and lower LDL, they did not experience a significant improvement in nonfatal myocardial infarction, death from coronary causes, stroke, or arterial revascularization after a median follow-up of 3.9 years [65]. Similar lipid profile changes would have been expected to yield a 2% reduction in MACE overall and a 6% reduction in MACE for the highest quintile of Lp(a) [66]. Similarly, the Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH) trial tested whether extended-release niacin added to intensive statin therapy, as compared with statin therapy alone, would reduce the risk of cardiovascular events in patients with established atherosclerotic cardiovascular disease. The trial demonstrated that while niacin did increase HDL by 7 mg/dl and lower LDL by 12 mg/dl and triglycerides by 42 mg/dl, it did not have a significant impact on the primary composite endpoint of death from coronary heart disease, nonfatal myocardial infarction, ischemic stroke, hospitalization for an acute coronary syndrome, or symptom-driven coronary or cerebral revascularization [67]. The AIM-HIGH trial also showed high rates of adverse side effects due to niacin, including the typical flushing, itching, and raised blood glucose in addition to more serious effects of increased gastrointestinal symptoms and infections [68]. These studies combined have led practitioners to cease prescribing niacin to patients for primary cardiovascular disease prevention.
Proprotein convertase subtilisin/kexin 9 (PCSK9) inhibitors have been demonstrated to reduce Lp(a) levels. A pre-specified analysis of the ODYSSEY outcomes trial of alirocumab in 18,924 patients 1–12 months after acute coronary syndromes showed that PCSK9 inhibition resulted in a 5 mg/dl reduction in Lp(a), a 51.1 mg/dl reduction in LDL-C, and a 15% reduction in MACE [69]. After controlling for the reduction in LDL-C, reducing Lp(a) by 1 mg/dl was associated with a reduction in the hazard ratio for MACE to 0.994 (95% CI 0.990–0.999, p = 0.0081). This suggests that Lp(a) could serve as an independent biomarker for cardiovascular risk. The FOURIER (Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk) trial measured Lp(a) levels in 25,096 patients for a median follow-up of 2.2 years and found that subjects with fourth quartile Lp(a) had a hazard ratio of 1.22 for coronary heart disease death, myocardial infarction, and urgent revascularization compared to those in the first quartile. Evolocumab was compared to placebo in the FOURIER trial, and it was found to reduce coronary heart disease death, myocardial infarction, or urgent revascularization by 23% in patients who had higher than median Lp(a) at baseline and by 7% in patients who had less than median Lp(a) at baseline. The absolute reduction in the composite endpoint was 2.49% and the number needed to treat was 40 over 3 years [70].
Antisense oligonucleotides (ASO) are sequences targeted to reduce expression of specific RNA molecules. One such agent is Mipomersen, an ASO targeted to B-100 that has been demonstrated to reduce Lp(a) levels but is not currently indicated for use outside of LDL reduction [71]. Akcea Pharmaceuticals (formerly Ionis Pharmaceuticals) has developed Apo(a)LRx, an ASO targeted to hepatocytes via conjugation with a triantennary N-acetylgalactosamine (GalNAc3) moiety, a high-affinity ligand for the asialoglycoprotein receptor on the surface of hepatocytes, to reduce hepatic synthesis of Lp(a). A randomized double-blinded controlled phase I trial of efficacy demonstrated that a single dose of the ASO did not result in reductions in Lp(a) but six doses reduced Lp(a) by 39–77% in a dose-dependent manner [72]. The phase 2 trial in 286 patients with cardiovascular disease demonstrated that Apo(a)LRx reduced Lp(a) in a dose-dependent manner, with the highest dose (20 mg every week) reducing Lp(a) by 80% over 27 weeks. The agent appears to be well tolerated, with no significant derangements in liver or renal measures and no reductions in platelet counts. The two deaths in the study were from a car accident and suicide from previously diagnosed depression.
Conclusions
There have been no studies examining the role of targeted reduction of Lp(a) in primary or secondary prevention of cardiovascular disease. The numerous Mendelian randomization studies noted above suggest a causal relationship between elevated Lp(a) and increased cardiovascular disease—reverse causation is unlikely due to the fact that CVD does not change a subject’s genetic profile—but without a dedicated trial it is not possible to say definitively that Lp(a) is a risk factor in the same way that elevated LDL or triglycerides are. Additionally, until very recently, there were no drugs developed to specifically lower Lp(a), and thus dedicated studies were not feasible. With the advent of antisense oligonucleotide therapies to reduce Lp(a) mRNA translation, it will be possible and indeed necessary to determine if reducing Lp(a) results in a reduction in cardiovascular disease burden.
Lp(a) is an emerging biomarker that may offer a promising target to reduce residual cardiovascular risk. Lp(a) now has an increasing armamentarium of treatment options available, and thus now it is important to develop standardized measurements of this biomarker in individuals with family history of premature cardiovascular disease or stroke. Patients of African descent in particular stand to gain from increased awareness of this highly atherogenic lipid fraction, and health equity may be improved by concerted effort on this front. Future studies will need to be completed to evaluate the benefit of reducing Lp(a) in primary and secondary prevention of cardiovascular disease in an ethnically diverse population.
References
Chan L. Apolipoprotein B, the major protein component of triglyceride-rich and low density lipoproteins. J Biol Chem. 1992;267(36):25621–4.
Koschinsky: Structure, function and regulation of… - Google Scholar [Internet]. [cited 2020 Mar 24]. https://scholar.google.com/scholar_lookup?title=Structure%2C%20function%20and%20regulation%20of%20lipoprotein%28a%29&journal=Curr%20Pharm%20Design&volume=2&pages=121-121&publication_year=1996&author=Koschinsky%2CML&author=Ramharack%2CR. Accessed 24 Mar 2020.
Utermann G. The mysteries of lipoprotein(a). Science. 1989;246(4932):904–10.
Anglés-Cano E, de la Peña Díaz A, Loyau S. Inhibition of fibrinolysis by lipoprotein(a). Ann NY Acad Sci. 2001;936:261–75.
Topçiu-Shufta V, Begolli L, Kryeziu E. Lipoprotein(a) as an acute-phase reactant in patients on chronic hemodialysis. Bosn J Basic Med Sci. 2010;10(1):19–25.
Lp(a): an acute-phase reactant? - ScienceDirect [Internet]. [cited 2020 Feb 18]. https://www.sciencedirect.com/science/article/pii/0009308494901643. Accessed 18 Feb 2020.
Hoover-Plow J, Hart E, Gong Y, Shchurin A, Schneeman T. A physiological function for apolipoprotein(a): a natural regulator of the inflammatory response. Exp Biol Med (Maywood). 2009;234(1):28–34.
Taleb A, Witztum JL, Tsimikas S. Oxidized phospholipids on apoB-100-containing lipoproteins: a biomarker predicting cardiovascular disease and cardiovascular events. Biomark Med. 2011;5(5):673–94.
Tsimikas S, Willeit P, Willeit J, Santer P, Mayr M, Xu Q, et al. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J Am Coll Cardiol. 2012;60:2218–29.
Sung K-C, Wild SH, Byrne CD. Lipoprotein(a), metabolic syndrome and coronary calcium score in a large occupational cohort. Nutr Metab Cardiovasc Dis. 2013;23(12):1239–46.
Silaste M-L, Rantala M, Alfthan G, Aro A, Witztum JL, Kesäniemi YA, et al. Changes in dietary fat intake alter plasma levels of oxidized low-density lipoprotein and lipoprotein(a). Arterioscler Thromb Vasc Biol. 2004;24(3):498–503.
Cardoso GC, Posadas C, Orvanaños OO, Peniche C, Zamora J, Aguilar R, et al. Long distance runners and body-builders exhibit elevated plasma levels of lipoprotein(a). Chem Phys Lipid. 1994;1(67–68):207–21.
Holme I, Urdal P, Anderssen S, Hjermann I. Exercise-induced increase in lipoprotein(a). Atherosclerosis. 1996;122(1):97–104.
Cohen LI, Hartford CG, Rogers GG. Lipoprotein(a) and cholesterol in body builders using anabolic androgenic steroids. Med Sci Sports Exerc. 1996;28(2):176–9.
Ponjee GAE, Janssen EME, van Wersch JWJ. Long-term physical exercise and lipoprotein(a) Levels in a previously sedentary male and female population. Ann Clin Biochem. 1995;32(2):181–5.
Hubinger L, Mackinnon LT. The effect of endurance training on lipoprotein(a) [Lp(a)] levels in middle-aged males. Med Sci Sports Exerc. 1996;28(6):757–64.
Hubinger L, Mackinnon LT, Lepre F. Lipoprotein(a) [Lp(a)] levels in middle-aged male runners and sedentary controls. Med Sci Sports Exerc. 1995;27(4):490–6.
Mankowitz K, Seipa R, Semenkovich CF, Daugherty A, Schonfeld G. Short-term interruption of training affects both fasting and post-prandial lipoproteins. Atherosclerosis. 1992;95(2):181–9.
Haskell WL, Alderman EL, Fair JM, Maron DJ, Mackey SF, Superko HR, et al. Effects of intensive multiple risk factor reduction on coronary atherosclerosis and clinical cardiac events in men and women with coronary artery disease. The Stanford Coronary Risk Intervention Project (SCRIP). Circulation. 1994;89(3):975–90.
Lobo RA, Notelovitz M, Bernstein L, Khan FY, Ross RK, Paul WL. Lp(a) lipoprotein: relationship to cardiovascular disease risk factors, exercise, and estrogen. Am J Obstet Gynecol. 1992;166(4):1182–90.
Seip RL, Moulin P, Cocke T, Tall A, Kohrt WM, Mankowitz K, et al. Exercise training decreases plasma cholesteryl ester transfer protein. Arterioscler Thromb. 1993;13(9):1359–67.
Oyelola OO, Rufai MA. Plasma lipid, lipoprotein and apolipoprotein profiles in Nigerian university athletes and non-athletes. Br J Sports Med. 1993;27(4):271–4.
Halle M, Berg A, von Stein T, Baumstark MW, König D, Keul J. Lipoprotein(a) in endurance athletes, power athletes, and sedentary controls. Med Sci Sports Exerc. 1996;28(8):962–6.
Israel RG, Sullivan MJ, Marks RHL, Cayton RS, Chenier TC. Relationship between cardiorespiratory fitness and lipoprotein(a) in men and women. Med Sci Sports Exerc. 1994;26(4):425–31.
Mackinnon LT, Hubinger LM. Effects of exercise on lipoprotein(a). Sports Med 1999. 1999;28(1):11–24.
Davies NM, Holmes MV, Smith GD. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ [Internet]. 2018 Jul 12 [cited 2020 Apr 7];362. https://www.bmj.com/content/362/bmj.k601. Accessed 7 Apr 2020.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–9.
Schaefer EJ, Lamon-Fava S, Jenner JL, McNamara JR, Ordovas JM, Davis CE, et al. Lipoprotein(a) levels and risk of coronary heart disease in men: the lipid research clinics coronary primary prevention trial. JAMA. 1994;271(13):999–1003.
Langsted A, Kamstrup PR, Nordestgaard BG. High lipoprotein(a) and high risk of mortality. Eur Heart J. 2019;40(33):2760–70.
Madsen CM, Kamstrup PR, Langsted A, Varbo A, Nordestgaard BG. Lipoprotein(a)-lowering by 50 mg/dl (105 nmol/l) may be needed to reduce cardiovascular disease 20% in secondary prevention. Arteriosclerosis, Thromb Vasc Biol. 2020;40(1):255–66.
Gudbjartsson DF, Thorgeirsson G, Sulem P, Helgadottir A, Gylfason A, Saemundsdottir J, et al. Lipoprotein(a) concentration and risks of cardiovascular disease and diabetes. J Am Coll Cardiol. 2019;74(24):2982–94.
Wang J, Zhang C, Han A, Gong J, Li K. Percutaneous coronary intervention results in acute increases in native and oxidized lipoprotein(a) in patients with acute coronary syndrome and stable coronary artery disease. Clin Biochem. 2010;43(13):1107–11.
Konishi H, Miyauchi K, Tsuboi S, Ogita M, Naito R, Dohi T, et al. Plasma lipoprotein(a) predicts major cardiovascular events in patients with chronic kidney disease who undergo percutaneous coronary intervention. Int J Cardiol. 2016;15(205):50–3.
Konishi H, Miyauchi K, Shitara J, Endo H, Wada H, Doi S, et al. Impact of lipoprotein(a) on long-term outcomes in patients with diabetes mellitus who underwent percutaneous coronary intervention. Am J Cardiol. 2016;118(12):1781–5.
O’Donoghue ML, Morrow DA, Tsimikas S, Sloan S, Ren AF, Hoffman EB, et al. Lipoprotein(a) for risk assessment in patients with established coronary artery disease. J Am Coll Cardiol. 2014;63(6):520–7.
Langsted A, Nordestgaard BG, Kamstrup PR. Elevated lipoprotein(a) and Risk of ischemic stroke. J Am Coll Cardiol. 2019;74(1):54–66.
Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331–9.
Goldstein LB, Bushnell CD, Adams RJ, Appel LJ, Braun LT, Chaturvedi S, et al. Guidelines for the Primary prevention of stroke. Stroke. 2011;42(2):517–84.
Meschia JF, Bushnell C, Boden-Albala B, Braun LT, Bravata DM, Chaturvedi S, et al. Guidelines for the primary prevention of stroke. Stroke. 2014;45(12):3754–832.
Afshar M, Kamstrup PR, Williams K, Sniderman AD, Nordestgaard BG, Thanassoulis G. Estimating the population impact of Lp(a) lowering on the incidence of myocardial infarction and aortic stenosis—brief report. Arterioscler Thromb Vasc Biol. 2016;36(12):2421–3.
Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503–12.
Zheng KH, Tsimikas S, Pawade T, Kroon J, Jenkins WSA, Doris MK, et al. Lipoprotein(a) and oxidized phospholipids promote valve calcification in patients with aortic stenosis. J Am Coll Cardiol. 2019;73(17):2150–62.
Banerjee D, Wong EC, Shin J, Fortmann SP, Palaniappan L. Racial and ethnic variation in lipoprotein(a) levels among Asian Indian and Chinese Patients. J Lipids. 2011;2011:291954.
Enkhmaa B, Anuurad E, Berglund L. Lipoprotein(a): impact by ethnicity and environmental and medical conditions. J Lipid Res. 2016;57(7):1111–25.
Parra HJ, Luyéyé I, Bouramoué C, Demarquilly C, Fruchart JC. Black-white differences in serum Lp(a) lipoprotein levels. Clin Chim Acta. 1987;168(1):27–31.
Guyton JR, Dahlen GH, Patsch W, Kautz JA, A M Gotto J. Relationship of plasma lipoprotein Lp(a) levels to race and to apolipoprotein B. Arteriosclerosis: An Official Journal of the American Heart Association, Inc [Internet]. 1985 Jun [cited 2020 Feb 15]; https://www.ahajournals.org/doi/abs/10.1161/01.atv.5.3.265. Accessed 15 Feb 2020.
Nordestgaard BG, Chapman MJ, Ray K, Borén J, Andreotti F, Watts GF, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844–53.
Knapp RG, Schreiner PJ, Sutherland SE, Keil JE, Gilbert GE, Klein RL, et al. Serum lipoprotein(a) levels in elderly black and white men in the Charleston Heart Study. Clin Genet. 1993;44(5):225–31.
Srinivasan SR, Dahlen GH, Jarpa RA, Webber LS, Berenson GS. Racial (black-white) differences in serum lipoprotein(a) distribution and its relation to parental myocardial infarction in children. Bogalusa Heart Study. Circulation. 1991;84(1):160–7.
Steffen BT, Thanassoulis G, Duprez D, Stein JH, Karger AB, Tattersall MC, et al. Race-Based Differences in lipoprotein(a)-associated risk of carotid atherosclerosis. Arterioscler Thromb Vasc Biol. 2019;39(3):523–9.
Gaillard T, Osei K. Ethnic differences in serum lipids and lipoproteins in overweight/obese African-American and white American women with pre-diabetes: significance of NMR-derived lipoprotein particle concentrations and sizes. BMJ Open Diabetes Res Care. 2016;4(1):e000246.
Helmhold M, Bigge J, Muche R, Mainoo J, Thiery J, Seidel D, et al. Contribution of the apo[a] phenotype to plasma Lp[a] concentrations shows considerable ethnic variation. J Lipid Res. 1991;32(12):1919–28.
Marcovina SM, Albers JJ, Wijsman E, Zhang Z, Chapman NH, Kennedy H. Differences in Lp[a] concentrations and apo[a] polymorphs between black and white Americans. J Lipid Res. 1996;37(12):2569–85.
Deo RC, Wilson JG, Xing C, Lawson K, Kao WHL, Reich D, et al. Single-nucleotide polymorphisms in LPA explain most of the ancestry-specific variation in Lp(a) levels in African Americans. PLoS One [Internet]. 2011 Jan 24 [cited 2020 Feb 15];6(1). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3025914/. Accessed 15 Feb 2020.
Virani SS, Brautbar A, Davis BC, Nambi V, Hoogeveen RC, Sharrett AR, et al. Associations between lipoprotein(a) Levels and cardiovascular outcomes in African Americans and Caucasians: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2012;125(2):241–9.
Grundy SM, Stone NJ, Bailey AL, Craig B, Birtcher KK, Blumenthal RS, et al. (2018) AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: a Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139(25):e1082–143.
Cook NR, Mora S, Ridker PM. Lipoprotein(a) and cardiovascular risk prediction among women. J Am Coll Cardiol. 2018;72(3):287–96.
Marcovina SM, Albers JJ, Gabel B, Koschinsky ML, Gaur VP. Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a). Clin Chem. 1995;41(2):246–55.
Scharnagl H, Stojakovic T, Dieplinger B, Dieplinger H, Erhart G, Kostner GM, et al. Comparison of lipoprotein(a) serum concentrations measured by six commercially available immunoassays. Atherosclerosis. 2019;1(289):206–13.
Wilson DP, Jacobson TA, Jones PH, Koschinsky ML, McNeal CJ, Nordestgaard BG, et al. Use of Lipoprotein(a) in clinical practice: a biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13(3):374–92.
Tsimikas S, Gordts PLSM, Nora C, Yeang C, Witztum JL. Statin therapy increases lipoprotein(a) levels. Eur Heart J [Internet]. [cited 2020 Feb 4]; Available from: https://academic.oup.com/eurheartj/advance-article/doi/10.1093/eurheartj/ehz310/5492355. Accessed 4 Feb 2020.
Thompson G, Parhofer KG. Current role of lipoprotein apheresis. Curr Atheroscler Rep [Internet]. 2019 [cited 2020 Apr 16];21(7). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6491397/. Accessed 16 Apr 2020.
Lui M, Garberich R, Strauss C, Davin T, Knickelbine T. Usefulness of lipid apheresis in the treatment of familial hypercholesterolemia. J Lipids [Internet]. 2014 [cited 2020 Apr 8];2014. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4217354/. Accessed 8 Apr 2020.
Schettler VJJ, Ringel J, Jacob S, Julius U, Klingel R, Heigl F, et al. Current insights into the German lipoprotein apheresis standard: PCSK9-inhibitors, lipoprotein apheresis or both? Atheroscler Suppl. 2017;1(30):44–9.
The HPS2-THRIVE Collaborative Group. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371(3):203–12.
Parish S, Hopewell JC, Hill MR, Marcovina S, Valdes-Marquez E, Haynes R, et al. Impact of apolipoprotein(a) isoform size on lipoprotein(a) lowering in the HPS2-THRIVE study. Circ Genom Precis Med. 2018;11(2):e001696.
AIM-HIGH Investigators, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–67.
Anderson TJ, Boden WE, Desvigne-Nickens P, Fleg JL, Kashyap ML, McBride R, et al. Safety profile of extended-release niacin in the AIM-HIGH trial. N Engl J Med. 2014;371(3):288–90.
Bittner VA, Szarek M, Aylward PE, Bhatt DL, Diaz R, Edelberg JM, et al. Effect of Alirocumab on lipoprotein(a) and cardiovascular risk after acute coronary syndrome. J Am Coll Cardiol. 2020;75(2):133–44.
O’Donoghue ML, Fazio S, Giugliano RP, Stroes ESG, Kanevsky E, Gouni-Berthold I, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk: insights from the FOURIER trial. Circulation. 2019;139(12):1483–92.
Santos RD, Raal FJ, Catapano AL, Witztum JL, Steinhagen-Thiessen E, Tsimikas S. Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein(a) in various populations with hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2015;35(3):689–99.
Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study| Elsevier Enhanced Reader [Internet]. [cited 2020 Mar 22]. https://reader.elsevier.com/reader/sd/pii/S0140673615612521?token=7F42DDEE825CFFF47784525B56C8D3D15F019936124FA0A2E1055BC650E91D805A71583F82498D3635D72B0F98860AB5. Accessed 20 Mar 2020.
Acknowledgements
Funding
Dr. Rodriguez was funded by a career development award from the National Heart, Lung, and Blood Institute (K01 HL 14460) and the American Heart Association/Robert Wood Johnson Harold Amos Medical Faculty Development Program. No funding was received for the publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Fatima Rodriguez has served as an advisor to the Medicines Company, Janssen, and 23andMe; she serves an event adjudicator for NovoNordisk and is editorial board member of the journal. Keon Pearson has no conflicts of interest to disclose.
Compliance with Ethical Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Open Access
This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12290483.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Pearson, K., Rodriguez, F. Lipoprotein(a) and Cardiovascular Disease Prevention across Diverse Populations. Cardiol Ther 9, 275–292 (2020). https://doi.org/10.1007/s40119-020-00177-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40119-020-00177-4