
Abstract
The severe acute respiratory syndrome COVID-19 declared a global pandemic by WHO has become the present wellbeing worry to the whole world. There is an emergent need to search for possible medications. We report in this study a molecular docking study of eighteen Oroxylum indicum molecules with the main protease (Mpro) responsible for the replication of SARS-CoV-2 virus. The outcome of their molecular simulation and ADMET properties reveal four potential inhibitors of the enzyme (Baicalein-7-O-diglucoside, Chrysin-7-O-glucuronide, Oroxindin and Scutellarein) with preference of ligand Chrysin-7-O-glucuronide that has the second highest binding energy (− 8.6 kcal/mol) and fully obeys the Lipinski’s rule of five.
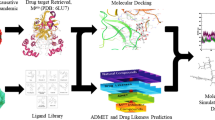
Graphical abstract

Similar content being viewed by others

1 Introduction
Corona viruses are enveloped and single-stranded ribonucleic acid (RNA) viruses which can infect humans as well as animal species (Wu et al. 2020a). These viruses are categorized into four genera as, α-corona virus (α-COV), β-corona virus (β-COV), δ-corona virus (δ-COV) and γ—corona virus (γ-COV). Till now, six human corona viruses are known; (CoV)—HCoV-NL63, HCoV-HKU1, HCoV-229E, HCoVOC43, Severe Acute Respiratory Syndrome Corona virus (SARS-CoV) and Middle East Respiratory Syndrome Corona virus (MERS-CoV). SARS-CoV and MERS-CoV have been reported to cause severe respiratory symptom and large number of deaths in past few years across globe (Wang et al. 2020). As on 06 November, 2020, the WHO has reported 48,534,508 confirmed cases and 1,231,017 death cases worldwide (WHO 2020) https://covid19.who.int/. Structural and non-structural proteins of SARS-CoV-2 have been proposed as potential drug targets. Various active targets of SARS-CoV-2 includes 3-chymotrypsin—like protease (3CLpro), papain like protease (PLpro), RNA-dependent RNA polymerase, spike (S) proteins and human Angiotensin-converting enzyme (ACE) 2 (Wu et al. 2020b). Main protease (Mpro) or 3CLPro of corona virus is conserved among the corona viruses and it is mainly responsible for the viral replication. The mediation of nonstructural viral proteins and maturation by main protease makes Mpro a very attractive target for the development of anti-corona virus drugs (Vlachakis et al. 2020).
Currently, no recognized antiviral therapies or preventive vaccines are available for this pandemic disease. Anti-malarial drugs like Chloroquine, Hydroxychloroquine sulfate and the antiviral drugs such as Remdesivir, Lopinavir/Ritonavir, Favipiravir were found to be efficacious in the treatment of patients infected with this virus and is a possible therapeutic option for COVID-19 (Philippe, Rolain Jean-Marc 2020; Zhang and Xie 2020). Now a day, plant extracts and isolated phytoconstituents are broadly used to evaluate their in vitro antiviral activities. The natural products such as conventional herbal medicines and phytochemicals are the rich sources of potential antiviral drugs (Liu and Du 2012). Some of the medicinal plants reported for its antiviral potential against notable viral pathogens include Azadirachta indica, Artemisia annua, Pyrrosia lingua, Terminalia chebula, Piper longum, etc. (Parida et al. 2002; Lin et al. 2014).
Oroxylum indicum (family: Bignoniaceae) commonly known as Shyonaka and Indian Trumpet tree due to resemblance of its flower to trumpet (Kirtikar and Basu 2001). It is an imperative herb in Ayurvedic medicine and has been used as an ingredient of polyherbal formulations like Dasamularistha, Amartarista, Dantyadyarista, Narayantaila, Dhanawantaraghrita, Brahmarasayan and Chyavanaprash. Pharmacologically, it has been found to possess antimicrobial, antidiabetic, antihyperlipidemic, hepatoprotective, analgesic and anti-inflammatory, anticarcinogenic, immunomodulatory, nephroprotective, antitussive cardioprotective, antiallergic, antibronchitic, antirheumatic activities (Ahad et al. 2012). Flavonoids, alkaloids, glycosides, essential oils and phenolic compounds has been reported as an important phytoconstituents (Lawania et al. 2010). Flavonoids such as baicalein, baicalin, chrysin, oroxylin-A, scutellarin, acacetin, hispidulin, isorhamnetin, and Isoquercetin, quercetin-3-o-α-l-arabinopyranoside, 1-(2-hydroxyethyl) cyclohexane-1,4-diol, apigenin, 2,5-dihydroxy-6,7-dimethoxy flavones, 3,7,3′,5′-tetramethoxy-4′-hydroxyflavone, pterocarpans, etc., has been isolated previously (Ahad et al. 2012; Deka et al. 2013). Other chemical constituents such as Prunetin, β-Sitosterol, Stigmasterol glucoside, Ellagic acid, Triterpene Carboxylic acid, Ursolic Acid, Lupeol, p-Coumaric Acid, Napthquinones, Anthraquinone, Phenylethanoid glycosides, and Cyclohexylethanoids are also extracted from this plant (Yan et al. 2011; Dinda et al. 2015).
Drug discovery is a time consuming and challenging process. Moreover, to screen out large number phytoconstituents from herbs with antiviral activity against corona virus will be a challenge in very short period. Considering the consequence of increase in the number of active cases and death cases in current time and deficient effective treatment, computer aided drug design is an important approach to be preferred. In silico process of drug design will minimize the time and expenditure require in the drug research (Kiran et al. 2020).
Literature suggest that there is no study reported for Oroxylum indicum as antiviral drug although owning reach source of constituents, makes it possible herbal contender to interfere with the corona virus life cycle. Hence, the aim of the current study is to identify the potential antiviral phytoconstituents from Oroxylum indicum against target main protease (PDB ID: 6LU7) using molecular simulation study.
2 Materials and methods
2.1 Protein preparation
In silico analysis of phytoconstituents of Oroxylum indicum was performed on 2.16 Å crystal structure of COVID-19 Mpro: main protease in complex with an inhibitor N3 (PDB ID: 6LU7) also a key CoV enzyme which plays a pivotal role in mediating viral replication and transcription, making it an attractive drug target for this virus (Isah 2019), which was retrieved from protein data bank (https://www.rcsb.org). The 6LU7 protein contains two chains, A and B, which form a homodimer. Chain A was used for macromolecule preparation (Fig S1).
The protein was visualised using PyMol (http://www.pymol.org) and unwanted chain residues, bound ligand N3, water molecules were removed. Additionally to prepare protein, charges added and minimized the energy of the protein using Autodock Vina in PyRx open source software and subsequently converting it to pdbqt format.
2.2 Ligand preparations
For this investigation, chemical structures of chemical constituents of Oroxylum indicum were retrieved in a SDF format from chemical database PubChem available on NCBI website (https://pubchem.ncbi.nlm.nih.gov/). The ligand N3 (N-[(5-methylisoxazol-3yl) carbonyl] alanyl-l-valyl-n ~ 1 ~ -((1R, 2Z)-4-(benzyloxy)-4-oxo-1-{[(3R)-2-oxopyrrolidin-3-yl] methyl} but-2-enyl)-l-leucinamide) was obtained from database of chemspider. CSID:4883311, http://www.chemspider.com/ChemicalStructure.4883311.html (accessed 04:56, May 12, 2020). Using OpenBabel control all ligands were loaded into the PyRx virtual screening software. The OpenBabel software converts the SDF files of the ligands to PDB format (O’Boyle et al. 2011). Further, the ligands are prepared by detecting the torsion root, correcting the torsion angles, assigning charges, optimizing using UFF (Universal force field) (Jaillet et al. 2017) and finally converting them to pdbqt format to generate 3D atomic coordinates of the molecules. The chemical structures of all the ligands is depicted in Fig. 1.

Chemical structure of all selected ligand molecules in docking studies
2.3 Receptor grid generation and molecular docking
A primary objective in molecular docking was the ability to estimate the scoring function and to evaluate interactions between a protein and small molecules based on the geometry in order to predict the binding affinity and activity of the ligand molecule (Verdonk et al. 2003; Leach et al. 2006). In this study, docking was performed using Autodock Vina in PyRx virtual screening open source software (Leach et al. 2006). The protein and ligand molecules to be docked are selected under the Vina wizard control. A grid appears over the protein structure. The grid size can be adjusted according to the active site residues that are selected and Autodock Vina is run. A grid box with the size 32.803 × 27.870 × 26.639 with coordinates (x,y,z) of − 9.8336, 11.1638, 69.5594 having the grid spacing of 0.375 Å was used. The docking Lamarckian Genetic Algorithm (LGA) was allowed to produce 10 docked positions for each ligand (Fuhrmann et al. 2010). The docking results can be viewed under ‘analyse results’ tab. Although, the final results were analyzed and visualized on the basis of docking scores using Discovery Studio 2020 Client (Biovia 2016) and PyMol software’s (DeLano 2002).
2.4 Molecular dynamics (MD) studies
To perform molecular dynamics simulations, CABS Flex 2.0 server were used. It is based on coarse-grained simulations of protein motion (Kurcinski et al. 2019) for 50 cycles, 50 trajectory frames for 10 ns with some additional distance restraints with a global weight of 1.0, built with Poisson-Boltzmann/Generalized Born (PB/GB) molecular mechanics the solvent probe radius was set as 1.4 Å, minimum atomic radius 1 Å, salt radius 2 Å, ionic strength 0.15, temperature of simulation 1.4, in order to analyze the conformational stability of the receptor-ligand complex system.
2.5 ADME and toxicity studies
The ligands were further screened for Lipinski rule of five by following certain criteria such as molecular weight ≤ 500, logP ≤ 5, hydrogen bond donor ≤ 5, hydrogen bond acceptor ≤ 10 and topological polar surface area ≤ 500. During drug development, safety is always the most important issue, therefor Toxicology prediction of small molecules is important to predict amount of tolerability of the small molecule before being ingested into the human and animal models. Two main approaches for assessment of phytoconstituents toxicity, webserver based and quantitative structure activity relationship.
pkCSM (http://biosig.unimelb.edu.au/pkcsm/prediction) is an online server database in which the small molecule can be analyzed for calculating pharmacokinetic and toxicological properties by uploading the SMILES of molecules. We can obtain the details of toxicological effects in the fields of AMES Toxicity, human maximum tolerance dose, hERG-I inhibitor, hERG-II inhibitor, LD50, LOAEL, Hepatotoxicity, Skin Toxicity, T. pyriformis toxicity and Minnow toxicity(Pires et al. 2015).
VEGA-QSAR ((http://www.vega-qsar.eu/) is integrating In silico QSAR models and read-across method for a number of toxicological data outcomes(Rogiers et al. 2020). The molecules can be analyzed for calculating toxicological properties by either inserting SMILES notations or uploading SDF files and then selecting toxicological models for generating numerous information about structure related effects. The results also show structural alerts in chemical structure based on known mutagenic and carcinogenic structural analogy (Benfenati et al. 2019).
3 Result and discussion
3.1 Molecular docking studies
The main aim of the study was to prospect active chemical constituents of Oroxylum indicum to a highly conserved protein, Mpro of SARS-CoV-2, therefore we performed molecular docking studies of all chemical constituents of Oroxylum indicum followed by identification of top hits which is discussed below. Furthermore, to investigate pharmacokinetics and toxicity properties of the molecules showing promising results so that they can be proposed as potential drugs candidates.
Autodock Vina is used to determine molecular interactions and binding energy between phytoconstituents of Oroxylum indicum and COVID-19 Mpro. Phytoccosntituents of Oroxylum indicum has shown antiviral actions (Mohamat et al. 2018; Antonio et al. 2020). Therefore we selected 18 chemical constituents from this plant to investigate their inhibition potential for COVID-19 Mpro. Remdesivir is shown potential in treatment of Marburg virus, Paramyxoviridae (such as parainfluenza type 3, Nipah, Hendra, measles and mumps viruses) and Pneumoviridae (respiratory syncytial virus) (Ko et al. 2020) that can represent possible treatment options against corona (Mahmood et al. 2020) it was selected as standard drug (Pizzorno et al. 2020).
The binding potential energy or Vina fitness score obtained from docking 6LU7 with the native ligand (N3) Fig S2, Remdesivir and Oroxylum indicum compounds are given in Table S1. The binding energy of the best docked pose of ligand is compared with co-crystalized ligand and reference standard drug. The docked binding energy of the N3 and Remdesivir is placed in the fifth and sixth position, a bit lower than those of the four best ligands. The binding affinity values range from − 9.1 to − 6.2 kcal/mol. There is no doubt that the Baicalein-7-O-diglucoside exhibits the higher binding affinity (− 9.1 kcal/mol), higher to Chrysin-7-O-glucuronide (− 8.6 kcal/mol), Oroxindin (− 8.1 kcal/mol) and Scutellarein (− 8.0 kcal/mol).
According to the analysis of docking results, the interactions (listed in Table 1) between Baicalein-7-O-diglucoside, Chrysin-7-O-glucuronide, Oroxindin and Scutellarein are highly consistent with that of Remdesivir and even they represent the most promising inhibitors of the SARS-CoV-2 Mpro. The results of the molecular docking showed that the tested compound baicalein-7-O-diglucoside gives the lowest binding energy (− 9.1 kcal/mol) in complex with 6LU7, which is the best score when compared to other docked compounds. However, it is reasonably disapproving to obey Lipinski’s rule of five as it is characterized by two violations (Table 2).
The docking of Chrysin-7-O-glucuronide with main protease is accompanied by an affinity of − 8.6 kcal/mol by forming five hydrogen bonds with THR 26 A, LEU 141 A, GLY 143A, SER 144A, CYS 145A along with hydrophobic interaction with MET 165 A, GLN 189 A. It shows favorable drug-like properties by following Lipinski's rule of five without any violation. Both Oroxindin and Scutellarein exhibit drug-likeness as they were found to obey Lipinski’s rule of five. Oroxindin was found to exhibit a hydrogen bond with THR 26 A, ASN 142A, GLY 143A, CYS 145A, ASP 187A and residue of the main protease with an affinity of − 8.1 kcal/mol. It also forms pi–pi T-shape interactions with His 41A, pi-alkyl interactions with PRO 168A (Fig S3). Similarly, Scutellarein can be recognized as promising drug candidate on the basis of qualifying Lipinski’s rule five. Scutellarein was found to show carbon-hydrogen bonding with TYR 54A, HIS 163A, HIS 164A, GLU 166A, ASP 187A and pi-alkyl interaction with MET 49A.
Hydrogen bonds formed by OH and C=O groups stabilizes ligand-receptor complex in which ligand plays dual role of acceptor and donor (Matondo et al. 2018) (Fig. 3 and Table 1). Also, stability of complexes are directed by dispersion forces (Trujillo and Sánchez-Sanz 2016), π–π interactions(Kasende et al. 2017), and hydrophobic interactions (Muya et al. 2019) types of interactions. It is the interaction (both lipophilic and H-bonds) between polar amino acid and hydrophobic residues are responsible to inhibit COVID-19 Mpro (Figs. 2, 3) (Khaerunnisa et al. 2020).

Docked pose of a Baicalein-7-O-diglucoside b Chrysin-7-O-glucuronide and c Oroxindin, d Scutellarein, e N3, f Remdesivir against Mpro protease (PDB ID: 6LU7). The ligand is shown in ball and stick representation whereas residues forming binding pocket of Mpro are shown as colored sticks. Hydrogen bond interactions are shown with black dotted lines

Hydrogen-bonds parameters (distances and angles) a Baicalein-7-O-diglucoside, b Chrysin-7-O-glucuronide and c Oroxindin, d Scutellarein, e N3, f Remdesivir
3.2 Molecular dynamics studies
Molecular dynamics studies are performed to validate docking results of phytoconstituents in complex with target protein 6LU7 using CABS-flex 2.0 server. The input of docking poses of phytoconstiutents complexed with protein (PDB) added with default parameters to study fluctuations of the individual amino acid residues (Jamroz et al. 2013). The conformational stability of docked poses are represented by the root mean square fluctuation (RMSF) values (Figs. 4, 5). The highest RMSF value in MD simulations reflects more flexibility of protein, whereas the lowest value implies the limited motion of the system during the simulation process (Zhao et al. 2015). Chrysin-7-O-glucuronide shows lowest RMSF values indicating limited motion of the system over period of simulation trajectories which also validates that Autodock Vina docking results are not obtained by chance. The presence of amino acid residues with fitting α-helix and β-sheet in secondary structures reflects minimal fluctuation with efficient constraints to the all atom. All the selected molecules (Baicalein-7-O-diglucoside, Chrysin-7-O-glucuronide, Oroxindin and Scutellarein) maintained the molecular interactions with the protein.

Root Mean Square Fluctuations plots of protein structure with compounds. No abrupt fluctuations were observed in any region of the protein with the proposed ligands. a N3, b Baicalein-7-O-diglucoside (10077207), c Chrysin-7-O-glucuronide (14135335), d Oroxindin (3084961) and e Scutellarein (5281697)

Stable structures of protein generated after MD Simulation of compound. a N3, b Baicalein-7-O-diglucoside, c Chrysin-7-O-glucuronide, d Oroxindin and e Scutellarein
3.3 ADME and toxicity studies
The results of the pkCSM webserver shows that selected screened phytoconstituents does not have AMES toxicity, does not inhibit hERG-I, does not produce hepatotoxicity, it does not cause skin sensitivity. Only Baicalein-7-O-diglucoside inhibit hERG-II other do not. From the Lipinski and predicted ADME/tox filtering analyses (Table 2), we obtained four non-toxic, herbal compounds that bind to the receptor-binding site and catalytic site of SARS-CoV-2 Mpro.
Additionally to confirm reliability of toxicological data, QSAR modelling method performed using VEGA-QSAR (Table 3). The software incorporated algorithm provides evaluation of reliability prediction as Applicability domain index (ADI) value. We used positive results with ADI > 0.5, as indicators of reliability effect; low (0.5 < ADI < 0.6), medium (0.6 < ADI < 0.8) and high (0.8 < ADI < 1). The four phytoconstituents does not have AMES toxicity (CONSENSUS model, assessment and prediction) (Votano et al. 2004), does not have carcinogenicity (CAESAR model, assessment and prediction) (Fjodorova et al. 2010), no adverse health effects to humans and ecological species (IRFMN/COMPARA, assessment and prediction) (Kamel et al. 2020) and found to be inactive for Thyroid hormone receptor α/β (NRMEA, assessment and prediction). Unlike Scutellarine all compounds does not have skin sensitivity (CAESAR model, assessment and prediction) (Chaudhry et al. 2010). However, all compounds controversial to webserver predictions have very low reliability of hepatotoxic potential (IRFMN, assessment and prediction).
A literature review supports the docking result and reveals that extract of selected plant were reported to possess antimicrobial activities (Mat Ali et al. 1998; Suresh Babu et al. 2005) and act as antioxidant by inhibiting lipid-peroxidation (IC50 0.08 μg/ml) (Siriwatanametanon et al. 2010; Rajkumar et al. 2012). Baicalein extensively investigated with respect to their antiviral activity. It possess potent anti-DENV activity (Moghaddam et al. 2014), impaired H5N1 virus replication, prolong death rate in individual infected with H1N1 and Sendai viruses, suppress replication of HIV-I virus. Computational studies also promises effectiveness against chikungunya virus (CHIKV) nsP3 and NS5 protein (Zakaryan et al. 2017). In vitro, scutellarein found to inhibit the SARS-CoV helicase protein by affecting the ATPase activity, nsP13 (Yu et al. 2012). Scutellarin and Chrysin-7-O-glucuronide showed high antioxidant capacities in DPPH, ABTS and CAA assays, antiviral activity of Chrysin derivative against Coxsackievirus B3 (CVB3) (Li et al. 2018).
Thus, we anticipate that this selected phytoconstituents has the potential to enhance immunity along with the inhibition of COVID-19 infections (Lin and Weng 2006). Furthermore, Combination therapy often results in superior outcome in antiviral treatments (Mucsi et al. 1992; Chang et al. 2011). The screened phytoconstituents with antiviral activity, displayed higher docking scores, stronger binding energies, better interaction with the conserved catalytic residues and that may cause inhibition/blockade of the SARS-CoV-host protein pathways could be potential supportive and therapeutic candidates (Fuzimoto and Isidoro 2020).
4 Conclusion
Oroxylum indicum, a medicinal plant traditionally uses as antiviral, contains chemical constituents which has potential for prevention and prophylaxis of SARS-CoV-2. Computational algorithmic screening of the selected phytoconstituents of the plant were highly selective, shown strong binding potential and have strong interactions with main protease Mpro of SARS-CoV-2 virus. The evidence of highest docking score, positioning of ligands at site of inhibition, interaction profile with catalytic residues and acceptable ADMET parameters by QSAR model suggest probability of Chrysin-7-O-glucuronide may be potent against SARS-CoV-2 Mpro. These results encourage further in vitro and in vivo investigations and also encourage traditional use of Oroxylum indicum preventively and will provide vital information on novel scaffolds for further lead optimization.
Data availability
The authors have no financial or proprietary interests in any material discussed in this article.
Abbreviations
- AMES:
-
Salmonella typhimurium Reverse mutation assay
- WHO:
-
World health organization
- ADMET:
-
Absorption, distribution, metabolism, excretion and toxicological
- RNA:
-
Ribonucleic acid
- SDF:
-
Spatial data file
- NCBI:
-
National Center for Biotechnology Information
- UFF:
-
Universal force field
- SMILES:
-
Simplified Molecular Input Line Entry System
- hERG:
-
Human ether-a-go-go-related gene
- LOAEL:
-
Lowest-observed-adverse-effect level
- LD:
-
Lethal dose
- ATPase:
-
Adenosine triphosphatase
- DPPH:
-
2,2-Diphenyl-1-picrylhydrazyl
- ABTS:
-
2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid
- CAA:
-
Cellular antioxidant activity
- DENV:
-
Dengue virus
- QSAR:
-
Quantitative structure activity relationship
- CAESAR:
-
Computer assisted evaluation of industrial chemical substances according to regulation
- CoMPARA:
-
Collaborative modeling project for androgen receptor activity
- IRFMN:
-
Estrogen receptor relative binding affinity model
- NRMEA:
-
Nuclear receptor-mediated endocrine activity model
- ADI:
-
Applicability domain index
- SMILES:
-
Simplified molecular-input line-entry system
References
Ahad A, Ganai A, Sareer O et al (2012) Therapeutic potential of Oroxylum indicum: a review. J Pharm Res Opin 2:163–172
Antonio AS, Wiedemann LSM, Veiga-Junior VF (2020) Natural products’ role against COVID-19. RSC Adv 10:23379–23393. https://doi.org/10.1039/D0RA03774E
Benfenati E, Chaudhry Q, Gini G, Lou DJ (2019) Integrating in silico models and read-across methods for predicting toxicity of chemicals: a step-wise strategy. Environ Int 131:105060. https://doi.org/10.1016/j.envint.2019.105060
Biovia DS (2016) Discovery studio modeling environment, release 2017, San Diego. In: Dassault Systèmes
Chang J, Schul W, Butters TD et al (2011) Combination of α-glucosidase inhibitor and ribavirin for the treatment of dengue virus infection in vitro and in vivo. Antiviral Res 89:26–34. https://doi.org/10.1016/j.antiviral.2010.11.002
Chaudhry Q, Piclin N, Cotterill J et al (2010) Global QSAR models of skin sensitisers for regulatory purposes. Chem Cent J 4(Suppl 1):S5–S5. https://doi.org/10.1186/1752-153X-4-S1-S5
DE Pires V, Blundell TL, Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58:4066–4072. https://doi.org/10.1021/acs.jmedchem.5b00104
Deka DC, Kumar V, Prasad C, Kumar K, Gogoi BJ, Singh LSRB (2013) Oroxylum indicum—a medicinal plant of North East India: an overview of its nutritional, remedial, and prophylactic properties. J Appl Pharm Sci 3:S104–S112
DeLano LW (2002) Pymol: an open-source molecular graphics tool. Newsl Protein Crystallogr 400:82–92
Dev LR, Ranjeeta P, Anurag M, Rajiv G (2010) Pharmacognostic and phytochemical studies of bark of Oroxylum indicum. Pharmacogn J 2:297–303. https://doi.org/10.1016/S0975-3575(10)80120-8
Dinda B, SilSarma I, Dinda M, Rudrapaul P (2015) Oroxylum indicum (L.) Kurz, an important Asian traditional medicine: from traditional uses to scientific data for its commercial exploitation. J Ethnopharmacol 161:255–278. https://doi.org/10.1016/j.jep.2014.12.027
Fjodorova N, Vracko M, Novic M et al (2010) New public QSAR model for carcinogenicity. Chem Cent J 4(Suppl 1):S3–S3. https://doi.org/10.1186/1752-153X-4-S1-S3
Fuhrmann J, Rurainski A, Lenhof H-P, Neumann D (2010) A new Lamarckian genetic algorithm for flexible ligand-receptor docking. J Comput Chem 31:1911–1918. https://doi.org/10.1002/jcc.21478
Fuzimoto AD, Isidoro C (2020) The antiviral and coronavirus-host protein pathways inhibiting properties of herbs and natural compounds—additional weapons in the fight against the COVID-19 pandemic? J Tradit Complement Med 10:405–419. https://doi.org/10.1016/j.jtcme.2020.05.003
Isah T (2019) Stress and defense responses in plant secondary metabolites production. Biol Res 52:39. https://doi.org/10.1186/s40659-019-0246-3
Jaillet L, Artemova S, Redon S (2017) IM-UFF: Extending the universal force field for interactive molecular modeling. J Mol Graph Model 77:350–362. https://doi.org/10.1016/j.jmgm.2017.08.023
Jamroz M, Orozco M, Kolinski A, Kmiecik S (2013) Consistent view of protein fluctuations from all-atom molecular dynamics and coarse-grained dynamics with knowledge-based force-field. J Chem Theory Comput 9:119–125. https://doi.org/10.1021/ct300854w
Kasende OE, Matondo A, Muya JT, Scheiner S (2017) Interactions between temozolomide and guanine and its S and Se-substituted analogues. Int J Quantum Chem 117:157–169. https://doi.org/10.1002/qua.25294
Khaerunnisa S, Kurniawan H, Awaluddin R et al (2020) Potential inhibitor of COVID-19 main protease (Mpro) from several medicinal plant compounds by molecular docking study. Prepr. https://doi.org/10.20944/PREPRINTS202003.0226.V1
Kiran G, Karthik L, Shree Devi MS et al (2020) In Silico computational screening of Kabasura Kudineer—official siddha formulation and JACOM against SARS-CoV-2 spike protein. J Ayurveda Integr Med. https://doi.org/10.1016/j.jaim.2020.05.009
Kirtikar KR, Basu BD (2001) Indian medicinal plants, 2nd edn. Oriental Enterprises, Dehradun
Ko W-C, Rolain J-M, Lee N-Y et al (2020) Arguments in favour of remdesivir for treating SARS-CoV-2 infections. Int J Antimicrob Agents 55:105933. https://doi.org/10.1016/j.ijantimicag.2020.105933
Kurcinski M, Oleniecki T, Ciemny MP et al (2019) CABS-flex standalone: a simulation environment for fast modeling of protein flexibility. Bioinformatics 35:694–695. https://doi.org/10.1093/bioinformatics/bty685
Leach AR, Shoichet BK, Peishoff CE (2006) Prediction of protein−ligand interactions. Docking and scoring: successes and gaps. J Med Chem 49:5851–5855. https://doi.org/10.1021/jm060999m
Li K, Fan H, Yin P et al (2018) Structure-activity relationship of eight high content flavonoids analyzed with a preliminary assign-score method and their contribution to antioxidant ability of flavonoids-rich extract from Scutellaria baicalensis shoots. Arab J Chem 11:159–170. https://doi.org/10.1016/j.arabjc.2017.08.002
Lin JK, Weng MS (2006) Flavonoids as nutraceuticals. Sci Flavonoids 7:213–238. https://doi.org/10.1007/978-0-387-28822-2_8
Lin L-T, Hsu W-C, Lin C-C (2014) Antiviral natural products and herbal medicines. J Tradit Complement Med 4:24–35. https://doi.org/10.4103/2225-4110.124335
Liu A-L, Du G-H (2012) Antiviral Properties of phytochemicals BT. In: Patra AK (ed) Dietary phytochemicals and microbes. Springer Netherlands, Dordrecht, pp 93–126
Mahmood K, Rashed ER, Oliveros E et al (2020) Predisposition or protection?: COVID-19 in a patient on LVAD support with HIV/AIDS. JACC Case Rep 2:1337–1341. https://doi.org/10.1016/j.jaccas.2020.05.015
Mansouri K, Kleinstreuer N, Abdelaziz AM et al (2020) CoMPARA: collaborative modeling project for androgen receptor activity. Environ Health Perspect 128:27002. https://doi.org/10.1289/EHP5580
Mat Ali R, Houghton PJ, Raman A, Hoult JRS (1998) Antimicrobial and antiinflammatory activities of extracts and constituents of Oroxylum indicum (L.) Vent. Phytomedicine 5:375–381. https://doi.org/10.1016/S0944-7113(98)80020-2
Matondo A, Mukeba CT, Muzomwe M et al (2018) Unravelling syn- and anti-orientation in the regioselectivity of carbonyl groups of 5-fluorouracil an anticancer drug toward proton donors. Chem Phys Lett 712:196–207. https://doi.org/10.1016/j.cplett.2018.09.074
Moghaddam E, Teoh B-T, Sam S-S et al (2014) Baicalin, a metabolite of baicalein with antiviral activity against dengue virus. Sci Rep 4:5452. https://doi.org/10.1038/srep05452
Mohamat SA, Shueb RH, Che Mat NF (2018) Anti-viral activities of Oroxylum indicum extracts on chikungunya virus infection. Indian J Microbiol 58:68–75. https://doi.org/10.1007/s12088-017-0695-8
Mucsi I, Gyulai Z, Béládi I (1992) Combined effects of flavonoids and acyclovir against herpesviruses in cell cultures. Acta Microbiol Hung 39:137–147
Muya JT, Mwanangombo DT, Tsalu PV et al (2019) Conceptual DFT study of the chemical reactivity of four natural products with anti-sickling activity. SN Appl Sci 1:1457. https://doi.org/10.1007/s42452-019-1438-8
O’Boyle NM, Banck M, James CA et al (2011) Open babel: an open chemical toolbox. J Cheminform 3:33. https://doi.org/10.1186/1758-2946-3-33
Parida MM, Upadhyay C, Pandya G, Jana AM (2002) Inhibitory potential of neem (Azadirachta indica Juss) leaves on Dengue virus type-2 replication. J Ethnopharmacol 79:273–278. https://doi.org/10.1016/S0378-8741(01)00395-6
Philippe C, Rolain Jean-Marc RD (2020) Chloroquine for the 2019 novel coronavirus SARS-CoV-2. Int J Antimicrob Agents 55:105923
Pizzorno A, Padey B, Dubois J et al (2020) In vitro evaluation of antiviral activity of single and combined repurposable drugs against SARS-CoV-2. Antiviral Res 181:104878. https://doi.org/10.1016/j.antiviral.2020.104878
Rajkumar V, Guha G, Kumar RA (2012) Isolation and bioactivity evaluation of two metabolites from the methanolic extract of Oroxylum indicum stem bark. Asian Pac J Trop Biomed 2:S7–S11. https://doi.org/10.1016/S2221-1691(12)60120-8
Rogiers V, Benfenati E, Bernauer U et al (2020) The way forward for assessing the human health safety of cosmetics in the EU—workshop proceedings. Toxicology 436:152421. https://doi.org/10.1016/j.tox.2020.152421
Siriwatanametanon N, Fiebich BL, Efferth T et al (2010) Traditionally used Thai medicinal plants: in vitro anti-inflammatory, anticancer and antioxidant activities. J Ethnopharmacol 130:196–207. https://doi.org/10.1016/j.jep.2010.04.036
Suresh Babu K, Hari Babu T, Srinivas PV et al (2005) Synthesis and in vitro study of novel 7-O-acyl derivatives of Oroxylin A as antibacterial agents. Bioorg Med Chem Lett 15:3953–3956. https://doi.org/10.1016/j.bmcl.2005.05.045
Trujillo C, Sánchez-Sanz G (2016) A study of π–π stacking interactions and aromaticity in polycyclic aromatic hydrocarbon/nucleobase complexes. ChemPhysChem 17:395–405. https://doi.org/10.1002/cphc.201501019
Verdonk ML, Cole JC, Hartshorn MJ et al (2003) Improved protein–ligand docking using GOLD. Proteins Struct Funct Bioinforma 52:609–623. https://doi.org/10.1002/prot.10465
Vlachakis D, Papakonstantinou E, Mitsis T et al (2020) Molecular mechanisms of the novel coronavirus SARS-CoV-2 and potential anti-COVID19 pharmacological targets since the outbreak of the pandemic. Food Chem Toxicol 146:111805. https://doi.org/10.1016/j.fct.2020.111805
Votano JR, Parham M, Hall LH et al (2004) Three new consensus QSAR models for the prediction of Ames genotoxicity. Mutagenesis 19:365–377. https://doi.org/10.1093/mutage/geh043
Wang L, Wang Y, Ye D, Liu Q (2020) Review of the 2019 novel coronavirus (SARS-CoV-2) based on current evidence. Int J Antimicrob Agents 55:105948
WHO (2020) No Title. https://www.who.int/emergencies/diseases/novel-coronavirus-2019
Wu A, Peng Y, Huang B et al (2020a) Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 27:325–328. https://doi.org/10.1016/j.chom.2020.02.001
Wu C, Liu Y, Yang Y et al (2020b) Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B 10:766–788. https://doi.org/10.1016/j.apsb.2020.02.008
Yan R, Cao Y, Chen C et al (2011) Antioxidant flavonoids from the seed of Oroxylum indicum. Fitoterapia 82:841–848. https://doi.org/10.1016/j.fitote.2011.04.006
Yu M-S, Lee J, Lee JM et al (2012) Identification of myricetin and scutellarein as novel chemical inhibitors of the SARS coronavirus helicase, nsP13. Bioorg Med Chem Lett 22:4049–4054. https://doi.org/10.1016/j.bmcl.2012.04.081
Zakaryan H, Arabyan E, Oo A, Zandi K (2017) Flavonoids: promising natural compounds against viral infections. Arch Virol 162:2539–2551. https://doi.org/10.1007/s00705-017-3417-y
Zhang J, Xie B (2020) Current status of potential therapeutic candidates for the COVID-19 crisis. Brain Behav Immun J 87:59–73
Zhao Y, Zeng C, Massiah MA (2015) Molecular dynamics simulation reveals insights into the mechanism of unfolding by the A130T/V mutations within the MID1 Zinc-binding Bbox1 domain. PLoS ONE 10:e0124377
Acknowledgements
The authors are thankful to Dr. A. M. Ittadwar, Principal, Gurunanak College of Pharmacy for giving moral supports throughout the work.
Funding
The authors have no relevant financial or non-financial interests to disclose.
Author information
Authors and Affiliations
Contributions
In the present research all authors have contributed and has given cooperation throughout to approved the manuscript. Sapan Shah, Dinesh Chaple and Sumit Arora has selected research theme. Sapan Shah, Subhash Yende and Govind Lohiya carried out docking method and ADMET studies. Sapan Shah and Keshav Moharir analysed, interpreted results and prepared Figures. Sapan Shah and Dinesh Chaple written manuscript. All authors than reviewed the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Ethics approval
All authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.
Consent for publication
All authors agree to publish article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
13721_2020_279_MOESM1_ESM.docx
Supporting Information: It includes Author information, Corresponding author: shah.sapan@rediffmail.com (Sapan Shah), Effector domain structure (Fig S1), Structure of native ligand N3 (Fig S2), Ligands binding interaction parameter with the main protease of SARS-CoV-2 (Table S1), 2D Interaction of ligands against Mpro protease (Fig S3), Highlights of the study and Graphical abstract. (DOCX 8270 KB)
Rights and permissions
About this article

Cite this article
Shah, S., Chaple, D., Arora, S. et al. Exploring the active constituents of Oroxylum indicum in intervention of novel coronavirus (COVID-19) based on molecular docking method. Netw Model Anal Health Inform Bioinforma 10, 8 (2021). https://doi.org/10.1007/s13721-020-00279-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13721-020-00279-y