
Abstract
Purpose
The coronavirus outbreak emerged as a severe pandemic, claiming more than 0.8 million lives across the world and raised a major global health concern. We survey the history and mechanism of coronaviruses, and the structural characteristics of the spike protein and its key residues responsible for human transmissions.
Methods
We have carried out a systematic review to summarize the origin, transmission and etiology of COVID-19. The structural analysis of the spike protein and its disordered residues explains the mechanism of the viral transmission. A meta-data analysis of the therapeutic compounds targeting the SARS-CoV-2 is also included.
Results
Coronaviruses can cross the species barrier and infect humans with unexpected consequences for public health. The transmission rate of SARS-CoV-2 infection is higher compared to that of the closely related SARS-CoV infections. In SARS-CoV-2 infection, intrinsically disordered regions are observed at the interface of the spike protein and ACE2 receptor, providing a shape complementarity to the complex. The key residues of the spike protein have stronger binding affinity with ACE2. These can be probable reasons for the higher transmission rate of SARS-CoV-2. In addition, we have also discussed the therapeutic compounds and the vaccines to target SARS-CoV-2, which can help researchers to develop effective drugs/vaccines for COVID-19.
Summary
The overall history and mechanism of entry of SARS-CoV-2 along with structural study of spike-ACE2 complex provide insights to understand disease pathogenesis and development of vaccines and drugs.
Similar content being viewed by others

Introduction
The SARS-CoV-2 is a single, positive-strand RNA virus that causes severe respiratory syndrome in humans [1]. The Coronavirus disease 2019 (COVID-19) has emerged as a severe pandemic, claiming more than 0.8 million lives worldwide between December 2019 and August 2020 [2, 3]. Compared to SARS-CoV, SARS-CoV-2 human-to-human infection is more readily transmitted and spread to almost all continents leading to the WHO’s declaration of a Public Health Emergency of International Concern (PHEIC) on January 30, 2020 [3,4,5]. Generally, coronaviruses can cause respiratory, gastrointestinal, and central nervous system diseases in humans and animals, threatening humans life and causing economic loss [6, 7]. These viruses also have the capacity to adapt to a new environment through mutations and are programmed to modify host tropism; thus, the threats are constant and long-term [6, 8, 9].
Including SARS-CoV-2, other coronaviruses cross the species barrier into humans, which lead to outbreaks of severe and fatal respiratory diseases. The SARS-CoV was first identified in bats, and spread to other animals in different geographic regions. The SARS-CoV outbreak was first emerged in humans in 2003, through transmissions from animals in open-air markets in China [10, 11]. Thereafter, a higher number of genetically related viruses were also identified in Chinese Horseshoes bats (Rhinolophus sinicus) [11,12,13]. Coronaviruses belong to the family Coronaviridae and are divided into alpha (α-CoV), beta (β-CoV), gamma (γ-CoV), and delta (δ-CoV) coronaviruses. The alpha and betacoronaviruses can infect mammals, and the viruses found in humans are genetically similar to β-CoV genus. The β-CoVs are further divided into different lineages (A, B, C, and D lineages): SARS-CoV and SARS-CoV-2 are grouped in lineage B, which has approximately 200 published virus sequences, whereas MERS-CoV belongs to lineage C, which has ~ 500 viral sequences [11].
The HCoV-229E and HCoV-NL63 belong to the Alphacoronavirus family, whereas HCoV-OC43, HCoV-HKU1 and SARS-CoV are Betacoronaviruses [14,15,16]. The phylogenetic analysis shows that SARS-CoV-2 protein is firmly rooted in the β-genus lineage of bat coronaviruses [14]. The whole genome of SARS-CoV-2 shares 80% identity with that of SARS-CoV and is 96% identical to the bat coronavirus BatCoV-RaTG13 [17]. The spike protein sequence similarity between SARS-CoV-2 and SARS-CoV is around 76–78%. The RBD alone shares a similarity of 73–76%, and RBM shares 50–53%. In contrast, the human MERS-CoV is related to Tylonycteris bat coronavirus HKU4, shares less sequence similarity (~ 54%) and recognizes DPP4 as their receptor. The sequence similarity between SARS-CoV-2 and SARS-CoV spike proteins explains the possibility of binding to the same receptor angiotensin converting enzyme 2 (ACE2) in the host cell [14].
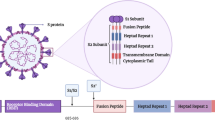
Coronavirus is one of the largest genomes among all RNA viruses ranging from 27 to 32 kb. Receptor-mediated endocytosis is the main process of virus entry to the host cells. SARS-CoV-2 uses ACE2, a cell-surface receptor that is present in the kidney, blood vessels, heart, and importantly, in the lung AT2 alveolar respiratory tract epithelial cells for viral infection [18]. The spike protein, which is responsible for the viral entry, has N-terminal and C-terminal domains, and two major subunits S1 and S2 are present in almost all coronaviruses [6]. One of these S1 or S2 subunits binds with the host receptors and acts as a receptor-binding domain (RBD) (Fig. 1a).

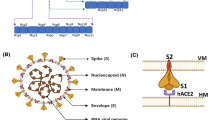
a Domain arrangement of SARS-CoV-2 spike protein. SS signal sequence, NTD N-terminal transactivation domain, RBD receptor binding domain, SD subdomain, FP fusion peptides, HR1 heptad repeat 1, HR2 heptad repeat 2, CD connector domain, S1/S2 and S2′ protease cleavage sites, TM transmembrane domain, CT cytoplasmic tail. b Host cell entry and replication of SARS-CoV-2. SARS-CoV-2 infection starts with the binding of spike protein with ACE2 receptor and the invasion process is triggered by host cell proteases (furin, trypsin, TMPRSS2 and cathepsin). SARS-CoV-2 releases RNA into the host cell, and the RNA is translated into viral replicase polyproteins pp1a and pp1ab, and subsequently cleaves into NSPs. The full-length negative strand RNA copies of the viral genome are produced by the enzyme replicase using the full-length positive-strand RNA genome as a template. During transcription, RNA polymerase produces a series of subgenomic mRNAs and translates into viral proteins [S (Spike), E (Envelope), N (Nucleocapsid), and M (Membrane)]. The viral proteins and the genome RNA are assembled into virions in Golgi and ER (endoplasmic reticulum), which are budding into ERGIC (ER–Golgi intermediate compartment) and released out of the cell via vesicles
Next-generation sequencing technology (NGS) revolutionized the biological sciences, including virus discovery. The NGS made it easy to recognize thousands of novel virus sequences from wild animal and human populations around the world. Despite these vast coronavirus sequences are published, very little work has been performed for further studies. Further, the lack of tools to test these novel viruses and their ability to infect humans hindered the efforts to predict the subsequent zoonotic viral outbreaks [11]. Understanding the virology of coronaviruses and the methods to control their spread is currently a necessary task to maintain the global health and economic stability.
In this review, we discuss the history of coronaviruses in both humans and animals, their transmissions, mechanism of host cell entry and the structural studies, explaining active and inactive receptor binding of spike protein and the key residues playing an important role in the receptor binding. The drug repurposing and the therapeutic targets for SARS-CoV-2 are also discussed.
History known so far
Until the first identification of human coronaviruses 229E and OC43, in the late 1960s, coronavirus infections were witnessed as harmless for humans [14, 15]. The outbreak of SARS-CoV in southern China in the winter of 2002, took a fatality rate of 10% of the infected patients [18,19,20]. The virus had been rapidly spreading throughout the world, especially in Asia, and controlled after July 2003 [21]. Viral analysis of the outbreak of SARS showed that bats are natural reservoirs for SARS-CoVs, and civet cats and raccoon dogs are the intermediate hosts. In the year 2012, a novel highly pathogenic Middle East Respiratory Syndrome coronavirus (MERS-CoV) was identified in humans, demonstrating that the coronaviruses are transmitted from animals to humans at any time and with unexpected consequences for the public health [22]. MERS-CoV, the slow-spreading virus, has affected ~ 1700 people with a fatality rate of ~ 36% [6, 22]. The animal sources of SARS-CoV-2 infections are bats and SARS-CoV-2 can be transmitted to cats, pangolins, and dogs [23].
Other than humans, the coronaviruses have a big impact on the animal kingdom. Animal coronaviruses can cause severe threat to their hosts; since 1984, an unrecognized infection massively spread among the swine population in Europe, and later it was identified as porcine epidemic diarrhea coronavirus (PEDV), which is derived from the porcine enteric coronavirus TGEV [24]. In 2013, the same PEDV in less than a year had caused a 100% fatality rate in piglets and rubbing out more than 10% of America’s pig population [5, 25, 26]
Therefore, the total number of human coronaviruses identified has been increasing throughout the years. HCoV-229E and HCoV-OC43 are the first two discovered human coronaviruses in the 1960s, HCoV-NL63 and HCoV-HKU1 human coronaviruses identified after the SARS-CoV outbreak in 2003. The sixth coronavirus MERS-CoV was identified in Saudi Arabia in 2012 [22]. However, the story continues with the new identification of SARS-CoV-2 in December 2019 at the seafood wholesale market in Wuhan, China. SARS-CoV-2 is the seventh member of the family of coronaviruses that infects humans and it is different from both MERS-CoV and SARS-CoV.
Although some infections caused by human coronaviruses are mild and associated with common colds, certain animal and human coronaviruses can make a severe impact on the human population. Especially in young children, elderly people, and immune-deficient patients, the infections can be lethal [9, 27,28,29]. Therefore, it is important to understand the mechanism of invading viruses to theirs hosts, transmission and prevention of these processes.
Transmission and epidemiology
The respiratory droplets are the main routes of transmissions; SARS-CoV-2 can be transmitted to a healthy person if he happens to have contact with the infected person or any of his belongings, including clothes, doorknobs, etc. Studies have been reported that aerosol transmission (Airborne transmission) is also possible for SARS-CoV-2, but there is no clear study on neonatal infections (mother to child) [30,31,32,33,34,35,36]. However, the transmission can be avoided by keeping a distance of 2 m between two people, wearing masks while going out, and the isolation of infected people.
During the initial phase of the COVID-19 outbreak, a dataset was obtained from 1099 patients with laboratory-confirmed COVID-19 from 552 hospitals in 30 provinces of China on January 29, 2020. Only 2% of the patients had a history of contact with animals; more than three quarters have either visited the Wuhan city or are residents. Hence, the outbreak patterns or the source of infection could not be predicted from their study. The incubation period of the SARS-CoV-2 was from 1 to 12 days; however, the median incubation period was 4 days [32]. The most common symptoms are fever (43% on admission, and 88.7% during hospitalization), cough (67.8%), diarrhea (3.8%), and fatigue [32, 37]. The SARS-CoV-2 was detected in saliva, blood, sputum, and urine before the development of viral pneumonia, and some patients do not develop pneumonia at all. Asymptomatic persons are potential sources of SARS-CoV-2 infection, which control the transmission dynamics of the current outbreak [32, 38].
The SARS-CoV-2 first identified in Wuhan, China has spread all over the globe. As of August 20, 2020, more than 22 million confirmed infection cases and 0.8 million deaths had been reported across the world, including almost all the countries. The rate of infection or the average number of people getting infected by an individual (R0) was 2.75 in the case of SARS pandemic in 2003. The R0 value of Ebola 2014 was in the range of 1.51–2.53, and H1N1 influenza 2009 was from 1.46 to 1.48, and for MERS, it was around 1. The SARS-CoV-2 R0 value was estimated to be in the range of 1.5–3.5. The comparison of R0 values of various coronaviruses shows that the difference is minimal. However, the difficulties arising for SARS-CoV-2 infection are due to the following: (1) basic properties of the viral infection and the infection periods are uncertain, (2) most of the infected individuals do not show symptoms, but are capable of spreading the infection, (3) changing susceptibility of the population in affecting the spread of infection remains unanswered. In addition, there are no control measures for this spread [39].
Spike protein structure and mechanism of SARS-CoV-2 entry
Coronaviruses consist of four structural proteins: the nucleocapsid protein (N) forms the helical capsid to accommodate its genome. The whole structure is further surrounded by a lipid envelope, which is made of S (spike), E (envelope) and M (membrane) proteins. The membrane and the envelope proteins are needed for the virus assembly and the spike protein is for virus entry and host cell recognition [6]. The spike protein forms large protrusions (Peplomers) on the virus surface (looks like the virus has crowns), and hence it is named as “CORONA” (corona is a Latin word which means crown). It comprises three segments: (1) large ectodomain, (2) transmembrane domain and (3) intracellular tail. The receptor-binding subunits S1 and S2 are placed in the ectodomain region. During the infection, the S1 binds with the host receptor, and S2 fuses the host and viral membranes, thereby releasing the viral genome into the cell (Fig. 1). The spike protein is a clove-shaped trimer with three S1 heads and a trimeric S2 stalk [6].
During viral infection, spike protein (~ 1300 amino acid residues) is cleaved by host proteases into receptor binding subunit S1 and membrane fusion subunit S2. During cell entry, the S1 subunit binds directly to the sugar receptors [40] and ACE2 of the host cell surface, and the S2 subunit undergoes conformational changes and obtains post-fusion state [41]. During this state, the three pairs of heptad repeat region HR-N and HR-C in trimeric S2 form a six-helix bundle structure [42]. The buried hydrophobic fusion peptides become exposed and insert into the target host membrane. These fusion peptides and the transmembrane anchors are positioned at the end of a six-helix bundle structure, bringing the viral and host membranes to fuse [42, 43]. During this process, a large amount of energy is released, which accelerates the membrane fusion forward. Along with this, receptor binding and low pH can also trigger this membrane fusion [6].
Since the spike protein has a good binding affinity for sugar receptors of human cells, it uses them as a mechanism of cell entry [6]. Notably, the SARS-CoV-2 has a higher affinity to human ACE2 than the SARS-CoV virus strain. The ectodomain of the SARS-CoV-2 spike protein binds to the peptidase domain (PD) of ACE2 with a Kd (equilibrium dissociation constant) of ~ 15 nM [4]. Spike protein priming is done by transmembrane protease serine 2 (TMPRSS2), which is also essential for the entry of SARS-CoV-2 [44]. Generally, SARS-CoV enters host cells through endocytosis, where its spike protein is processed by cathepsin L and cathepsin B lysosomal proteases. The extracellular proteases, including elastase in the respiratory tract and TMPRSS2 on the surface of flung cells are also known to activate spike membrane fusion [6].
The viral entry to the host cells can be via: (1) the endocytic pathway and (2) non-endosomal pathway [45] (Fig. 1b). The endocytic pathway, especially clathrin-dependent endocytosis is extensively studied for SARS-CoV and MERS-CoV viral entry. Since the SARS-CoV-2 also uses the same receptor as SARS-CoV, it is reported that SARS-CoV-2 also uses the same viral entry mechanism. Wang et al. [46] reported clathrin- and caveolae-independent endocytic pathways for SARS-CoV entry. Despite the common use of endocytic pathway as a viral entry mechanism, the discrepancies about the same are unavoidable. Thus, the exact nature of the viral entry is context-dependent, including the type of the virus and the host cells [47].
In addition, the macrophages can also act as a viral reservoir and support minimally for the SARS-CoV-2 entry and its replication. Although the dendritic cells and other immune cells are not infected by SARS-CoV-2, they may serve as a transporter for the viruses, which is also responsible for the pathogenesis [48, 49].
Active and inactive state of spike protein
The S1 subunit of SARS-CoV spike glycoprotein is predominantly composed of β-strand structures, composed of an N-terminal domain (NTD, residues 14–294) and three C-terminal domains (CTD1, residues 320–516; CTD2, residues 517–578 and CTD3, residues 579–663). The NTD is linked with CTD1 through the linker residues 295–319, where CTD1 acts as a potential RBD for SARS-CoV and binds explicitly with the ACE2 receptor.
Gui et al. [43] reported that the SARS-CoV spike glycoprotein trimer could obtain different conformations, which are necessary for effective binding with ACE2. All these conformations are termed based on the position of CTD1 of the spike glycoprotein (Fig. 2). The three spike glycoprotein monomers intertwine with each other and form densely packed homotrimer. The head portion of this trimer is taken place by NTDs and CTD1s of S1 subunits, where the CTD1s are located at the center and the NTDs are located outside of this triangular head. The S2 subunits represented as a stem for this trimer, which is further surrounded by CTD2s and CTD3s of the S1 subunits (Fig. 2). In the inactive state (Fig. 2a, b), S2 subunits (stem portion) are completely covered by the “down” position of CTD1s (head portion), which causes steric clashes for the binding between spike protein and ACE2. In the active state (Fig. 2c), two CTD1s adapt the “down” conformation and one CTD1 rotates outward and obtains “up” conformation, which does not cover the S2 subunit and allows the interactions between spike protein and ACE2. The “up” position also paves the way for the S2 subunit to expose and insert its fusion peptides into the host cell membrane [43] (Fig. 2).

Inactive and active state conformations of spike protein with its receptor binding. a, b Represent the inactive state conformations in which S2 subunits (stem portion) are completely covered by the “down” position of CTD1s (head portion). It causes steric clashes and inhibits the binding between spike protein and ACE2. c Represents the active state conformation, in which one of the CTD1s is in open state (shown in red stars) and facilitates the binding between spike protein and ACE2. TM and CT stands for transmembrane domain and cytoplasmic tail, respectively
Key residues of SARS-CoV-2 and SARS-CoV
The sequence similarity between SARS-CoV-2 and SARS-CoV spike protein explains the possibility of having the same receptor ACE2 in the host cell [14]. The sequence and the structural studies revealed the key residues, which are involved in spike–ACE2 interactions. The key residue at position 493 in RBD of SARS-CoV-2 is Gln, wherein SARS-CoV (479 is the corresponding residue in SAR-CoV) of civets and humans, it is Lys and Asn, respectively. Since the residue 479 of RBD is near to the virus-binding hotspot residue Lys31 of human ACE2, the Lys residue present in civet causes steric clashes and not favoring human ACE2 receptor binding. However, the Lys479Asn mutation revealed that the Asn present in 479 of human SARs-CoV enhances the viral binding with human ACE2 receptors. The Gln493 in SARS-CoV-2 RBD is compatible with the human ACE2 receptor hotspot residue Lys31, which explains its target cell identification [14].
Similarly, the residue 501 in SARS-CoV-2 RBD is Asn, wherein civet RBD, it is Ser (487 is the corresponding residue in SARS-CoV), and for humans, it is Thr. This residue also plays an important role in making interaction with the hot spot residue Lys353 of receptor ACE2. Ser487Thr mutation analysis shows encouraging results upon human ACE2 receptor binding and plays a role in human-to-human transmission. Thus, the interactions with Lys353 will be favorable for threonine (human SARS-CoV) and Asn (SARS-CoV-2) than serine (civet) [14].
The residues Lue455, Phe486, and Ser494 in SARS-CoV-2 RBD (Tyr442, Leu472, and Asp480 in human and civet SARS-CoV) are considered as important for the human ACE2 receptor binding. Tyr442 of human and civet SARS-CoV RBDs shows unfavorable interactions with hotspot residue Lys31 of human ACE2; however, Lue455 of SARS-CoV-2 provides favorable interactions. Compared with Leu472 of human civet SARS-CoV RBD, Phe486 of SARS-CoV-2 provides better interaction with hotspot residue Lys31 of human ACE2. Although Ser494 provides positive support for the human ACE2 receptor hotspot residue Lys353, the SARS-CoV Asp480 also makes favorable interaction with hotspot residue Lys353. Throughout this viral entry process, the Lys31 and Lys 353 of human ACE2 receptors are termed as “hot spot” residues, which consist of a salt bridge buried in a hydrophobic environment and contribute critically for the virus and host cell receptor binding. Thus, this key residue comparison of SARS-CoV-2 with the civet and human SARs-CoV explains how actively SARS-CoV-2 is choosing and binding with the human ACE2 receptors, which is likely to cause the human-to-human transmission [14]. The heterogeneity of amino acids in ACE2 receptors is also responsible for the wavering binding affinities between the host and the virus, which is associated with the viral transmission. However, the variants in the host cell receptors (ACE2) can confer resistance against the invading pathogens. Hussain et al. reported that the mutations S19P and E329G in ACE2 disrupt the intermolecular interactions and have low binding affinity with viral spike protein. In addition to the variations in the viral spike protein, ACE2 allelic variants can also drive the potential resistance against SARS-CoV-2 infection [50].
Intrinsic disorder propensities in coronaviruses
Intrinsically disordered proteins (IDPs) and intrinsically disordered regions (IDRs) also play major roles in a number of biological functions, including DNA/RNA binding, protein binding, and facilitating access to the binding sites between the binding partners [51, 52]. The RNA–protein recognition often needs conformational changes in both RNA and protein, which is facilitated by the structural flexibility of disordered residues [51]. Also, the functional importance of intrinsically disordered regions in proteins includes transcription, translation, post-translational modifications, and cell signaling [51, 53]. The categorization of coronaviruses, based on the intrinsic disorder propensities, can represent useful identification for the viral life cycle and its pathogenicity.
The IDRs in SARS-CoV nucleocapsid protein comprise three segments, such as 1–44, 182–247 and 366–422 [54, 55]. The highly flexible intrinsic disordered linker region, which connects the NTD and CTD is rich in serine and arginine residues. An intrinsically disordered domain that flanks the CTD (C-terminal tail peptide) plays a significant role in dimer–dimer association in human coronavirus 229E (hCoV-229E). Likewise, the coronavirus HKU1, has a partially disordered conserved linker loop (amino acids 428–587) structure [56]. HKU1 S2 subunit also shows the presence of disordered residues at its protease cleavage site [57].
In SARS-CoV-2, the attachment of spike protein with the host cell is activated by the host cell enzymes trypsin, cathepsin L, furin and TMPRSS2 (Fig. 1b). The sequence comparison of SARS-CoV-2 against other lineage B betacoronaviruses shows that the unique amino acid pattern “RRAR” is present at the S1/S2 junction of the spike protein, which is cleaved by the furin enzyme. However, the structure reported for SARS-CoV-2 spike protein (PDB code: 6VSB), shows that S1/S2 junction is in a disordered, solvent-exposed loop [4]. Hence, it has been hypothesized that the unique amino acid sequence “RRAR” present in SARS-CoV-2 is responsible for their effective transmission [58,59,60].
The binding with ACE2 is governed by the intrinsically flexible receptor binding motif, and the binding interfaces along with the key residues are reported in the literature [61, 62]. In addition, based on our analysis (Fig. 3), the monomeric structure of the spike protein in the free form (PDB ID: 6VSB; green color) [4] shows a number of missing residues in the structure. These missing residues of the spike protein attain a stable conformation upon binding to the ACE2 receptor and hence, they are termed as disorder-to-order transition (DOT) residues (PDB ID: 6LZG; light golden color). Specifically, the regions, Leu455 to Pro491 and Asn501 to Val503 show disorder-to-order transitions [61]. These disordered-to-ordered residues are facilitating a better shape complementary and affinity between ACE2 and spike protein. Interestingly, the key residues responsible for transmission, and interaction with ACE2 receptors (already discussed in section “Key residues of SARS-CoV-2 and SARS-CoV”) are overlapping with these mentioned disordered-to-ordered residues. Based on these observations, the disorder-to-order conformational change is necessary to facilitate the spike protein binding with its receptor. Thus, an in-depth analysis of these disordered residues will shed additional insights on the viral recognition and transmission mechanism.

Structure of the monomeric spike protein (green)—ACE2 receptor (blue) complex. The interface residues are shown in light golden. The disordered-to-ordered transition residues (Leu455 to Pro491 and Asn501 to Val503) have been marked in the figure
The studies by Goh et al. [63] on 1918 H1N1 and H5N1 explain that it is very likely that disordered regions are important for host specificity and recognition, e.g., across species of birds. They also explained the changes created by disorder in crucial regions, increasing the virulence of both the H5N1 and the 1918 H1N1 viruses. Therefore, the increasing reports for the intrinsically disordered regions in coronaviruses need to be pointed out. The importance and functional role of intrinsically disordered regions need to be thoroughly studied. This could identify additional targets for drugs to combat coronavirus through the disruption of their packing and assembly process.
Therapeutic targets
The receptor binding, along with its membrane fusion, is the initial and important step in the coronavirus infection and serves as primary targets for inhibiting the viral entry.
The genome sequencing of SARS-CoV-2 and the comparison of the genomes of related virus proteins suggested the anti-HIV lopinavir plus ritonavir combination can be likely effective [15, 64, 65]. SARS-CoV-2 uses the ACE2 receptor of AT2 cells in the lung as its primary targets. Since the viral entry is governed by receptor-mediated endocytosis, AP2-associated protein kinase 1 (AAK1), a known regulator of endocytosis, can be a good target to interrupt the virus entry. The Janus kinase inhibitor baricitinib, and its binding with the cyclin G-associated kinase (endocytosis regulator) is sufficient to inhibit AAK1 [66, 67]. Similarly, sunitinib and erlotinib, the oncology drugs, have been shown to inhibit viral infection of cells through the inhibition of AAK1 [68]. However, these compounds bring serious side effects and cannot be considered for a safe therapy.
Based on the pathogenicity studies of SARs-CoV and MERS-CoV, an increased amount of inflammatory cytokines in serum is associated with the inflammation and extensive lung damage [69]. Similarly, SARS-CoV-2 also has a high amount of IL1B, IFNγ, IP10, and MCP1/CCL2, which lead to T-helper-1 (Th1) activation. However, SARS-CoV-2 is shown to increase the T-helper-2 (Th2) cytokines (IL4 and IL10) that are known to suppress inflammation, which differs from SARs and MERS-CoV infection. In view of cytokines induced by SARS-CoV-2 and other SARS-CoV, MERS-CoV, corticosteroids were frequently used to reduce the inflammation-induced lung injury [2].
Host-and-viral-targeted drugs
Arabi et al. [70] examined the combination of interferon beta-1b, lopinavir, and ritonavir combination for the MERS infection in Saudi Arabia. The antiviral nucleotide prodrug remdesivir showed a potent efficacy on MERS-CoV and SARS-CoV infections. Since SARS-CoV-2 is an emerging virus, no effective treatment has been developed so far, yet the already available combination of lopinavir and ritonavir is being used [2].
Since the SARS-CoV enters the cell through endocytosis and the lysosomal protease is priming the spike protein, targeting/inhibiting the endosomal acidification or lysosomal cysteine protease can block the SARS-CoV entry [6]. The view of the activation of SARS-CoV and SARS-CoV-2 by TMPRSS2 might also have therapeutic suggestions. A protease inhibitor, camostat mesylate, which is studied as a TMPRSS2 inhibitor and has been used to treat humans, is available and could be employed as a defense against the respiratory viruses [71, 72].
Autophagy has been implicated in viral replications, which is also responsible for the formation of double-membrane vesicles (DMV) in the host cells [73]. Since the autophagosomes are degraded by lysosomes, inhibitors like lysosomotropic agents have been proposed for SARS-CoV-2 [74]. The antimalarial drugs hydroxychloroquine (HCQ) and chloroquine (CQ) have been demonstrated for SARS-CoV-2 antiviral activity. However, data to support the use of HCQ and CQ for COVID-19 are incomplete [75]. These lysosomotropic agents are helpful for neutralizing the endosome-lysosomal acidic pH, thereby blocking protease activity and subsequently blocking the viral entry. However, how autophagy is implicated in the infection of CoVs is still under debate [47, 74, 76]. Table 1 describes the different inhibitors used for blocking the viral entry. The most studied and the common pathway proposed for all coronaviruses is the endocytic pathway, so blocking that pathway is a big hallmark for treating the disease.
Identification of potential drugs for SARS-CoV-2 is a necessary task, so drug repurposing can also work for this scenario. The small molecules, which are already in clinical trials or used for some other diseases and the molecules from the expert’s opinion are also listed in Table 2. The table is categorized into three groups: (1) compounds involving drug repurposing, (2) compounds in clinical and pre-clinical study, (3) compounds targeting the mechanism pathway.
Researchers in government and private sectors are making huge efforts to develop effective vaccines for SARS-CoV-2. The vaccine development approaches are mainly based on inactivated and attenuated viral protein particles, viral vectors and viral DNA/RNAs. A novel RNA-based vaccine uses a part of the genetic code of spike protein mRNA-1273 (ClinicalTrials.gov: NCT04283461) [139]. Several other mRNA-based vaccines by CureVac (Tübingen, Germany), BNT162 by BioNTech (Mainz, Germany), Pfizer (New York, NY, USA) and BioNTech mRNA vaccine (Mainz, Germany) are in different stages of development [139, 140]. CanSino Biologics (Tianjin, China), the company that developed the vaccine for Ebola is also developing a vaccine named Ad5-nCoV for SARS-CoV-2. It is a spike protein-based vaccine that is undergoing phase I clinical trials in healthy individuals in Wuhan china (ClinicalTrials.gov: NCT04313127) [141, 142]. The current status of the vaccines under development is available at the Milken Institute Treatment and Vaccine Tracker:
https://milkeninstitute.org/sites/default/files/2020-03/Covid19%20Tracker%20032020v3-posting.pdf [141].
Summary and perspectives
SARS-CoV-2 is a continuously growing life-threatening disease. This coronavirus has rapidly evolved and spread all over the world, having a mortality of more than 0.8 million lives so far. The exact origin and mechanism of attack and spatial distribution are still not completely explored.
The viral and the host cell proteins assisting in invasion process can be a target for treating the infections. The available crystal structures and the binding mechanism proposed by other coronaviruses can help to study the SARS-CoV-2. The critical structural studies of the viral particles are also helpful to identify the drug targets. The positional changes (up and down conformation) of spike protein trimer determine the active to inactive state. Thus, targeting these conformations and developing small molecules or peptides can also stop the viral entry. In addition, phylogenetic analysis and structural studies revealed that the hot spot residues, the role of Gln493 in human ACE2 receptor binding and the critical residue Asn501 of RBD explain the human-to-human transmission. The intrinsic disorder region and a precise furin-like cleavage site can be responsible for viral cycle and pathogenicity. However, in-depth studies are needed to address this issue, which may represent a potential antiviral strategy.
The small molecules, which are in clinical trials, the drug compounds, which are currently in use for treating different diseases can also help to identify (screen) potential drug candidates. Development of suitable mice models to understand this novel virus infection and comparison with the other coronavirus will speed up the drug discovery. Drug testing techniques are also necessary to be accelerated. Recognizing the risks and commercial benefits, researchers are developing effective vaccines for SARS-CoV-2 infection.
Abbreviations
- ACE2:
-
Angiotensin-converting enzyme 2
- CCL2:
-
Chemokine (C–C motif) ligand 2
- CTD:
-
C-terminal domain
- DPP4:
-
Dipeptidyl peptidase-4
- IDPs:
-
Intrinsic disordered proteins
- IDRs:
-
Intrinsic disordered regions
- IL1B:
-
Interleukin 1 beta
- IFN‐γ:
-
Interferon‐gamma
- IP10:
-
Interferon gamma-induced protein 10
- MERS:
-
Middle east respiratory syndrome
- MCP1:
-
Monocyte chemoattractant protein-1
- NTD:
-
N-terminal domain
- NSPs:
-
Non-structural proteins
- PD:
-
Peptidase domain
- PEDV:
-
Porcine epidemic diarrhea virus
- RBD:
-
Receptor-binding domain RBD
- SARS:
-
Severe acute respiratory syndrome
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- TMPRSS2:
-
Transmembrane protease, serine 2
- TGEV:
-
Transmissible gastroenteritis virus
References
Holmes KV. SARS coronavirus: a new challenge for prevention and therapy. J Clin Investig. 2003;111:1605–9.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506.
Chan JFW, Yuan S, Kok KH, To KKW, Chu H, Yang J, Xing F, Liu J, Yip CCY, Poon RWS, Tsoi HW. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395:514–23.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–3.
Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, Ren R, Leung KS, Lau EH, Wong JY, Xing X. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med. 2020;382:1199–207.
Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. 2016;3:237–61.
Perlman S, Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol. 2009;7:439–50.
Li F. Receptor recognition and cross-species infections of SARS coronavirus. Antivir Res. 2013;100:246–54.
Graham RL, Baric RS. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J Virol. 2010;84:3134–46.
Wang M, Yan M, Xu H, Liang W, Kan B, Zheng B, Chen H, Zheng H, Xu Y, Zhang E, Wang H. SARS-CoV infection in a restaurant from palm civet. Emerg Infect Dis. 2005;11:1860.
Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5:562–9.
Ge XY, Li JL, Yang XL, Chmura AA, Zhu G, Epstein JH, Mazet JK, Hu B, Zhang W, Peng C, Zhang YJ. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503:535–8.
Hu B, Zeng LP, Yang XL, Ge XY, Zhang W, Li B, Xie JZ, Shen XR, Zhang YZ, Wang N, Luo DS. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017;13:e1006698.
Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 2020;94:e00127–e220.
Simmons G, Zmora P, Gierer S, Heurich A, Pöhlmann S. Proteolytic activation of the SARS-coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antivir Res. 2013;100:605–14.
Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, Gulyaeva AA, Haagmans BL, Lauber C, Leontovich AM, Neuman BW, Penzar D. The species severe acute respiratory syndrome related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–44.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–3.
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–7.
Peiris JSM, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nat Med. 2004;10:S88–S97.
Stadler K, Rappuoli R. SARS: understanding the virus and development of rational therapy. Curr Mol Med. 2005;5:677–97.
Cheng VC, Lau SK, Woo PC, Yuen KY. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Cin Microbiol Rev. 2007;20:660–94.
Zaki AM, Van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814–20.
Leroy EM, Gouilh MA, Brugère-Picoux J. The risk of SARS-CoV-2 transmission to pets and other wild and domestic animals strongly mandates a one-health strategy to control the COVID-19 pandemic. One Health. 2020. https://doi.org/10.1016/j.onehlt.2020.100133.
Laude H, Van Reeth K, Pensaert M. Porcine respiratory coronavirus: molecular features and virus-host interactions. Vet Res. 1993;24:125–50.
Mole B. Deadly pig virus slips through US borders. Nature. 2013;499:388.
Stevenson GW, Hoang H, Schwartz KJ, Burrough ER, Sun D, Madson D, Cooper VL, Pillatzki A, Gauger P, Schmitt BJ, Koster LG. Emergence of porcine epidemic diarrhea virus in the United States: clinical signs, lesions, and viral genomic sequences. J Vet Diagn Investig. 2013;25:649–54.
Jean A, Quach C, Yung A, Semret M. Severity and outcome associated with human coronavirus OC43 infections among children. Pediatr Infect Dis J. 2013;32:325–9.
Chiu SS, Hung Chan K, Wing Chu K, Kwan SW, Guan Y, Man Poon LL, Peiris JSM. Human coronavirus NL63 infection and other coronavirus infections in children hospitalized with acute respiratory disease in Hong Kong. China Clin Infect Dis. 2005;40:1721–9.
Jevšnik M, Uršič T, Žigon N, Lusa L, Krivec U, Petrovec M. Coronavirus infections in hospitalized pediatric patients with acute respiratory tract disease. BMC Infect Dis. 2012;12:365.
Phelan AL, Katz R, Gostin LO. The novel coronavirus originating in Wuhan, China: challenges for global health governance. JAMA. 2020;323:709–10.
Zhu H, Wang L, Fang C, Peng S, Zhang L, Chang G, Xia S, Zhou W. Clinical analysis of 10 neonates born to mothers with 2019-nCoV pneumonia. Transl Pediatr. 2020;9:51.
Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of Coronavirus disease in 2019 in China. N Engl J Med. 2020;382:1708–20.
Setti L, Passarini F, De Gennaro G, Barbieri P, Perrone MG, Borelli M, Palmisani J, Di Gilio A, Piscitelli P, Miani A. Airborne transmission route of COVID-19: why 2 meters/6 feet of inter-personal distance could not be enough. Int J Environ Res Public Health. 2020;17:2932.
Anderson EL, Turnham P, Griffin JR, Clarke CC. Consideration of the aerosol transmission for COVID-19 and public health. Risk Anal. 2020;40:902–7.
Lee S, Meyler P, Mozel M, Tauh T, Merchant R. Asymptomatic carriage and transmission of SARS-CoV-2: what do we know? Can J Anaesth. 2020. (In press).
Somsen GA, van Rijn C, Kooij S, Bem RA, Bonn D. Small droplet aerosols in poorly ventilated spaces and SARS-CoV-2 transmission. Lancet Respir Med. 2020;8:658–9.
Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, Xiong Y, Zhao Y. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA. 2020;323:1061–9.
Rothe C, Schunk M, Sothmann P, Bretzel G, Froeschl G, Wallrauch C, Zimmer T, Thiel V, Janke C, Guggemos W, Seilmaier M. Transmission of 2019-nCoV infection from an asymptomatic contact in Germany. N Engl J Med. 2020;382:970–1.
https://labblog.uofmhealth.org/rounds/how-scientists-quantify-intensity-of-an-outbreak-like-covid-19. Accessed 30 June 2020
Huang X, Dong W, Milewska A, Golda A, Qi Y, Zhu QK, Marasco WA, Baric RS, Sims AC, Pyrc K, Li W. Human coronavirus HKU1 spike protein uses O-acetylated sialic acid as an attachment receptor determinant and employs hemagglutinin-esterase protein as a receptor-destroying enzyme. J Virol. 2015;89:7202–13.
Millet JK, Whittaker GR. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015;202:120–34.
Hofmann H, Pöhlmann S. Cellular entry of the SARS coronavirus. Trends Microbiol. 2004;12:466–72.
Gui M, Song W, Zhou H, Xu J, Chen S, Xiang Y, Wang X. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017;27:119–29.
Gralinski LE, Menachery VD. Return of the Coronavirus: 2019-nCoV. Viruses. 2020;12:135.
Zumla A, Chan JF, Azhar EI, Hui DS, Yuen KY. Coronaviruses—drug discovery and therapeutic options. Nat Rev Drug Discov. 2016;15:327–47.
Wang H, Yang P, Liu K, Guo F, Zhang Y, Zhang G, Jiang C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008;18:290–301.
Yang N, Shen HM. Targeting the endocytic pathway and autophagy process as a novel therapeutic strategy in COVID-19. Int J Biol Sci. 2020;16:1724–31.
Abassi Z, Knaney Y, Karram T, Heyman SN. The lung macrophage in SARS-CoV-2 infection: a friend or a foe? Front Immunol. 2020;11:1312.
Yilla M, Harcourt BH, Hickman CJ, McGrew M, Tamin A, Goldsmith CS, Bellini WJ, Anderson LJ. SARS-coronavirus replication in human peripheral monocytes/macrophages. Virus Res. 2005;107:93–101.
Hussain M, Jabeen N, Raza F, Shabbir S, Baig AA, Amanullah A, Aziz B. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J Med Virol. 2020. https://doi.org/10.1002/jmv.25832.
Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Bio. 2005;6:197–208.
Srivastava A, Yesudhas D, Ramakrishnan C, Ahmad S, Gromiha MM. Role of disordered regions in transferring tyrosine to its cognate tRNA. Int J Biol Macromol. 2020;150:705–13.
McBride R, Van Zyl M, Fielding BC. The coronavirus nucleocapsid is a multifunctional protein. Viruses. 2014;6:2991–3018.
Huang Q, Yu L, Petros AM, Gunasekera A, Liu Z, Xu N, Hajduk P, Mack J, Fesik SW, Olejniczak ET. Structure of the N-terminal RNA-binding domain of the SARS CoV nucleocapsid protein. Biochemistry. 2004;43:6059–63.
Chang CK, Hou MH, Chang CF, Hsiao CD, Huang TH. The SARS coronavirus nucleocapsid protein—forms and functions. Antivir Res. 2014;103:39–50.
Li F. Receptor recognition mechanisms of coronaviruses: a decade of structural studies. J Virol. 2015;89:1954–64.
Kirchdoerfer RN, Cottrell CA, Wang N, Pallesen J, Yassine HM, Turner HL, Corbett KS, Graham BS, McLellan JS, Ward AB. Pre-fusion structure of a human coronavirus spike protein. Nature. 2016;531:118–21.
Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir Res. 2020;176:104742.
Xia S, Lan Q, Su S, Wang X, Xu W, Liu Z, Zhu Y, Wang Q, Lu L, Jiang S. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transduct Target Ther. 2020;5:92.
Hasan A, Paray BA, Hussain A, Qadir FA, Attar F, Aziz FM, Sharifi M, Derakhshankhah H, Rasti B, Mehrabi M, Shahpasand K. A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. J Biomol Struct Dyn. 2020. (In press).
Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, Lu G, Qiao C, Hu Y, Yuen KY, Wang Q. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181:894–904.
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–8.
Goh GKM, Dunker AK, Uversky VN. Protein intrinsic disorder and influenza virulence: the 1918 H1N1 and H5N1 viruses. Virol J. 2009;6:69.
Muralidharan N, Sakthivel R, Velmurugan D, Gromiha MM. Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 Protease against COVID-19. J Biomol Struct Dyn. 2020. (In press).
Anusuya S, Muralidharan N, Velmurugan D, Gromiha MM. Therapeutic targets and computational approaches on drug development for COVID-19. Curr Top Med Chem. 2020. (In press).
Richardson P, Griffin I, Tucker C, Smith D, Oechsle O, Phelan A, Stebbing J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet. 2020;395:e30–e3131.
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y, Yu T. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–13.
Pu SY, Xiao F, Schor S, Bekerman E, Zanini F, Barouch-Bentov R, Nagamine CM, Einav S. Feasibility and biological rationale of repurposing sunitinib and erlotinib for dengue treatment. Antivir Res. 2018;155:67–75.
Wong CK, Lam CWK, Wu AKL, Ip WK, Lee NLS, Chan IHS, Lit LCW, Hui DSC, Chan MHM, Chung SSC, Sung JJY. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. 2004;136:95–103.
Arabi YM, Alothman A, Balkhy HH, Al-Dawood A, AlJohani S, Al Harbi S, Kojan S, Al Jeraisy M, Deeb AM, Assiri AM, Al-Hameed F. Treatment of Middle East Respiratory Syndrome with a combination of lopinavir-ritonavir and interferon-β1b (MIRACLE trial): study protocol for a randomized controlled trial. Trials. 2018;19:81.
Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol. 2012;86:6537–45.
Meyer D, Sielaff F, Hammami M, Böttcher-Friebertshäuser E, Garten W, Steinmetzer T. Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. Biochem J. 2013;452:331–43.
Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem. 2004;279:10136–41.
Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, Seidah NG, Nichol ST. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J. 2005;2:69.
Yazdany J, Kim AH. Use of hydroxychloroquine and chloroquine during the COVID-19 pandemic: what every clinician should know. Ann Intern Med. 2020;172:754–5.
Huang IC, Bosch BJ, Li F, Li W, Lee KH, Ghiran S, Vasilieva N, Dermody TS, Harrison SC, Dormitzer PR, Farzan M. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J Biol Chem. 2006;281:3198–203.
McLure KG, Gesner EM, Tsujikawa L, Kharenko OA, Attwell S, Campeau E, Wasiak S, Stein A, White A, Fontano E, Suto RK. RVX-208, an inducer of ApoA-I in humans, is a BET bromodomain antagonist. PLoS ONE. 2013;8:e83190.
Pierre F, Chua PC, O’Brien SE, Siddiqui-Jain A, Bourbon P, Haddach M, Michaux J, Nagasawa J, Schwaebe MK, Stefan E, Vialettes A. Discovery and SAR of 5-(3-chlorophenylamino) benzo [c][2, 6] naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J Med Chem. 2011;54:635–54.
Janeczko M, Orzeszko A, Kazimierczuk Z, Szyszka R, Baier A. CK2α and CK2α’ subunits differ in their sensitivity to 4,5,6,7-tetrabromo-and 4,5,6,7-tetraiodo-1H-benzimidazole derivatives. Eur J Med Chem. 2012;47:345–50.
Siddiqui-Jain A, Drygin D, Streiner N, Chua P, Pierre F, O'Brien SE, Bliesath J, Omori M, Huser N, Ho C, Proffitt C. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010;70:10288–98.
Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–9.
Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–71.
Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, Sleeman JP, Coco FL, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–78.
Krämer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I, Heinzel T, Göttlicher M. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003;22:3411–20.
Fass DM, Shah R, Ghosh B, Hennig K, Norton S, Zhao WN, Reis SA, Klein PS, Mazitschek R, Maglathlin RL, Lewis TA. Effect of inhibiting histone deacetylase with short-chain carboxylic acids and their hydroxamic acid analogs on vertebrate development and neuronal chromatin. ACS Med Chem Lett. 2010;2:39.
Nissinen E, Linden IB, Schultz E, Pohto P. Biochemical and pharmacological properties of a peripherally acting catechol-O-methyltransferase inhibitor entacapone. Naunyn Schmiedebergs Arch Pharmacol. 1992;346:262–6.
De Santi C, Giulianotti PC, Pietrabissa A, Mosca F, Pacifici GM. Catechol-O-methyltransferase: variation in enzyme activity and inhibition by entacapone and tolcapone. Eur J Clin Pharmacol. 1998;54:215–9.
Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231:232–5.
Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014;3:e02242.
Asano N, Ishii S, Kizu H, Ikeda K, Yasuda K, Kato A, Martin OR, Fan JQ. In vitro inhibition and intracellular enhancement of lysosomal α-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. Eur J Biochem. 2000;267:4179–86.
Carter SB, Franklin TJ, Jones DF, Leonard BJ, Mills SD, Turner RW, Turner WB. Mycophenolic acid: an anti-cancer compound with unusual properties. Nature. 1969;223:848–50.
Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–32.
Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046.
Priebe W, Krawczyk M, Kuo MT, Yamane Y, Savaraj N, Ishikawa T. Doxorubicin-and daunorubicin-glutathione conjugates, but not unconjugated drugs, competitively inhibit leukotriene C4Transport mediated byMRP/GS-XPump. Biochem Biophys Res Commun. 1998;247:859–63.
Perrotton T, Trompier D, Chang XB, Di Pietro A, Baubichon-Cortay H. (R)-and (S)-verapamil differentially modulate the multidrug-resistant protein MRP1. J Biol Chem. 2007;282:31542–8.
Toral-Barza L, Zhang WG, Lamison C, LaRocque J, Gibbons J, Yu K. Characterization of the cloned full-length and a truncated human target of rapamycin: activity, specificity, and enzyme inhibition as studied by a high capacity assay. Biochem Biophys Res Commun. 2005;332:304–10.
Fonseca BD, Jia JJ, Hollensen AK, Pointet R, Hoang HD, Niklaus MR, Pena IA, Lahr RM, Smith EM, Hearnden J, Wang XD. LARP1 is a major phosphorylation substrate of mTORC1. bioRxiv. 2018;PPR64205.
Tesei A, Cortesi M, Zamagni A, Arienti C, Pignatta S, Zanoni M, Paolillo M, Curti D, Rui M, Rossi D, Collina S. Sigma receptors as endoplasmic reticulum stress “gatekeepers” and their modulators as emerging new weapons in the fight against cancer. Front Pharmacol. 2018;9:711.
Phadke M, Remsing Rix LL, Smalley I, Bryant AT, Luo Y, Lawrence HR, Schaible BJ, Chen YA, Rix U, Smalley KS. Dabrafenib inhibits the growth of BRAF-WT cancers through CDK16 and NEK9 inhibition. Mol Oncol. 2018;12:74–88.
Díaz JL, Cuberes R, Berrocal J, Contijoch M, Christmann U, Fernández A, Port A, Holenz J, Buschmann H, Laggner C, Serafini MT. Synthesis and biological evaluation of the 1-Arylpyrazole Class of σ1 receptor antagonists: identification of 4-{2-[5-Methyl-1-(naphthalen-2-yl)-1 H-pyrazol-3-yloxy] ethyl} morpholine (S1RA, E-52862). J Med Chem. 2012;55:8211–24.
Akunne HC, Whetzel SZ, Wiley JN, Corbin AE, Ninteman FW, Tecle H, Pei Y, Pugsley TA, Heffner TG. The pharmacology of the novel and selective sigma ligand, PD 144418. Neuropharmacology. 1997;36:51–62.
Prezzavento O, Campisi A, Ronsisvalle S, Li Volti G, Marrazzo A, Bramanti V, Cannavo G, Vanella L, Cagnotto A, Mennini T, Ientile R. Novel sigma receptor ligands: synthesis and biological profile. J Med Chem. 2007;50:951–61.
Azzariti A, Colabufo NA, Berardi F, Porcelli L, Niso M, Simone GM, Perrone R, Paradiso A. Cyclohexylpiperazine derivative PB28, a σ2 agonist and σ1 antagonist receptor, inhibits cell growth, modulates P-glycoprotein, and synergizes with anthracyclines in breast cancer. Mol Cancer Ther. 2006;5:1807–16.
Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino) ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–72.
Sintchak MD, Nimmesgern E. The structure of inosine 5′-monophosphate dehydrogenase and the design of novel inhibitors. Immunopharmacology. 2000;47:163–84.
Koltun ES, Tsuhako AL, Brown DS, Aay N, Arcalas A, Chan V, Du H, Engst S, Ferguson K, Franzini M, Galan A. Discovery of XL413, a potent and selective CDC7 inhibitor. Bioorg Med Chem Lett. 2012;22:3727–31.
Leung L, Niculescu-Duvaz D, Smithen D, Lopes F, Callens C, McLeary R, Saturno G, Davies L, Aljarah M, Brown M, Johnson L. Anti-metastatic inhibitors of lysyl oxidase (LOX): design and structure–activity relationships. J Med Chem. 2019;62:5863–84.
Llona-Minguez S, Hoglund A, Jacques SA, Johansson L, Calderon-Montano JM, Claesson M, Loseva O, Valerie NC, Lundback T, Piedrafita J, Maga G. Discovery of the first potent and selective inhibitors of human dCTP pyrophosphatase 1. J Med Chem. 2016;59:1140–8.
Llona-Minguez S, Hoglund A, Wiita E, Almlof I, Mateus A, Calderon-Montano JM, Cazares-Korner C, Homan E, Loseva O, Baranczewski P, Jemth AS. Identification of triazolothiadiazoles as potent inhibitors of the dCTP pyrophosphatase 1. J Med Chem. 2017;60:2148–54.
Llona-Minguez S, Häggblad M, Martens U, Johansson L, Sigmundsson K, Lundbäck T, Loseva O, Jemth AS, Lundgren B, Jensen AJ, Scobie M. Diverse heterocyclic scaffolds as dCTP pyrophosphatase 1 inhibitors. Part 2: Pyridone-and pyrimidinone-derived systems. Bioorg Med Chem Lett. 2017;27:3219–25.
McIver EG, Bryans J, Birchall K, Chugh J, Drake T, Lewis SJ, Osborne J, Smiljanic-Hurley E, Tsang W, Kamal A, Levy A. Synthesis and structure–activity relationships of a novel series of pyrimidines as potent inhibitors of TBK1/IKKε kinases. Bioorg Med Chem Lett. 2012;22:7169–73.
Seitzberg JG, Knapp AE, Lund BW, Mandrup Bertozzi S, Currier EA, Ma JN, Sherbukhin V, Burstein ES, Olsson R. Discovery of potent and selective small-molecule PAR-2 agonists. J Med Chem. 2008;51:5490–3.
Cheng RK, Fiez-Vandal C, Schlenker O, Edman K, Aggeler B, Brown DG, Brown GA, Cooke RM, Dumelin CE, Doré AS, Geschwindner S. Structural insight into allosteric modulation of protease-activated receptor 2. Nature. 2017;545:112–5.
Barry GD, Suen JY, Le GT, Cotterell A, Reid RC, Fairlie DP. Novel agonists and antagonists for human protease activated receptor 2. J Med Chem. 2010;53:7428–40.
Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, Zhang L, Bui MH, Sheppard GS, Wang L, Sehgal V. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature. 2020;578:306–10.
Xu L, Chen Y, Mayakonda A, Koh L, Chong YK, Buckley DL, Sandanaraj E, Lim SW, Lin RYT, Ke XY, Huang ML. Targetable BET proteins-and E2F1-dependent transcriptional program maintains the malignancy of glioblastoma. Proc Natl Acad Sci USA. 2018;115:E5086–E50955095.
Zengerle M, Chan KH, Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem Biol. 2015;10:1770–7.
Albrecht BK, Gehling VS, Hewitt MC, Vaswani RG, Côté A, Leblanc Y, Nasveschuk CG, Bellon S, Bergeron L, Campbell R, Cantone N. Identification of a benzoisoxazoloazepine inhibitor (CPI-0610) of the bromodomain and extra-terminal (BET) family as a candidate for human clinical trials. J Med Chem. 2016;59:1330–9.
Schenone S, Brullo C, Musumeci F, Radi M, Botta M. ATP-competitive inhibitors of mTOR: an update. Curr Med Chem. 2011;18:2995–3014.
Thompson PA, Eam B, Young NP, Fish S, Chen J, Barrera M, Howard H, Sung E, Parra A, Staunton J, Chiang GG. eFT226, a potent and selective inhibitor of eIF4A, is efficacious in preclinical models of lymphoma. Cancer Res. 2019;79:2698.
Jorquera PA, Mathew C, Pickens J, Williams C, Luczo JM, Tamir S, Ghildyal R, Tripp RA. Verdinexor (KPT-335), a selective inhibitor of nuclear export, reduces respiratory syncytial virus replication in vitro. J Virol. 2019;93:e01684–e1718.
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–6.
Pua KH, Stiles DT, Sowa ME, Verdine GL. IMPDH2 is an intracellular target of the cyclophilin A and sanglifehrin A complex. Cell Rep. 2017;18:432–42.
Carelli JD, Sethofer SG, Smith GA, Miller HR, Simard JL, Merrick WC, Jain RK, Ross NT, Taunton J. Ternatin and improved synthetic variants kill cancer cells by targeting the elongation factor-1A ternary complex. Elife. 2015;4:e10222.
Cencic R, Desforges M, Hall DR, Kozakov D, Du Y, Min J, Dingledine R, Fu H, Vajda S, Talbot PJ, Pelletier J. Blocking eIF4E-eIF4G interaction as a strategy to impair coronavirus replication. J Virol. 2011;85:6381–9.
Reich SH, Sprengeler PA, Chiang GG, Appleman JR, Chen J, Clarine J, Eam B, Ernst JT, Han Q, Goel VK, Han EZ. Structure-based design of pyridine-aminal eFT508 targeting dysregulated translation by selective mitogen-activated protein kinase interacting kinases 1 and 2 (MNK1/2) inhibition. J Med Chem. 2018;61:3516–40.
Xu Y, Poggio M, Jin HY, Shi Z, Forester CM, Wang Y, Stumpf CR, Xue L, Devericks E, So L, Nguyen HG. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat Med. 2019;25:301–11.
Mackman RL, Steadman VA, Dean DK, Jansa P, Poullennec KG, Appleby T, Austin C, Blakemore CA, Cai R, Cannizzaro C, Chin G. Discovery of a potent and orally bioavailable cyclophilin inhibitor derived from the sanglifehrin macrocycle. J Med Chem. 2018;61:9473–99.
Rutaganira FU, Fowler ML, McPhail JA, Gelman MA, Nguyen K, Xiong A, Dornan GL, Tavshanjian B, Glenn JS, Shokat KM, Burke JE. Design and structural characterization of potent and selective inhibitors of phosphatidylinositol 4 kinase IIIβ. J Med Chem. 2016;59:1830–9.
Shah PS, Link N, Jang GM, Sharp PP, Zhu T, Swaney DL, Johnson JR, Von Dollen J, Ramage HR, Satkamp L, Newton B. Comparative flavivirus-host protein interaction mapping reveals mechanisms of dengue and Zika virus pathogenesis. Cell. 2018;175:1931–45.
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, O’meara MJ, Guo JZ, Swaney DL, Tummino TA, Huttenhain R, Kaake RM, et al. A SARS-CoV-2-human protein–protein interaction map reveals drug targets and potential drug-repurposing. Nature. 2020;583:459–68.
Ma J, Wu S, Zhang X, Guo F, Yang K, Guo J, Su Q, Lu H, Lam P, Li Y, Yan Z. Ester prodrugs of IHVR-19029 with enhanced oral exposure and prevention of gastrointestinal glucosidase interaction. ACS Med Chem Lett. 2017;8:157–62.
Ma J, Zhang X, Soloveva V, Warren T, Guo F, Wu S, Lu H, Guo J, Su Q, Shen H, Solon E. Enhancing the antiviral potency of ER α-glucosidase inhibitor IHVR-19029 against hemorrhagic fever viruses in vitro and in vivo. Antivir Res. 2018;150:112–22.
Michaud A, Williams TA, Chauvet MT, Corvol P. Substrate dependence of angiotensin i-converting enzyme inhibition: captopril displays a partial selectivity for inhibition of N-acetyl-seryl-aspartyl-lysyl-proline hydrolysis compared with that of angiotensin I. Mol Pharmacol. 1997;51:1070–6.
Natesh R, Schwager SL, Sturrock ED, Acharya KR. Crystal structure of the human angiotensin-converting enzyme–lisinopril complex. Nature. 2003;421:551–4.
Hoffmann M, Kleine-Weber H, Krüger N, Mueller MA, Drosten C, Pöhlmann S. The novel coronavirus 2019 (2019-nCoV) uses the SARS-coronavirus receptor ACE2 and the cellular protease TMPRSS2 for entry into target cells. bioRxiv. 2020. https://doi.org/10.1101/2020.01.31.929042.
Yamamoto M, Matsuyama S, Li X, Takeda M, Kawaguchi Y, Inoue JI, Matsuda Z. Identification of nafamostat as a potent inhibitor of Middle East respiratory syndrome coronavirus S protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob Agents Chemother. 2016;60:6532–9.
Nagiec EE, Wu L, Swaney SM, Chosay JG, Ross DE, Brieland JK, Leach KL. Oxazolidinones inhibit cellular proliferation via inhibition of mitochondrial protein synthesis. Antimicrob Agents Chemother. 2005;49:3896–902.
Amanat F, Krammer F. SARS-CoV-2 vaccines: status report. Immunity. 2020;52:583–9.
Hodgson J. The pandemic pipeline. Nat Biotechnol. 2020;38:523–32.
Uddin M, Mustafa F, Rizvi TA, Loney T, Suwaidi HA, Al-Marzouqi AHH, Eldin AK, Alsabeeha N, Adrian TE, Stefanini C, Nowotny N. SARS-CoV-2/COVID-19: Viral genomics, epidemiology, vaccines, and therapeutic interventions. Viruses. 2020;12:526.
Le TT, Andreadakis Z, Kumar A, Roman RG, Tollefsen S, Saville M, Mayhew S. The COVID-19 vaccine development landscape. Nat Rev Drug Discov. 2020;19:305–6.
Acknowledgements
We thank Indian Institute of Technology Madras, Department of Biotechnology for the computational facilities. The work is partially supported by the Department of Biotechnology, Government of India to MMG (no. MSC/2020/000319).
Author information
Authors and Affiliations
Contributions
DY designed the outline of the work. DY and AS wrote the manuscript and prepared the figures, and MMG revised the work and edited the manuscript. All authors read and finalized the manuscript.
Corresponding authors
Ethics declarations
Conflict of interests
The authors declare that they have no conflicts of interest with the contents of this article.
Rights and permissions
About this article

Cite this article
Yesudhas, D., Srivastava, A. & Gromiha, M.M. COVID-19 outbreak: history, mechanism, transmission, structural studies and therapeutics. Infection 49, 199–213 (2021). https://doi.org/10.1007/s15010-020-01516-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s15010-020-01516-2