
Abstract
Community-acquired pneumonia is primarily caused by Streptococcus pneumoniae and Klebsiella pneumoniae, two pathogens that have high morbidity and mortality rates. This is largely due to bacterial resistance development against current antibiotics and the lack of effective vaccines. The objective of this work was to develop an immunogenic multi-epitope subunit vaccine capable of eliciting a robust immune response against S. pneumoniae and K. pneumoniae. The targeted proteins were the pneumococcal surface proteins (PspA and PspC) and choline-binding protein (CbpA) of S. pneumoniae and the outer membrane proteins (OmpA and OmpW) of K. pneumoniae. Different computational approaches and various immune filters were employed for designing a vaccine. The immunogenicity and safety of the vaccine were evaluated by utilizing many physicochemical and antigenic profiles. To improve structural stability, disulfide engineering was applied to a portion of the vaccine structure with high mobility. Molecular docking was performed to examine the binding affinities and biological interactions at the atomic level between the vaccine and Toll-like receptors (TLR2 and 4). Further, the dynamic stabilities of the vaccine and TLRs complexes were investigated by molecular dynamics simulations. While the immune response induction capability of the vaccine was assessed by the immune simulation study. Vaccine translation and expression efficiency was determined through an in silico cloning experiment utilizing the pET28a(+) plasmid vector. The obtained results revealed that the designed vaccine is structurally stable and able to generate an effective immune response to combat pneumococcal infection.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Streptococcus pneumoniae and Klebsiella pneumoniae are responsible for numerous infections, including pneumonia, bacteremia, meningitis, otitis media, bronchitis, sinusitis, and endocarditis (Tumbarello et al. 2015). Children under 2 years and adults over 65 years of age are the most likely population groups to become infected, and disease conditions are commonly preceded by nasopharyngeal bacterial colonization (Von Mollendorf et al. 2017). Both bacteria have high morbidity and mortality rates and have developed resistance against current antibiotics, making them a significant public health concern. Targeting both bacteria in a single vaccine can provide broad-spectrum protection against pneumonia caused by these pathogens, potentially reducing the burden of disease and the need for multiple vaccines. Additionally, there may overlap in the antigenic epitopes targeted by the vaccine, allowing for more efficient and effective immunization.
Currently, pneumococcal vaccines are divided into two categories: pneumococcal conjugate vaccine (PCV) and pneumococcal polysaccharide vaccine (PPV). However, these vaccines have several limitations, such as being specific to certain serotypes, manufacturing complexities, high production expenses, refrigeration necessities, and requiring multiple injections to generate an efficient immune response (Dorosti et al. 2019; Weinberger et al. 2011). To overcome these problems, the development of a peptide-based vaccine that targets a diverse combination of virulence proteins having conserved conformation may represent an alternate solution.
A comprehensive understanding of bacterial colonization and pathogenic mechanisms is crucial for the successful design and development of a peptide-based vaccine. The group of virulence factors associated with S. pneumoniae comprises choline-binding proteins (CBPs), for example, pneumococcal surface proteins (PspA and PspC), pneumococcal histidine triad (Pht) proteins (PhtD and PhtE), pneumolysin (PLY) toxin, LPXTG motif-anchored surface proteins containing hyaluronidase (neuraminidase, and serine protease PrtA), and the lipoprotein components of iron uptake ATP-binding cassette transporters (ABC) transporters (PiuA, PiaA, and PsaA) (Berry and Paton 2000; Khan and Pichichero 2012). These virulence factors might be directly involved in provoking diverse levels of protection in animal models in response to immune challenges using S. pneumoniae serotypes (Paton 2004). The bacterial colonization and adherence to nasopharyngeal cells are mediated by an interaction between bacterial CbpA and human polymeric immunoglobin receptor (hpIgR), which facilitates the bacterial invasion of epithelial cells. PspA is located on the surface of bacterial cells and interacts with the host cell wall through the interface between lipoteichoic acid, C-terminal choline-binding repeats (CBRs), and phosphorylcholine moieties (Brooks-Walter et al. 1999). Many reports have described that during S. pneumoniae infection, PspA upregulation can influence complement deposition through alternative pathways (Yuste et al. 2005). This antigen stimulated a defensive immune response in a mouse model challenged with S. pneumoniae. In a rat model, PspA elicited a protective response against otitis media, and immunization with the wild-type, full-length PspA eliminates nasal carrying of S. pneumonia (White et al. 1999). These findings indicate PspA as a potential vaccine candidate, although a high degree of variations were identified inside the protein family. One study described PspA as being highly immunogenic based on serum obtained from recovering patients since a majority of patients have acquired antibodies against this protein. Pneumococcal surface protein C (PspC), the major adhesion factor for pneumococcus, is involved in pneumococcal colonization and adhesion to the nasopharynx through the binding of hpIgR (Kerr et al. 2006). PspC has been shown to protect an animal model against invasive pneumococcal disease by promoting a protective immune response against a peptide epitope (Chen et al. 2015). The fusion between PspC peptides and L460D pneumolysin can induce specific protection against pneumococcal infection. CbpA is a well-characterized choline-binding protein expressed by S. pneumoniae that contributes to the pneumococcus complement attack, plays a significant role in various disease states, and has been demonstrated to elicit a protective effect against pneumococcal infection in various animals models (Chen et al. 2015).
The gram-negative bacterium K. pneumoniae belongs to the Enterobacteriaceae family and causes infections of the nosocomial respiratory tract and urinary tract (Huang et al. 2015). Hypervirulent K. pneumoniae strains have been considered fundamental threats associated with increased mortality and morbidity. The enhanced virulence exhibited by these strains appears to be due to the overproduction of capsular polysaccharides (CPS) and anti-phagocytosis characteristics, leading to several infections, such as pneumonia and endophthalmitis (Huang et al. 2015). Several virulence factors have been associated with K. pneumoniae infections, including lipopolysaccharide (LPS), adhesins (OMPs, pilli, fimbriae), and exotoxins (Cryz et al. 1986; Yadav et al. 2005). The mechanism of host system infection is primarily governed by bacterial surface antigens, including LPS, CPS, and OMPS, which are commonly involved in developing immunity. A previous vaccine has been developed against K. pneumoniae based on LPS and CPS, but this vaccine only provides type-specific protection. A CPS-based vaccine has been developed that is immunogenic and nontoxic and covers at least 77 capsular antigen types (K-types) (Cryz et al. 1986). Outer membrane proteins (OMPs) are surface proteins acting as water-filled channels allowing hydrophilic molecules to cross the membrane. These proteins are required for proper cell function and growth, are associated with antibiotic resistance mechanisms, and serve as virulence factors during pathogenesis (Sun et al. 2014). Several studies have reported that the sequences of enterobacterial OMP family members are highly conserved (Lin et al. 2002). These represent convenient candidates for vaccine development against K. pneumoniae, and OMPs are essential for inducing robust cellular and humoral immune responses (Kurupati et al. 2006). OmpA is the primary OMP found in gram-negative bacteria and has been shown to elicit anti-tumor cytotoxic and humoral responses when accompanied by tumor antigens, displaying adjuvant properties (Jeannin et al. 2002). OmpA also provokes the inflammatory molecules to be expressed in a Toll-like receptor 2 (TLR2)-dependent manner in numerous cell types (Jeannin et al. 2003, 2000). OmpA has been shown to activate host immune responses that result in the elimination of K. pneumonia (Jeannin et al. 2002).
The use of computational tools and strategies represents an effective and rational method for developing an accurate and stable peptide-based vaccine against virulent antigens (Khan et al. 2018). Currently, various immunological datasets, advanced software programs, and genomic information are available for researchers to use in the prediction of potential pathogenic epitopes that could be utilized to construct effective subunit vaccines (Gupta et al. 2020). Subunit vaccines can trigger specific immunity against the target pathogen. Recently, immunoinformatics approaches have been applied to the design of vaccine candidates against multiple viruses and bacteria (Rafi et al. 2022a; Samad et al. 2022a, b). This method reduces the number of necessary experiments in vitro, which at the same time can be cost-efficient and time saving for successful vaccine development.
The objective of this study is to explore the proteomes of S. pneumonia and K. pneumoniae to identify potent antigenic proteins and screen potential cytotoxic T cell and helper T cell epitopes against major histocompatibility complex (MHC) alleles. The study aims to analyze the immunogenic, allergenic, and antigenic properties of the identified epitopes and design a vaccine by adding appropriate linker sequences and adjuvants. The designed vaccine will be validated through in silico methods, including the analysis of antigenicity, toxicity, immunogenicity, and allergenicity. The study also aims to investigate the binding affinity and the stability of molecular interaction between the vaccine and human pathogenic receptors through docking study and molecular dynamic simulation analysis. Finally, the immune simulation will be performed, and codon adaptation will be assessed to understand the immune response and expression profiling.
2 Materials and methods
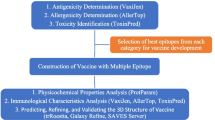
A visual representation of the study methodology is provided in Fig. 1, outlining the key steps and processes utilized in this research.

Schematic illustration of the study. The overall method is displayed in several steps: the selection of protein sequences from Streptococcus pneumoniae and Klebsiella pneumoniae, including the prediction of helper T lymphocyte (HTL)-, cytotoxic T lymphocyte (CTL)- and interferon (IFN)-γ-inducing epitopes in the target proteins; the vaccine construction and feature assessment; molecular docking with Toll-like receptor (TLR)4 and TLR2 immune receptors; molecular dynamics (MD) simulations to evaluated the real dynamic stability of the vaccine and receptor complexes; and immune simulation and codon optimization to understand the translation and immunogenicity of the designed vaccine
2.1 Sequence retrieval, antigenicity prediction, and structural analysis
Outer membrane proteins (OMPs) or surface proteins are typically suitable candidates for the design of peptide-based vaccines (Post et al. 2017). We selected PspA, PspC, and CbpA from S. pneumonia and OmpA and OmpW from K. pneumoniae as the basis for vaccine design. We adopted the National Center for Biotechnology Information (NCBI) (publicly available database) for retrieving the amino acid sequences of our target proteins in FASTA format. In the following step, the sequences were subjected to the VaxiJen v2.0 tool for identifying antigenicity relative to a threshold value (0.4) (Doytchinova and Flower 2007). To determine whether a targeted bacterial protein is suitable for use in the design of a peptide vaccine, several physicochemical and structural properties of the target protein should be evaluated. The ExPASy ProtParam tool and the SOPMA server were used for these purposes (Gasteiger et al. 2005; Geourjon and Deleage 1995).
2.2 Prediction of T cell epitope
2.2.1 CTL epitopes
NetCTL.1.2 server (http://www.cbs.dtu.dk/services/NetCTL/) was hired to predict the 9-mer CTL epitopes, where the thresholds set at 0.05, 0.15, and 0.75 are for TAP transport efficiency, C-terminal cleavage, and epitope recognition, respectively (Larsen et al. 2005, 2007). The NetCTL 1.2 server uses an artificial neural network system that includes the TAP transport efficiency and proteasomal C-terminal cleavage for predicting MHC class I-binding peptides, in which the TAP score is estimated using a weight matrix. Epitope prediction was performed based on commonly occurring human leukocyte antigen (HLA) allele 12 class I supertypes in the human population. The predicted epitopes were subjected to the Immune Epitope Database (IEDB) consensus method for assessing the binding affinity with the following MHC class I alleles: A-0101, A-0201, A-0301, A-2403, B-0702, B-3501, B-4403, B-5101, B-5401, and B-5801 (Kim et al. 2012). The MHC class I epitopes identified in this study are likely to cover over 90% of the global community (Moise et al. 2009). Those epitopes consensus scores less than 2 in the IEDB server were considered for additional analysis.
2.2.2 HTL epitopes
The 15-mer HTL was predicted via NetMHCIIpan 3.2 server (http://www.cbs.dtu.dk/services/NetMHCIIpan-3.2/) which identifies the major histocompatibility complex class II DRB1 alleles: 01:01, 03:01, 04:01, 07:01, 08:01, 08:03, 10:01, 11:01, 12:01, 13:01 13:02, 14:01 and 15:01 (Jensen et al. 2018; Wang et al. 2010). The NetMHCIIpan 3.2 (artificial neural network based-server) predicts the human HLA-DQ, HLA-DP, HLA-DR, and mouse MHC class II binding peptides, and each of the outputs was assigned a percentile rank by the server. Based on the assigned percentile rank, the epitope sequences were divided into strong binders (SBs), weak/intermediate binders (WBs), and non-binders (NBs), at threshold values of 2%, 10%, and greater than 10%, respectively. The MHC class II epitopes identified in our analysis are projected to be found in more than 95% of the global community (Moise et al. 2009).
2.3 Overlapping, promiscuous, and nontoxic T cell epitope prediction and assessment
Promiscuous epitopes have high binding affinities with multiple MHC alleles and can generate effective immune responses in the host. Therefore, T-cell epitopes with multiple HLA allele binding capacities were identified for further analysis. These epitopes, which consist of both HLA class II and class I peptide sequences and are able to promote the activation of both helper T cells and cytotoxic T cells, are referred to as overlapping epitopes (Chauhan et al. 2019). We used the IEDB server to identify the overlapping capacities of identified epitopes. Based on the promiscuity and overlapping nature, epitopes were nominated for vaccine construction. Nontoxic and nonallergenic epitopes are identified using the ToxinPred and AllerTOP v2.0 servers, respectively.
2.4 Prediction of IFN-gamma-inducing epitopes
Interferon-gamma, a cytokine that takes an important part in the activation of macrophages as well as natural killer cells, which exert immunity against bacterial and viral infection. The 15-mer IFN-γ-inducing epitopes in our target protein were predicted by submitting CD4+ epitope sequences to the IFNepitopes server (http://crdd.osdd.net/raghava/ifnepitope/) (Dhanda et al. 2013). This web server predicts and designs peptides that can provoke IFN-γ secretion from helper T cells based on the query sequences. The server was set up using a training dataset that includes 10,433 experimentally validated MHC class II binding peptides capable of activating CD4+ T cells (Dhanda et al. 2013). This web tool screens overlapped INF-gamma sequences derived from the query proteins, which are assigned numerical values.
2.5 Population coverage, peptide conservation, and human homology prediction
IEDB conservancy analysis tool (http://tools.iedb.org/conservancy/) was used to analyze the degree of the conservancy of the epitopes (Bui et al. 2007). The IEDB population coverage tool (http://tools.iedb.org/population/) was employed to evaluate the coverage of the population for the selected epitopes (Bui et al. 2006). The coverage of each epitope was assessed against the MHC I and MHC II binding alleles using the combined dataset. The epitopes were evaluated for the BLASTP comparison against the human proteome to reduce the possibility of eliciting an autoimmune response, and those sequences that were highly similar to any human protein sequences were removed.
2.6 Docking analysis between epitopes and MHC alleles
The binding affinity of the selected T cell epitopes was evaluated, molecular docking was performed with HLA alleles. Initially, the epitope sequences were analyzed by the PEP-FOLD v.3.5 server (Lamiable et al. 2016). The PEP-FOLD server was developed based on the taboo sampling backtrack algorithm, which generates a de novo protein structure based on sOPEP energy values using 200 simulations. The 3-dimensional (3D) crystal structures of HLA alleles, including HLA-A0201 (MHC class I) and HLA-DRB1-0101 (MHC class II), were retrieved from the RCSB PDB database using the PDB codes 1QEW and 2G9H, respectively. We considered to include the two HLA alleles in docking simulation, both considered as most common alleles and are deemed to be found in over 95% of the global community. Before the docking study, any ligands and water molecules from the PDB structure were eliminated. The epitopes were docked with each HLA allele using the ClusPro v.2.0 server (Comeau et al. 2004), where the epitope and the allele were considered to be the ligand and receptor, respectively. The ClusPro v.2.0 docking tool estimates the binding energy of the docked complex formed between the epitope and the allele, based on the desolvation contribution, shape complementarity, and decoys as reference states. The best-docked cluster was nominated on the basis of the lowest energy score and visualized by PyMOL.
2.7 Vaccine construction
Highly promiscuous, non-allergic, non-toxic, antigenic, and immunogenic epitopes were considered for designing the final vaccine sequence. The best CTL epitopes were combined via AAY linkers, and GPGPG linkers were added to link HTL- and IFN-γ inducing epitopes together. To generate long-lasting immunity, an EAAAK linker was utilized to link the cholera toxin B (CTB) as an adjuvant to the N-terminus of our vaccine structure.
2.8 Physicochemical evaluation of the vaccine
The antigenicity of the vaccine structure was estimated by the VaxiJen v2.0 web server at a threshold value of 0.4 (Doytchinova and Flower 2007). Allergenicity was determined using the AllergenFP v1.0 server and AllerTOP v2.0 (Dimitrov et al. 2013, 2014). To determine immunogenicity, we used the IEDB Class I immunogenicity prediction module (Calis et al. 2013). The identification of physicochemical properties is essential for the synthesis of the designed vaccine. Therefore, these properties of the construct, such as the aliphatic index (AI), estimated half-life, grand average of hydropathicity (GRAVY), instability index (II), theoretical isoelectric point (PI), and molecular weight (MW) were assessed using the ExPASy tool (Gasteiger et al. 2005). Moreover, the TMHMM and SignalP 4.1 servers were utilized to identify the existence of signal peptides and transmembrane helices, respectively, in the finalized vaccine structure (Krogh et al. 2001; Petersen et al. 2011).
2.9 Vaccine’s structural evaluation
The PSIPRED web server (http://bioinf.cs.ucl.ac.uk/psipred/) was adopted to examine the secondary structure of the linear vaccine (McGuffin et al. 2000). Whereas, the 3D structure of the final vaccine was predicted by the trRosetta server (https://yanglab.nankai.edu.cn/trRosetta) (Yang et al. 2020). The best 3D structure for the vaccine was refined by using the GalaxyRefine web-based tool (http://galaxy.seoklab.org/) (Heo et al. 2013), which relaxes the structure through MD simulations and uses CASP10 refinement techniques. The finalized model was validated by ProSA-web (Wiederstein and Sippl 2007). ProSA-web predicts the overall features of the model based on the Z-score and ERRAT values. Finally, the SWISS-MODEL workspace (https://swissmodel.expasy.org/assess) was implemented to determine the overall structure quality by analyzing the Ramachandran plot (Arnold et al. 2006).
2.10 B cell epitopes prediction
B-cell epitopes in the validated 3D vaccine structure were determined by the ElliPro web tool (http://tools.iedb.org/ellipro/), using the values set by default (maximum distance: 6 Å and minimum score: 0.5) (Ponomarenko et al. 2008). The ElliPro tool runs three algorithms that estimate the protein as an ellipsoid, compute the protrusion index of residues, and cluster neighboring residues based on their protrusion index values. For each of the output B cell epitopes, the ElliPro score described the protrusion index. A protrusion index score of 0.9 means that 90% of the protein residues are involved in the ellipsoid, whereas 10% of the residues lie outside of the ellipsoid. The ElliPro web server provides the best performance and is considered to be the preeminent algorithm among those currently available (Ponomarenko et al. 2008).
2.11 Disulfide engineering for vaccine stability
Disulfide bonds are a type of covalent interaction that provides protein stability by maintaining a precise geometric conformation. In the protein structure, disulfide engineering represents a unique strategy for creating a disulfide bond. We followed disulfide engineering, where the final structure of the vaccine was subjected to the Disulfide by Design 2.0 (DBD2) server (http://cptweb.cpt.wayne.edu/DbD2/index.php) to create disulfide bonds (Craig and Dombkowski 2013). Residue pairs were nominated based on energy (< 2.2) and χ3 values (between − 87 and + 97).
2.12 Molecular docking and refinement
The vaccine and immune cell receptor interaction are required for generating a stable immune response. To examine the interaction patterns, the structures of TLR2 (PDB: 2Z7X) and TLR4 (PDB: 3FXI) were obtained from the RCSB PDB database. Blind docking of the vaccine with these receptors was accomplished separately utilizing the PatchDock server (https://bioinfo3d.cs.tau.ac.il/PatchDock/) (Schneidman-Duhovny et al. 2005). The server builds and predicts the possible complex that forms between the vaccine and the TLR using three algorithms, including surface patch matching; shape representations; and scoring and filtering. The PatchDock server estimates the surface fix coordinating scores and separating scores and further PatchDock server portrays the atomic shape for protein–protein interactions. While, the FireDock server (Andrusier et al. 2007), which predicted an optimal complex using energy functions was utilized further to refine the structures of the best-docked complexes produced by the PatchDock server. The top-ranked complexes were nominated based on the lowest binding energy score. Finally, molecular interactions between the vaccine and receptors were visualized using the PDBsum server (http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/Generate.html) (Laskowski et al. 2018).
2.13 Molecular dynamics (MD) simulation
MD simulation of vaccine/receptor systems was implemented using the GROMACS package (Van Der Spoel et al. 2005). The topologies of complexes were built based on the CHARMM27 force field. The TIP3 water molecules were added, followed by energy minimization (Rafi et al. 2022c). For neutralizing the vaccine/receptor systems we added 0.15 M concentration of NaCl at a temperature of 300 K (Krieger et al. 2006). We evaluated the long-range electrostatic interactions by implementing the particle-mesh Ewald (PME) approach. A simulation box size was established at 20 Å. The MD simulation temperature was adjusted using a Berendsen thermostat, and energy minimization was performed using the simulated annealing method. The normal simulation time step (1.25 fs) was maintained, and simulated trajectories were set at 100-ps intervals for analysis (Krieger and Vriend 2015). Finally, MD simulations of the vaccine/TLR4 and vaccine/TLR2 complexes were run for 500 ns, and root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) were estimated to understand the conformational variations and stability of the complex (Rafi et al. 2022b). RMSD is calculated using the following formula:
where Ri represents the vector that links the positions of atom i (out of N atoms) in the reference snapshot to the current snapshot after optimal superposition.
The RMSF of atom i with j running from 1 to 3 for the x, y, and z coordinate of the atom position vector P and k running over the set of N evaluated snapshots is given by the following formula:
2.14 Immune simulation
For generating an in silico immune simulation profile for the designed vaccine, we hired the C-ImmSim server (https://kraken.iac.rm.cnr.it/C-IMMSIM/) at default parameters (Rapin et al. 2011). This server uses two methods: machine learning and the position-specific scoring matrix which predicts the immune interactions and immune epitopes. It simulates three different mammalian anatomical regions simultaneously, the tertiary lymphatic organs, the thymus, and the bone marrow. To assess immune simulations, three injections containing 1000 vaccines were administrated, with time steps set at 1, 84, and 168 (each time step represents 8 h); the doses were administered 4 weeks apart. We also tested 12 injections 4 weeks apart to simulate the repeated detection of antigens in a common endemic zone. Simpson’s Diversity Index, D, a measurement of diversity, was determined from the plot.
2.15 Codon adaptation and in silico cloning
for codon optimization and the translation of the vaccine in a suitable expression vector, we employed the JCat tool (http://www.jcat.de/) (Grote et al. 2005). The tool optimizes the ribosomal binding site and avoids the termination of rho-independent transcription. The desirable vaccine was expressed in an E. coli host system, and generated cDNA sequences, using an optimized CG content percentage and CAI value. In the next step, the optimized sequence of the vaccine was incorporated into the pET28(+) vector through the insertion of XhoI and NdeI restriction sites at the N- and C-terminal ends, respectively, by utilizing the SnapGene tool (Ali et al. 2017).
3 Results
3.1 Antigenicity prediction
To construct an epitope-based vaccine candidate, the complete sequences of the target proteins PspA, PspC, CbpA, OmpA, and OmpW were obtained from the NCBI database using accession numbers WP_061633550.1, SHD60938.1, ABI34558.1, AKN20218.1, and KXA22008.1, respectively. VaxiJen v2.0 server reported all of the retrieved sequences as being antigenic (Table 1). The physicochemical properties, such as the MW, theoretical PI, GRAVY, and index values, and the secondary structural characteristics of these sequences were analyzed (Table 1, Fig. S1). The MWs of the target proteins ranged from 24.3 to 83.4 kDa, and the instability index values were computed to be below 40, indicating that these sequences were stable. All of the targeted protein sequences returned negative GRAVY values, indicating their hydrophilic natures and the ability to engage in interactions in an aqueous environment. These data indicate that the target proteins represent suitable candidates for vaccine design to generate an appropriate immune response against S. pneumoniae and K. pneumoniae.
3.2 Prediction of T cell epitopes and assessment
For the generation of long-lasting immunity, CTL epitopes prime cytotoxic T cells to eliminate circulating pathogens, and HTL epitopes prime helper T cells, which are responsible for both cellular and humoral immune responses. Therefore, effective vaccine development should include both receptor-specific T-cell epitopes. In this study, 152 unique CTL (9-mer) epitopes and 66 unique HTL (15-mer) epitopes were predicted from the target protein by the NetCTL.1.2 and NetMHCIIpan 3.2 servers, respectively (Tables S1, S2). While the binding affinities of the epitopes with multiple MHC alleles were assessed by The IEDB consensus method. The epitope sequences were then subjected to numerous immune filters to examine immunogenicity, antigenicity, nonallergenicity, and nontoxicity, and, finally, promiscuous CTL and HTL epitopes were selected (Tables 2 and 3).
3.3 Vaccine construction and structural evaluation
The benchmarks for the epitopes used to construct a linear vaccine were the following characteristics: (a) immunogenic; (b) promiscuous; (c) coverage of a large proportion of the human population; (d) nonallergenic, nontoxic, and antigenic; (e) composed of overlapping CTL and HTL epitope sequences; and (f) no overlap found with any human protein. Based on the aforementioned properties, the final vaccine sequence comprised 11 CTL-, 12 HTL-, and 5 IFN-γ-inducing epitopes (Tables 2 and 3; Tables S3 and S4); for the prevention of junctional epitope formation, AAY and GPGPG linkers were included between epitopes. Further, EAAAK was the linker to attach the CTB adjuvant to the N-terminus of the vaccine sequence (Fig. 2). The vaccine contained 578 amino acid residues, with an MW of 59.8 kDa (Supplementary Materials SM1). The secondary structure evaluation showed that the vaccine was comprised of 15.57% alpha-helices, 20.76% beta-strands, and 63.66% coiled coils (Fig. 3). The 3D structure of the vaccine was predicted by the trRosetta online server, whereas the 3D model was refined via GalaxyRefine web tool. Among all refined vaccine structures, model 2 was selected for better quality (Fig. 4A). For structural validation, the finalized model was uploaded to ProSA-web, which returned a Z-score of − 7.17, which was within the score range of comparably sized wild-type proteins (Fig. 4B). Analysis of the Ramachandran plot indicated that 93.58% of the residues were within a preferred region, 1.39% within an outlier region, and 0.49% of the residues were rotamer outliers (Fig. 4C). The ERRAT value is for predicting the quality of protein structure (if the ERRAT score is more than 50, this indicates a good quality protein structure) (Messaoudi et al. 2013). The ERRAT value of the vaccine model was identified to be 54.74 (Fig. S2).

Schematic arrangement of the finalized vaccine construct. The vaccine was 578 amino acids long, comprising helper T lymphocyte (HTL)-, cytotoxic T lymphocyte (CTL)-, and interferon (IFN)-γ-inducing epitopes. EAAAK linker (black) at the end of the N-terminus used to link an adjuvant (green). AAY linkers (green) were used to attach CTL epitopes. And GPGPG linkers (yellow) utilized to link HTL and IFN-γ epitopes

Secondary structure features of the designed multi-epitope vaccine candidate. The alpha-helices, beta-strands, and coiled-coil residues are denoted in pink, yellow, and gray colors, respectively. The secondary structure evaluation revealed that the vaccine construct constitutes 15.57% alpha-helices, 20.76% beta-strands, and 63.66% coiled coils

A 3D structure of the final vaccine. B Structure validation, with a Z-score of − 7.17, as predicted using ProSA-web. C Ramachandran plot analysis using the SWISS-MODEL workspace, showing 93.58%, 1.39%, and 0.49% in the favored, outlier, rotamer outlier regions, respectively
3.4 Physicochemical, immunogenic, and antigenic assessments of the vaccine
Immunogenicity refers to the capacity of inducing cellular and humoral immune responses, whereas the capacity to identify a particular antigen molecule is referred to as antigenicity (Ilinskaya and Dobrovolskaia 2016). Thus, a suitable vaccine candidate must be antigenic and also be immunogenic. To predict the immunogenicity of the vaccine construct, the IEDB Class I Immunogenicity Web Server was used, and a score of 5.40534 was predicted; a higher score denotes a better chance of stimulating an immune response. The antigenicity score of the vaccine was reported as 0.7919 (VaxiJen v2.0 > 0.4 threshold). To confirm the vaccine as non-allergenic when presented to the body, allergenicity was checked using the AllergenFP v1.0 and AllerTOP v2.0 servers, and the results of both analyses showed that the vaccine construct was nonallergenic. Several physical and chemical characteristics must be analyzed for a vaccine construct, which were predicted using ExPASy (Supplementary Materials SM2). The AI and theoretical PI scores of the vaccine were reported as 77.32 and 9.51, respectively, which indicates a basic nature and high thermostability. The computed half-life was evaluated as 30 h in mammalian reticulocytes, 10 h in E. coli, and. higher than 20 h in yeast. The GRAVY score was − 0.079, which indicates the vaccine candidate is hydrophilic and confirms its ability to interact with water. The instability index value of the vaccine was 24.01, which classified the protein as stable. Generally, an instability index value less than 40 indicates stability. Vaccine structure was identified as signal peptide-free or transmembrane-able helix-free, indicating the absence of potential expression difficulties during production (Figs. S3 and S4).
3.5 Prediction of B cell epitopes
B lymphocytes are of paramount importance in the secretion of antibodies, providing long-term immunity (Chan et al. 2014). ElliPro server was used with default parameters to evaluate the presence of B cell epitopes in the vaccine, which identified 15 continuous (linear) and 5 discontinuous (conformational) epitopes (Tables 4 and 5; Figs. S5 and S6).
3.6 Population coverage
Successful vaccine development should consider the global HLA allele distribution. The population coverage of the designed vaccine structure was assessed by the population coverage tool of IEDB. The population coverage analysis exhibited that the T-cell epitopes included in our study covered 77.1% of the global human population (Table 6). The epitopes were reported to be associated with 88.85%, 64.07%, and 78.72% coverage in Europe, Southwest Asia, and North America, respectively (Table 6).
3.7 Disulfide engineering
Disulfide bonds (DB) are needed to provide conformational stability to folded proteins. For the stabilization of the vaccine structure, disulfide engineering was performed following vaccine construction using the DBD2 server. The server displayed 72 pairs of residues susceptible to disulfide engineering. Only four pairs of residues were selected according to energy and χ3 values. The energy values of the finalized residues were less than 2.2, and the χ3 scores ranged between − 87 and + 97 degrees. Thus, 8 mutations were generated at the residue pairs: Ala144–Pro195, Gly235–Lys252, Ala412–Arg434, and Val446–Ala469 (Fig. 5, Table S5).

Disulfide engineering in the finalized vaccine structure, showing a total of 4 mutated residues pairs in magenta color
3.8 Molecular docking analysis
To develop a stable immune response, a vaccine–receptor interaction is necessary. TLRs can detect pathogens and play significant roles in innate immunity. The binding affinities between the vaccine and the TLR4 and TLR2 immune receptors and the model of vaccine/TLR4 and TLR2 interactions were predicted by employing the protein–protein docking approach. The protein–protein docking approach was accomplished using the PatchDock server, which produced the vaccine and the TLR complexes between the vaccine and the TLR based on electrostatic complementary and the protein surface geometry. The obtained results were forwarded to the FireDock server, which refined the best complexes and exhibited the top 10 docked clusters. Among the top 10 generated complexes by the PatchDock server, solution 8 was reported as the best model with the following values: global energy (− 46.30), attractive van der Waals (VdW) energy (− 38.51), repulsive VdW energy (9.73), hydrogen bond (HB) energy (− 4.89), and atomic contact energy (ACE, − 4.89; Fig. 6A, Table S6, Fig. S7). Similarly, in the docking study examining the interaction between TLR2 and the vaccine, the global energy (− 12.89), attractive VdW energy (− 34.30), repulsive VdW energy (16.95), HB energy (1.49), and ACE (15.56) values for solution 9 were better than those for the other complexes (Fig. 7A, Table S7, Fig. S8). Docking results revealed that the vaccine–TLR4 and vaccine–TLR2 interactions were strong. The application of the PDBsum server provided insight into the interface residues and the intermolecular binding forces involved in the vaccine–TLR4 and vaccine–TLR2 interaction systems. Twelve amino acids in the vaccine interface interacted with 15 amino acids in TLR4 (Fig. 6B and C). Similarly, 24 interface residues in TLR2 interacted with 19 interface residues in the vaccine (Fig. 7B and C).

Interaction pattern of Vaccine/Toll-like receptor 4 (TLR4). A visualization of the interaction was performed using the PDBsum server. A Docked complex of designed vaccine (violet) and the TLR4 immune receptor (hot red). The predicted global energy value of the docked complex by was − 46.30 kcal/mol. B All interacting residues of the designed vaccine (Chain A) and TLR4 (Chain B). C Diagrammatic interaction of the vaccine/TLR4 complex

Interaction pattern of Vaccine/Toll-like receptor 2 (TLR2). A visualization of the interaction was generated using the PDBsum server. A Docked complex of designed vaccine (violet) and the TLR2 immune receptor (hot red). B All interacting residues of the designed vaccine (Chain A) and TLR2 (Chain B). C Diagrammatic interaction between vaccine-TLR2
The vaccine established a total of 3 HBs with TLR2: 249–522, 270–522, and 271–523. The interaction analysis showed that Ala249 and Ser522 generated an HB at a distance of 2.39 Å. Similarly, Ala270–Ser522 and Asn271–Gln523 developed HBs at distances of 1.45 Å and 2.88 Å, respectively. TLR4 formed 2 HBs [Chain A (Vaccine)–B (TLR4): 380–130 and 394–603] with the designed vaccine, including HBs between Pro380–Lys130 and Lys394–Glu603 at distances of 2.23 Å and 2.16 Å, respectively. The HBs in the vaccine–TLR4 and vaccine–TLR2 systems ranged from 2 to 3 Å, suggesting strong interactions. A strong interaction between our designed vaccine and TLR2 and TLR4 would indicate that the vaccine can induce immune pathway activation.
The selected T cell epitopes docked individually with commonly occurring human leukocyte antigen alleles: HLA-A0201 (MHC class I) and HLA-DRB1-0101 (MHC class II). Each epitope–allele cluster displayed a different energy score (Table S8). Molecular interaction patterns between each epitope and the MHC alleles were visualized, as shown in Figs. S9 and S10. The binding affinities (weighted scores) of the CTL epitopes for HLA-A0201 ranged from − 500 and − 749.1 kcal/mol, whereas the maximum and minimum binding affinities (weighted scores) of the HTL epitopes were − 1030.9 kcal/mol and − 621.2 kcal/mol, respectively. Based on this analysis, the epitopes displayed suitable binding efficiency with HLA alleles for use in a vaccine construct.
3.9 Molecular dynamics (MD) simulation
MD simulation is an effective strategy for examining the dynamic stability of a protein. The MD simulation study of the vaccine coupled with either TLR4 or TLR2 revealed that both complexes initially experienced a sharp increase in the RMSD values of backbone atoms within 30 ns (Fig. 8A). Thereafter, a sharp decrease was observed in the RMSD value of the TLR4/vaccine complex, which reached approximately 0.91 nm at 44 ns. The RMSD then increases again, dramatically, reaching 1.2 nm at 50 ns, followed by the stabilization of the RMSD profile. However, throughout the entire simulation time (500 ns), the RMSD value of the TLR2–vaccine complex did not display an over RMSD peak, confirming the structural integrity of the complex. The complex formed between TLR4 and the vaccine had slightly lower RMSD values than the complex formed between the vaccine and TLR2, suggesting the relatively more stable nature of the TLR4-containing complex. However, overall, both systems fluctuated similarly after 52 ns.

MD simulation of the complexes formed between the vaccine and Toll-like receptors (TLRs) over 500 ns. A RMSD plot. B RMSF of TLR2 during the MD simulation when bound to the designed vaccine. C The RMSF of TLR4 during the MD simulation when bound to the designed vaccine. D The RMSF plot for the vaccine when bound to TLR2 and TLR4
RMSF can provide insights into the dynamic behaviors and flexibility of amino acid residues. For the vaccine–TLR2 complex, Ser25 (beta-turn), Lys46 (beta-turn), Lys70 (beta-turn), Thr113 (loop), Asn137 (helix strand), Glu414 (helix strand), Gln463 (helix strand), Asp489 (loop), Arg510 (beta-turn), and Pro544 (helix strand) residues had more flexibility, revealing stronger RMSF profiles than the other residues in the complex (Fig. 8B). The residues Glu27 (beta-turn), Leu52 (loop), Ser76 (beta-turn), Lys541 (helix strand), His566 (loop), Gln588 (helix strand), and Ile625 (gamma turn) in the TLR4–vaccine complex showed less integrity and more mobility compared with other residues (Fig. 8C). The dynamic behavior of the vaccine was compared when bound to TLR2 and TLR4. The regions Met1 to Met38, Ala39 to Val83, Cys87 to Ala132, Pro302 to Leu367, Val387 to Arg455, Glu536 to Gly544, and Asp552 to Ala572 showed lower fluctuations when the vaccine was bound to TLR4 than when the vaccine was bound to TLR2. The RMSF values for most regions of the vaccine were lower when bound to TLR4, suggesting a more stable interaction (Fig. 8D).
3.10 Immune simulation
We calculated the immune simulation using the C-ImmSim server, which displays the immune response increased during the initiation of a secondary response. The primary response suggested a high level of immunoglobulin M, whereas the tertiary and secondary responses exhibited noticeable increments in the levels of IgM, IgG + IgM, and IgG1 + IgG2 antibodies, and an increment was noticed in the B-cell population, with corresponding mitigation in antigen concentration (Fig. 9A and B; Fig. S11A and B). Moreover, the cytotoxic and helper T-cell populations revealed a high response, which corresponded with memory cell development (Fig. 9C and D; Fig. S11C). This result specifies the improvement of immune memory, which subsequently increases antigen concentrations following consequent disclosures (Fig. 9D). A high level of interferon-gamma and interleukin-2 was produced later in the injection, and a low Simpson’s Diversity Index (D) revealed low diversity (Fig. 10). Enhanced macrophage activity, whereas a consistent dendritic cell activity was retained (Fig. S11G and I). This completes the evidence that the examined susceptibility peak is effectively due to α-relaxation, in the same sense as with any glass-forming liquid.

Immune simulation profile of the vaccine peptide. A Immunoglobulins and antigen; B B cell population; C Cytotoxic T cell population per state; D Helper T cell population per state

Cytokine production and interleukin expression provoked by three vaccine doses administered at 4 weeks apart. The cytokine levels after vaccination doses are shown in the main graph. The insert plot represents interleukin-2 levels. The Simpson’s Diversity Index, D, is denoted as the dotted line, which is a degree of diversity. A smaller D value designates lower diversity
3.11 In silico cloning and codon adaptation
In silico cloning, experiment was accomplished to examine if the vaccine is capable of expressing into the E. coli host system. To enhance the protein expression of the vaccine, the codon has to be optimized. The JCat tool was applied to produce a cDNA sequence, resulting in a 1467-nucleotide sequence that could be used for vaccine expression (Supplementary Material SM3). The codon adaptation index (CAI) score and the GC contents of the designed vaccine sequences were 0.9699 and 53.44%, respectively, which indicates a proficient and potentially stable expression capability in the E. coli K-12 strain (Fig. 11). When a GC content ranged from 30 to 70% and a CAI value over 0.8 are indicating that vaccine is a good protein expression in a host. In addition, the SnapGene tool was used to insert the nucleotide sequences into the plasmid vector pET28(+) by recombination using an appropriate restriction site (Fig. 12).

Codon optimization of the vaccine sequence. The GC content and CAI score of the optimized codon were 55.24% and 0.96, respectively, as shown in the graph

In silico cloning of the vaccine into a plasmid vector. The red portion specifies the sequences of the designed vaccine, and the black portion represents the pET28a(+) plasmid vector backbone. SnapGene tool was utilized to visualize the in silico cloning
4 Discussion
S. pneumoniae and K. pneumoniae are associated with various infections, including meningitis, bacteremia, and pneumonia, which are common causes of infant mortality. Currently available vaccines are unable to generate appropriate immunity due to high bacterial serotype specificity (Tumbarello et al. 2015; Von Mollendorf et al. 2017). A peptide-based multi-epitope vaccine might represent a promising alternative approach to combat bacterial infections. Based on analyses of the S. pneumoniae genome, several epitopes were found to be probable vaccine candidates, a few have been examined in clinical trials (Darrieux et al. 2015). PspA is a common virulence factor found in almost all pneumococcal strains. A capsular polysaccharide-protein conjugated to PspA and pneumococcal surface antigen A (PsaA) was able to induce efficient protection against the nasopharyngeal carriage of pneumococci (Meng et al. 2009). Wu et al. reported the protection of mice against systemic infection following intranasal immunization with PspA (Wu et al. 1997). Verhoeven et al. explored a multivalent recombinant vaccine against CbpA, PLY, and PhtD, which displayed high efficiency in an infant mouse model (Verhoeven et al. 2014). Outer membrane proteins are highly conserved among all K. pneumoniae serotypes and have a robust immune response nature. Vaccination with OMPs can induce a potent Th17 response against a broad spectrum of K. pneumoniae strains (Chen et al. 2011). According to Babu et al., targeting OMPs (OmpA and OmpK36) using antibodies acquired against these molecules could serve as a significant bactericidal defense mechanism against bacterial elimination from the system (Babu et al. 2017).
Modeling played a crucial role in the development of an immunogenic multi-epitope subunit vaccine against S. pneumoniae and K. pneumoniae in this study. Various computational approaches and immune filters were employed to design the vaccine, and physicochemical and antigenic profiles were used to evaluate its immunogenicity and safety. As part of this work, we applied a cutting-edge immunoinformatics approach, the sequences of the most recognizable antigenic virulence factors in S. pneumoniae, including pneumococcal surface proteins (PspA and PspC) and choline-binding protein A (CbpA), and K. pneumoniae, including OMPs (OmpA and OmpW), were targeted. The vaccine was designed using several computational tools to induce both cell-mediated and adaptive immunity. By using antigenic peptide fragments, an epitope-based vaccine candidate can generate a similar immune response to that against a whole genome or a large protein without promoting allergenic reactions in the host (Jabbar et al. 2018; Sette and Fikes 2003). The selection of the proper antigenic epitope is very important for vaccine development (Yin et al. 2016). Potential epitopes (CTL-, HTL-, and IFN-γ-inducing) in target proteins from S. pneumoniae and K. pneumoniae were identified using several immune filters to identify epitopes that are antigenic, immunogenic, able to interact with multiple HLA alleles (promiscuous), feature 100% conservancy among related protein sequences, share no overlap with the human proteome to reduce autoimmunity, and are incapable of inducing any allergic reactions. After various in silico analyses, a multi-epitope vaccine (CTL-, HTL-, IFN-γ-inducing) was constructed with an adjuvant (CTB; Fig. 2). The CTL epitopes were combined via AAY linkers, and GPGPG linkers were added to link HTL- and IFN-γ inducing epitopes together. The linker was developed based on specialized amino acid sequences which can improve vaccine construction. The AAY and GPGPG linker induces the binding immune response and immunogenicity of cytotoxic T lymphocytes and helper T lymphocytes epitopes, respectively, and also provide an impressive variety through simplified vaccine construction (Meza et al. 2017; Nishita et al. 2017). The EAAAK linker improves the bioactivity of fused protein which is added between vaccine and adjuvant for proper separation (Arai et al. 2001). The multiple epitopes (cytotoxic T lymphocytes, helper T lymphocytes, and IFN-gamma) and adjuvant are synchronized in such a way have been displayed to antigen-specific immune response stimulation (Bazhan et al. 2019; Foroutan et al. 2020), the adjuvant is attached in the N-terminal of the construct because binding of it with TLR displays enhanced results in producing immune-response whereas TLRs associated to the activation of the humoral and cellular immunity (Mizel and Bates 2010). Chen et al. reported that multivalent pneumococcal protein vaccines containing PspA and CbpA epitopes stimulated strong and broad protection against sepsis, meningitis, and focal pneumonia in various animal models (Chen et al. 2015). The use of an adjuvant can promote the development of long-lasting immunity and increase the intrinsic immunogenicity of an antigen. CTB, a nontoxic subunit of cholera toxin, showed a particular affinity for the ganglioside GM1 receptor on gut epithelial cells and other corresponding cell types, including antigen-presenting cells, like macrophages, B cells, and dendritic cells (Stratmann 2015). Kuipers et al. reported that CTB reduces the pneumococcal load in the nasopharynx, dependent on the activation of the macrophages, mucosal T cells, and caspase-1/11 inflammasome (Kuipers et al. 2016). In another study, Wiedinger et al. demonstrated that the immunization of CTB with PspA induced a cellular response that mediates significant defense against S. pneumoniae in mice (Wiedinger et al. 2017). The AllerTOP v2.0 and AllergenFP v1.0 servers were used to assess the allergenicity of the designed vaccine, which both indicated that the vaccine construct was nonallergenic. Various chemical and physical properties of the vaccine were assessed using the ExPASy ProtParam tool. The instability index value was 24.01, which was lower than 40, indicating a stable protein. The theoretical PI value was 9.51, indicating a basic nature, and the AI value was 77.32, indicating a thermostable product. The GRAVY value was calculated to be − 0.079, suggesting a hydrophilic product able to interact with an aqueous environment. Similarly, Foroutan et al. designed an epitope-based vaccine against Toxoplasma gondii, and the experimental validation of their candidate vaccine indicated that their vaccine could generate a strong immune response in mice (Foroutan et al. 2020). The physical and chemical parameters of our vaccine candidate indicate II and AI values that are more favorable than the candidate vaccine designed and tested by Foroutan et al. Dorosti et al. used PspA, CbpA, PiuA, and PhtD antigens as the basis for the development of a multi-epitope vaccine against S. pneumoniae, and they computationally evaluated the physicochemical and immunogenic characteristics of their vaccine candidate (Dorosti et al. 2019). The antigenic properties and various physicochemical values of our vaccine candidate are also more favorable than those reputed by the vaccine candidate developed by Dorosti et al. The analysis of the Ramachandran plot for our candidate vaccine structure demonstrated 93.58%, 1.39%, and 0.49% of amino acids in the favored, outlier, rotamer outlier regions, respectively. The Z-score of the vaccine model was − 7.17, signifying that the vaccine product is consistent with the range of Z-scores established for proteins with structures resolved using nuclear magnetic resonance and X-ray crystallographic experiments (Wiederstein and Sippl 2007). The structural features of our vaccine are better than those described for previously designed multi-epitope vaccines against these two bacteria. In one study, Dar et al. constructed a vaccine against K. pneumoniae (Dar et al. 2019), and the Z-score of their vaccine model was more positive than the Z-score for our vaccine model.
According to the findings of the study, the implementation of the disulfide engineering approach proved to be a successful strategy in stabilizing the vaccine structure. The success of this technique emphasizes its potential to be used in other vaccine candidates for enhancing their structural stability. The study results underline the significance of considering conformational stability in the design of vaccines to achieve efficacy against pneumococcal infections. By improving the structural stability of the vaccine using disulfide engineering, its immunogenicity can be enhanced, which may lead to its potential efficacy in fighting against pneumococcal infections.
TLRs were explored as a promising delivery platform for the vaccine. In several animal models, TLR binding with pneumococcal proteins showed protective efficiency and generated significant immune responses (Moffitt et al. 2014). TLR-4 is expressed in granulocytes, monocytes, macrophages, immature dendritic immune cells, and cholera Toxin B induces the activation of NF-kB cells by direct binding to this receptor (Vaure and Liu 2014). Direct interaction between TLR4 and CTB activates TLR4 (Phongsisay et al. 2015). According to a report by Regueiro et al., TLR4 and TLR2 expression are upregulated during K. pneumoniae infection in human airway epithelial cells (Regueiro et al. 2009). Molecular docking was performed to analyze molecular interactions and binding affinity patterns between our developed vaccine construct and TLR2 and TLR4. The total energies for the coupled complexes between the vaccine and TLR4 and TLR2 were − 46.30 kJ/mol and − 12.90 kJ/mol, respectively, indicating a favorable binding affinity. The docking analysis between the designed vaccine and TLR4 showed indicated the formation of 1 salt bridge, between Lys394 of the vaccine and Glu603 of TLR4, and 2 HBs, between Pro380 and Lys394 of the vaccine and Lys103 and Glu603 of TLR4, respectively. The molecular interaction pattern analysis for the docked vaccine–TLR2 complex showed the 3 HBs formed between Ala249 and Ala270 of the vaccine and Ser522 of TLR2 and between and Asn271 of the vaccine and Gln523 of TLR2. The docking study exhibited a significant interaction between the engineered vaccine and the innate immune receptor, hence indicating the vaccine’s ability to trigger TLR activation and improved immune responses against pneumococcal infection. The MD study revealed the stable nature of the vaccine–receptor complex. The RMSD values of TLR2 and TLR4 in the complex with the vaccine did not fluctuate and remained stable throughout the MD simulation. The beta-turn regions of both complexes were relatively flexible compared with other regions, but the degree of flexibility was lower than those of other regions. Based on all of the descriptors observed during the MD simulation, both complexes were relatively rigid. When designing a vaccine candidate, immune simulation is necessary to assess the immunogenic profile. The results of the immune simulation revealed steady and normal immune responses and the tertiary and secondary responses were considerably more immunogenic than the primary response. For expression in an E. coli host system, the vaccine sequence was reverse translated into cDNA using a codon optimization strategy. The CAI score and the GC content were recorded as 0.96 and 55.24%, respectively. Moreover, the cDNA sequence was successfully input into the pET28a(+) expression vector, suggesting that our vaccine can express in the bacterial host system.
The study has several advantages over the previously published paper. First, a wider range of protein sequences was utilized to identify antigenic epitopes, resulting in a more comprehensive and accurate representation of potential vaccine targets. Secondly, the use of computational techniques, such as molecular docking, MD simulations, and in silico immune simulations, enabled a more detailed analysis of the vaccine structure and its interactions with Toll-like receptors. This information can provide insights into the potential efficacy and safety of the vaccine prior to experimental assays. Additionally, the application of disulfide engineering to stabilize the vaccine structure is a novel approach that has the potential to enhance the vaccine's immunogenicity and efficacy. Overall, these advancements demonstrate the superiority of the proposed method.
The limitations of this study include the fact that it is entirely based on computational and in silico approaches, and lacks experimental validation. While the results suggest that the designed vaccine candidate is structurally stable and capable of inducing an immune response, these findings need to be verified through in vitro and in vivo experiments. Additionally, the study only focused on the two pathogens, S. pneumoniae and K. pneumoniae, and did not consider other possible pathogens causing community-acquired pneumonia. Therefore, the generalizability of the findings to other bacterial species or strains remains unclear. Finally, the study did not address the issue of potential side effects or adverse reactions of the vaccine candidate.
5 Conclusions
This study aimed to design a multi-epitope vaccine candidate against S. pneumoniae and K. pneumonia by utilizing an immunoinformatics approach. Total three categories of antigenic epitopes, including CTL-, HTL-, and IFN-γ-inducing epitopes, were identified from diverse protein sequences and utilized to build a vaccine structure. Several immunogenic, physicochemical, and antigenic profiles were computationally assessed for the designed vaccine. The interaction patterns of vaccine/TLR4 and vaccine/TLR2 complexes were analyzed using molecular docking and subsequently, these complexes were subjected to MD simulation, revealing the dynamical stable behaviors for under-studied complexes. Moreover, an in silico immune simulation study was used to investigate the ability of the vaccine to induce an immune response, and in silico cloning assures the vaccine’s ability to express in a bacterial host system. This study explains a novel approach for developing a vaccine candidate against community-acquired pneumonia, however, experimental assays exploring the effects of the vaccine could contribute to a better understanding of generating an immune response against pneumococcal disease. Taken together, these findings support strong recommendations to execute in vitro and in vivo experiments. Additionally, we propose further synthesis and biological activity of the designed vaccine candidate.
Availability of data and material
The datasets supporting the conclusions of this study are included within the article (and its additional files).
References
Ali M, Pandey RK, Khatoon N, Narula A, Mishra A, Prajapati VK (2017) Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci Rep 7:9232
Andrusier N, Nussinov R, Wolfson HJ (2007) FireDock: fast interaction refinement in molecular docking. Proteins: Struct Funct Bioinform 69:139–159
Arai R, Ueda H, Kitayama A, Kamiya N, Nagamune T (2001) Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng 14:529–532
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201
Babu L, Uppalapati SR, Sripathy MH, Reddy PN (2017) Evaluation of recombinant multi-epitope outer membrane protein-based Klebsiella pneumoniae subunit vaccine in mouse model. Front Microbiol 8:1805
Bazhan SI, Antonets DV, Karpenko LI, Oreshkova SF, Kaplina ON, Starostina EV, Dudko SG, Fedotova SA, Ilyichev AA (2019) In silico designed ebola virus T cell multi-epitope DNA vaccine constructions are immunogenic in mice. Vaccines 7:34
Berry AM, Paton JC (2000) Additive attenuation of virulence of Streptococcus pneumoniae by mutation of the genes encoding pneumolysin and other putative pneumococcal virulence proteins. Infect Immun 68:133–140
Brooks-Walter A, Briles DE, Hollingshead SK (1999) The pspC gene of Streptococcus pneumoniae encodes a polymorphic protein, PspC, which elicits cross-reactive antibodies to PspA and provides immunity to pneumococcal bacteremia. Infect Immun 67:6533–6542
Bui H-H, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A (2006) Predicting population coverage of T cell epitope-based diagnostics and vaccines. BMC Bioinform 7:1–5
Bui H-H, Sidney J, Li W, Fusseder N, Sette A (2007) Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinform 8:1–6
Calis JJ, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, Sette A, Keşmir C, Peters B (2013) Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol 9:e1003266
Chan J, Mehta S, Bharrhan S, Chen Y, Achkar JM, Casadevall A, Flynn J (2014) The role of B cells and humoral immunity in Mycobacterium tuberculosis infection, Seminars in immunology. Elsevier, pp 588–600
Chauhan V, Rungta T, Goyal K, Singh MP (2019) Designing a multi-epitope based vaccine to combat Kaposi Sarcoma utilizing immunoinformatics approach. Sci Rep 9:2517
Chen K, McAleer JP, Lin Y, Paterson DL, Zheng M, Alcorn JF, Weaver CT, Kolls JK (2011) Th17 cells mediate clade-specific, serotype-independent mucosal immunity. Immunity 35:997–1009
Chen A, Mann B, Gao G, Heath R, King J, Maissoneuve J, Alderson M, Tate A, Hollingshead SK, Tweten RK (2015) Multivalent pneumococcal protein vaccines comprising pneumolysoid with epitopes/fragments of CbpA and/or PspA elicit strong and broad protection. Clin Vaccine Immunol 22:1079–1089
Comeau SR, Gatchell DW, Vajda S, Camacho CJ (2004) ClusPro: a fully automated algorithm for protein–protein docking. Nucleic Acids Res 32:W96–W99
Craig DB, Dombkowski AA (2013) Disulfide by design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinform 14:1–7
Cryz S Jr, Mortimer P, Cross A, Fürer E, Germanier R (1986) Safety and immunogenicity of a polyvalentKlebsiella capsular polysaccharide vaccine in humans. Vaccine 4:15–20
Dar HA, Zaheer T, Shehroz M, Ullah N, Naz K, Muhammad SA, Zhang T, Ali A (2019) Immunoinformatics-aided design and evaluation of a potential multi-epitope vaccine against Klebsiella pneumoniae. Vaccines 7:88
Darrieux M, Goulart C, Briles D, Leite LCDC (2015) Current status and perspectives on protein-based pneumococcal vaccines. Crit Rev Microbiol 41:190–200
Dhanda SK, Vir P, Raghava GP (2013) Designing of interferon-gamma inducing MHC class-II binders. Biol Direct 8:1–15
Dimitrov I, Flower DR, Doytchinova I (2013) AllerTOP-a server for in silico prediction of allergens. BMC Bioinform 14:1–9
Dimitrov I, Naneva L, Doytchinova I, Bangov I (2014) AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics 30:846–851
Dorosti H, Eslami M, Negahdaripour M, Ghoshoon MB, Gholami A, Heidari R, Dehshahri A, Erfani N, Nezafat N, Ghasemi Y (2019) Vaccinomics approach for developing multi-epitope peptide pneumococcal vaccine. J Biomol Struct Dyn 37:3524–3535
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform 8:1–7
Foroutan M, Ghaffarifar F, Sharifi Z, Dalimi A (2020) Vaccination with a novel multi-epitope ROP8 DNA vaccine against acute Toxoplasma gondii infection induces strong B and T cell responses in mice. Comp Immunol Microbiol Infect Dis 69:101413
Gasteiger E, Hoogland C, Gattiker A, Duvaud SE, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. Springer
Geourjon C, Deleage G (1995) SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 11:681–684
Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn D (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 33:W526–W531
Gupta N, Regar H, Verma VK, Prusty D, Mishra A, Prajapati VK (2020) Receptor-ligand based molecular interaction to discover adjuvant for immune cell TLRs to develop next-generation vaccine. Int J Biol Macromol 152:535–545
Heo L, Park H, Seok C (2013) GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res 41:W384–W388
Huang Y, Li J, Gu D, Fang Y, Chan EW, Chen S, Zhang R (2015) Rapid detection of K1 hypervirulent Klebsiella pneumoniae by MALDI-TOF MS. Front Microbiol 6:1435
Ilinskaya AN, Dobrovolskaia MA (2016) Understanding the immunogenicity and antigenicity of nanomaterials: past, present and future. Toxicol Appl Pharmacol 299:70–77
Jabbar B, Rafique S, Salo-Ahen OM, Ali A, Munir M, Idrees M, Mirza MU, Vanmeert M, Shah SZ, Jabbar I (2018) Antigenic peptide prediction from E6 and E7 oncoproteins of HPV types 16 and 18 for therapeutic vaccine design using immunoinformatics and MD simulation analysis. Front Immunol 9:3000
Jeannin P, Renno T, Goetsch L, Miconnet I, Aubry J-P, Delneste Y, Herbault N, Baussant T, Magistrelli G, Soulas C (2000) OmpA targets dendritic cells, induces their maturation and delivers antigen into the MHC class I presentation pathway. Nat Immunol 1:502–509
Jeannin P, Magistrelli G, Goetsch L, Haeuw J-F, Thieblemont N, Bonnefoy J-Y, Delneste Y (2002) Outer membrane protein A (OmpA): a new pathogen-associated molecular pattern that interacts with antigen presenting cells—impact on vaccine strategies. Vaccine 20:A23–A27
Jeannin P, Magistrelli G, Herbault N, Goetsch L, Godefroy S, Charbonnier P, Gonzalez A, Delneste Y (2003) Outer membrane protein A renders dendritic cells and macrophages responsive to CCL21 and triggers dendritic cell migration to secondary lymphoid organs. Eur J Immunol 33:326–333
Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, Sette A, Peters B, Nielsen M (2018) Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 154:394–406
Kerr AR, Paterson GK, McCluskey J, Iannelli F, Oggioni MR, Pozzi G, Mitchell TJ (2006) The contribution of PspC to pneumococcal virulence varies between strains and is accomplished by both complement evasion and complement-independent mechanisms. Infect Immun 74:5319–5324
Khan MN, Pichichero ME (2012) Vaccine candidates PhtD and PhtE of Streptococcus pneumoniae are adhesins that elicit functional antibodies in humans. Vaccine 30:2900–2907
Khan A, Junaid M, Kaushik AC, Ali A, Ali SS, Mehmood A, Wei D-Q (2018) Computational identification, characterization and validation of potential antigenic peptide vaccines from hrHPVs E6 proteins using immunoinformatics and computational systems biology approaches. PLoS ONE 13:e0196484
Kim Y, Ponomarenko J, Zhu Z, Tamang D, Wang P, Greenbaum J, Lundegaard C, Sette A, Lund O, Bourne PE (2012) Immune epitope database analysis resource. Nucleic Acids Res 40:W525–W530
Krieger E, Vriend G (2015) New ways to boost molecular dynamics simulations. J Comput Chem 36:996–1007
Krieger E, Nielsen JE, Spronk CA, Vriend G (2006) Fast empirical pKa prediction by Ewald summation. J Mol Graph Model 25:481–486
Krogh A, Larsson B, Von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580
Kuipers K, Diavatopoulos DA, van Opzeeland F, Simonetti E, van den Kieboom CH, Kerstholt M, Borczyk M, van IngenSchenau D, Brandsma ET, Netea MG (2016) Antigen-independent restriction of pneumococcal density by mucosal adjuvant cholera toxin subunit B. J Infect Dis 214:1588–1596
Kurupati P, Teh BK, Kumarasinghe G, Poh CL (2006) Identification of vaccine candidate antigens of an ESBL producing Klebsiella pneumoniae clinical strain by immunoproteome analysis. Proteomics 6:836–844
Lamiable A, Thévenet P, Rey J, Vavrusa M, Derreumaux P, Tufféry P (2016) PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res 44:W449–W454
Larsen MV, Lundegaard C, Lamberth K, Buus S, Brunak S, Lund O, Nielsen M (2005) An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur J Immunol 35:2295–2303
Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M (2007) Large-scale validation of methods for cytotoxic T lymphocyte epitope prediction. BMC Bioinformatics 8:1–12
Laskowski RA, Jabłońska J, Pravda L, Vařeková RS, Thornton JM (2018) PDBsum: structural summaries of PDB entries. Protein Sci 27:129–134
Lin J, Huang S, Zhang Q (2002) Outer membrane proteins: key players for bacterial adaptation in host niches. Microbes Infect 4:325–331
McGuffin LJ, Bryson K, Jones DT (2000) The PSIPRED protein structure prediction server. Bioinformatics 16:404–405
Meng C, Lin H, Huang J, Wang H, Cai Q, Fang L, Guo Y (2009) Development of 5-valent conjugate pneumococcal protein A-Capsular polysaccharide pneumococcal vaccine against invasive pneumococcal disease. Microb Pathog 47:151–156
Messaoudi A, Belguith H, Ben Hamida J (2013) Homology modeling and virtual screening approaches to identify potent inhibitors of VEB-1 β-lactamase. Theor Biol Med Model 10:1–10
Meza B, Ascencio F, Sierra-Beltrán AP, Torres J, Angulo C (2017) A novel design of a multi-antigenic, multistage and multi-epitope vaccine against Helicobacter pylori: an in silico approach. Infect Genet Evol 49:309–317
Mizel SB, Bates JT (2010) Flagellin as an adjuvant: cellular mechanisms and potential. J Immunol 185:5677–5682
Moffitt K, Skoberne M, Howard A, Gavrilescu LC, Gierahn T, Munzer S, Dixit B, Giannasca P, Flechtner JB, Malley R (2014) Toll-like receptor 2-dependent protection against pneumococcal carriage by immunization with lipidated pneumococcal proteins. Infect Immun 82:2079–2086
Moise L, McMurry JA, Buus S, Frey S, Martin WD, De Groot AS (2009) In silico-accelerated identification of conserved and immunogenic variola/vaccinia T cell epitopes. Vaccine 27:6471–6479
Nishita M, Park S-Y, Nishio T, Kamizaki K, Wang Z, Tamada K, Takumi T, Hashimoto R, Otani H, Pazour GJ (2017) Ror2 signaling regulates Golgi structure and transport through IFT20 for tumor invasiveness. Sci Rep 7:1–15
Paton JC (2004) New pneumococcal vaccines: basic science developments. The pneumococcus. ASM Press, pp 382–402
Petersen TN, Brunak S, Von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786
Phongsisay V, Iizasa EI, Hara H, Yoshida H (2015) Evidence for TLR4 and FcRγ–CARD9 activation by cholera toxin B subunit and its direct bindings to TREM2 and LMIR5 receptors. Mol Immunol 66:463–471
Ponomarenko J, Bui H-H, Li W, Fusseder N, Bourne PE, Sette A, Peters B (2008) ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinform 9:1–8
Post DM, Slütter B, Schilling B, Chande AT, Rasmussen JA, Jones BD, D’Souza AK, Reinders LM, Harty JT, Gibson BW (2017) Characterization of inner and outer membrane proteins from Francisella tularensis strains LVS and Schu S4 and identification of potential subunit vaccine candidates. Mbio 8:e01592-e11517
Rafi MO, Al-Khafaji K, Sarker MT, Taskin-Tok T, Rana AS, Rahman MS (2022a) Design of a multi-epitope vaccine against SARS-CoV-2: immunoinformatic and computational methods. RSC Adv 12:4288–4310
Rafi MO, Al-Khafaji K, Tok TT, Rahman MS (2022b) Computer-based identification of potential compounds from Salviae miltiorrhizae against Neirisaral adhesion A regulatory protein. J Biomol Struct Dyn 40:4301–4313
Rafi MO, Bhattacharje G, Al-Khafaji K, Taskin-Tok T, Alfasane MA, Das AK, Parvez MAK, Rahman MS (2022c) Combination of QSAR, molecular docking, molecular dynamic simulation and MM-PBSA: analogues of lopinavir and favipiravir as potential drug candidates against COVID-19. J Biomol Struct Dyn 40:3711–3730
Rapin N, Lund O, Castiglione F (2011) Immune system simulation online. Bioinformatics 27:2013–2014
Regueiro V, Moranta D, Campos MA, Margareto J, Garmendia J, Bengoechea JA (2009) Klebsiella pneumoniae increases the levels of Toll-like receptors 2 and 4 in human airway epithelial cells. Infect Immun 77:714–724
Samad A, Ahammad F, Nain Z, Alam R, Imon RR, Hasan M, Rahman MS (2022a) Designing a multi-epitope vaccine against SARS-CoV-2: an immunoinformatics approach. J Biomol Struct Dyn 40:14–30
Samad A, Meghla NS, Nain Z, Karpiński TM, Rahman MS (2022b) Immune epitopes identification and designing of a multi-epitope vaccine against bovine leukemia virus: a molecular dynamics and immune simulation approaches. Cancer Immunol Immunother 71:2535–2548
Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ (2005) PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res 33:W363–W367
Sette A, Fikes J (2003) Epitope-based vaccines: an update on epitope identification, vaccine design and delivery. Curr Opin Immunol 15:461–470
Stratmann T (2015) Cholera toxin subunit B as adjuvant––an accelerator in protective immunity and a break in autoimmunity. Vaccines 3:579–596
Sun W-SW, Syu W-J, Ho W-L, Lin C-N, Tsai S-F, Wang S-H (2014) SitA contributes to the virulence of Klebsiella pneumoniae in a mouse infection model. Microbes Infect 16:161–170
Tumbarello M, Trecarichi EM, De Rosa FG, Giannella M, Giacobbe DR, Bassetti M, Losito AR, Bartoletti M, Del Bono V, Corcione S (2015) Infections caused by KPC-producing Klebsiella pneumoniae: differences in therapy and mortality in a multicentre study. J Antimicrob Chemother 70:2133–2143
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ (2005) GROMACS: fast, flexible, and free. J Comput Chem 26:1701–1718
Vaure C, Liu Y (2014) A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol 5:316
Verhoeven D, Xu Q, Pichichero ME (2014) Vaccination with a Streptococcus pneumoniae trivalent recombinant PcpA, PhtD and PlyD1 protein vaccine candidate protects against lethal pneumonia in an infant murine model. Vaccine 32:3205–3210
Von Mollendorf C, Tempia S, Von Gottberg A, Meiring S, Quan V, Feldman C, Cloete J, Madhi SA, O’Brien KL, Klugman KP (2017) Estimated severe pneumococcal disease cases and deaths before and after pneumococcal conjugate vaccine introduction in children younger than 5 years of age in South Africa. PLoS ONE 12:e0179905
Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, Peters B (2010) Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform 11:1–12
Weinberger DM, Malley R, Lipsitch M (2011) Serotype replacement in disease after pneumococcal vaccination. The Lancet 378:1962–1973
White P, Hermansson A, Svanborg C, Briles D, Prellner K (1999) Effects of active immunization with a pneumococcal surface protein (PspA) and of locally applied antibodies in experimental otitis media. ORL 61:206–211
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35:W407–W410
Wiedinger K, Pinho D, Bitsaktsis C (2017) Utilization of cholera toxin B as a mucosal adjuvant elicits antibody-mediated protection against S. pneumoniae infection in mice. Therap Adv Vaccines 5:15–24
Wu H-Y, Nahm MH, Guo Y, Russell MW, Briles DE (1997) Intranasal immunization of mice with PspA (pneumococcal surface protein A) can prevent intranasal carriage, pulmonary infection, and sepsis with Streptococcus pneumoniae. J Infect Dis 175:839–846
Yadav V, Sharma S, Harjai K, Mohan H, Chhibber S (2005) Lipopolysaccharide-mediated protection against Klebsiella pneumoniae-induced lobar pneumonia: intranasal vs. intramuscular route of immunization. Folia Microbiol 50:83–86
Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D (2020) Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci 117:1496–1503
Yin D, Li L, Song X, Li H, Wang J, Ju W, Qu X, Song D, Liu Y, Meng X (2016) A novel multi-epitope recombined protein for diagnosis of human brucellosis. BMC Infect Dis 16:1–8
Yuste J, Botto M, Paton JC, Holden DW, Brown JS (2005) Additive inhibition of complement deposition by pneumolysin and PspA facilitates Streptococcus pneumoniae septicemia. J Immunol 175:1813–1819
Author information
Authors and Affiliations
Contributions
Conceptualization: MOR, KA-K. Formal analysis: MOR, KA-K. Investigation: MOR, KA-K, MSR. Methodology: MOR, KA-K, NSM, SMM, PKB, MSR. Project administration: MSR. Resources: MSR. Software: MOR, KA-K. Supervision: MSR. Writing—original draft: MOR, KA-K. Writing—review & editing: MSR, NSM, SMM.
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest to declare.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rafi, M.O., Al-Khafaji, K., Mandal, S.M. et al. A subunit vaccine against pneumonia: targeting Streptococcus pneumoniae and Klebsiella pneumoniae. Netw Model Anal Health Inform Bioinforma 12, 21 (2023). https://doi.org/10.1007/s13721-023-00416-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13721-023-00416-3