
Abstract
Sperm cells are target cells for both estrogens and xenoestrogens. Due to the specific structure of spermatozoa, these hormonal compounds may act on sperm in a non-genomic mechanism only. However, the ESR-mediated signaling pathways are still poorly understood. In this study, we obtained 119 samples from male participants of Caucasian descent who donated semen for standard analysis. We analyzed gene expression of estrogen receptors (ESR1 and ESR2) and their coregulators—proline-, glutamic acid-, and leucine-rich protein 1 (PELP1), and cellular kinase c-Src (SRC). RNA level was established using reverse-transcribed RNA as a template, followed by a polymerase chain reaction. Proteins’ presence was confirmed by western blot and immunocytochemistry techniques. “Normal” values of semen parameters were defined as follows: > 32% sperm with progressive motility, > 4% sperm cells with normal morphology, > 15 × 106 sperm per mL, > 58% live spermatozoa and leukocyte amount < 106 cells per mL, according to WHO 2010 reference. Semen parameters that deviated from these “normal” values were labeled as “abnormal”. Gene expression ratios revealed significant, moderate, and negative correlations for ESR1/ESR2 and weak, negative ESR2/PELP1 correlations in the subgroup of patients with abnormal values of semen parameters. In addition, SRC/PELP1 was moderately and positively correlated in the subgroup with parameters within the reference values established by WHO 2010. Our study showed that both PELP1 scaffolding protein and SRC kinase might influence semen quality via ESRs. It seems that not the expression of a single gene may affect the sperm quality, but more gene-to-gene mutual ratio. Characterization of estrogen-signaling pathway-related genes’ modulated expression in sperm cells could aid in better understanding sperm biology and quality.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Reproductive medical care for men has lagged behind that of women. Even though progress has been made in the field of male reproductive health, idiopathic male infertility remains a challenging condition to diagnose and manage. Very often, it manifests with altered semen characteristics without an identifiable cause. Nearly, half of all infertility cases are due to a male factor, contributing to 30% of these cases and another 20% of cases if combined with female factors [1,2,3]. The interplay of thousands of genes, proteins, and metabolites contributes to male infertility problems. Etiological factors cannot be identified in ~ 70% of the male partners of infertile couples, with the cause of male infertility remaining elusive [1, 2]. Nearly, 10−15% of men in their prime reproductive age are affected by so-called idiopathic male infertility, whose etiology remains unknown. Various genomic studies suggest that more than 500 target genes may be associated with this disease condition [4].
Semen analysis remains a primary diagnostic tool for male infertility [5,6,7]. Given the high variability in standard semen parameters, identification of other additional useful biomarkers has been explored, including those for DNA damage, anti-sperm antibody testing, as well as gamete interaction tests (acrosome reaction testing, sperm penetration assays, sperm-zona pellucida binding assessments, and hyaluronan binding assays) [1, 8, 9]. However, semen analysis remains an imperfect test. Its results can be influenced by physiologic parameters, including medical comorbidities, abstinence intervals, systemic illness, or lifestyle factors such as work environment, physical activity, diet, and environmental exposures [10,11,12,13]. The listed factors can influence most of the results of medical tests performed in assessing the reproductive potential of men and women, not only semen analysis. Therefore, seminological diagnostics should be developed and extended using new biomarkers because the standard semen analysis is insufficient to explain the causes of infertility in many clinical cases.
A review of the published literature over the past few years makes it rather evident that current research concerning male infertility has become more focused on assessing sperm functions at the molecular level. It was proved that estrogens play an important role in spermatogenesis and that spermatozoa are the target cells for estrogens. Not only are sperm cells able to synthesize estrogen, but both estrogen receptors are also expressed in spermatogenic cells at different stages (from spermatogonia to spermatozoa). However, the cellular localization is not identical—both receptor types are located in the midpiece, while ESR2 is also expressed in the tail. In addition, in ejaculated sperm, 17beta-estradiol stimulates functions such as motility, vitality, acrosome reaction, and capacitation. Estrogen-induced biochemical changes occur rapidly, i.e., during capacitation. It suggests the non-genomic action of these hormones in spermatozoa [2, 14, 15].
Maintaining the balance between estrogens and androgens is essential for the proper functioning of the male reproductive system [16]. The observed levels of ESR1/ESR2 expression depend on the male’s tissue type and age [17, 18]. Studies indicate greater male exposure to endogenous and exogenous estrogens in recent years. It may influence male reproductive biology [19,20,21,22,23,24]. Exogenous estrogens are widely distributed in the environment and can be found in plastics, cosmetics, drugs, detergents, and food [16]. This category also includes endocrine-disrupting chemicals present in the environment. These estrogen-like compounds and anti-androgenic chemicals include e.g., dioxins, some pesticides, polychlorinated biphenyls, and dichlorodiphenyltrichloroethane. They can mimic the action of endogenous estrogens and interfere with any aspect of their function, which is crucial in maintaining homeostasis and regulating developmental processes. This way, they adversely affect the endocrine system and male reproductive health [25, 26]. However, the exact molecular mechanism of action remains vague [27,28,29,30,31]. The concept of “chemical mixture effects” has been widely discussed in this context to indicate that combined chemical action may induce different biological effects than an individual chemical’s influence [32,33,34,35].
It was reported that the biological effects of estrogens and xenoestrogens might occur via genomic and rapid, non-genomic pathways, which also involve a wide range of coregulators. Both types of classical estrogen receptors (ESRs; ESR1/ESR2) and G protein-coupled estrogen receptor 1 (GPER, GPR30) have been confirmed in different tissues of the male reproductive tract, spermatogenic cells, and spermatozoa. ESRs expression pattern was studied not only in samples obtained from male patients but also in murine models. ESR2 in the seminiferous tubule is expressed in all spermatogenic cells (i.e., spermatogonia, spermatocyte, round and elongated spermatids) and Sertoli cells. ESR1 expression is very similar, except for spermatids [36, 37]. ESRs are mostly involved in genomic and rapid signaling, while GPER mainly mediates non-genomic effects [28, 31, 38,39,40,41,42,43,44]. Apart from these well-known receptors, it has been suggested that other novel proteins may play a similar role in mediating estrogen signaling in males. These proteins are often characterized as putative estrogen receptors and include estrogen-related receptor, saxiphilin-binding protein, ER-X, and ER-x [45,46,47,48,49,50]. It is important to point out the role of estrogen-signaling coregulators that mediate the estrogen response at the tissue and cellular level, i.e., the novel scaffolding protein, proline-, glutamic acid-, and leucine-rich protein 1 (PELP1), also known as MNAR (modulator of non-genomic action of estrogen receptor). In addition to nuclear receptors and transcription factors (such as activator protein 1, specificity protein 1, or nuclear factor kappaB), PELP1 was proven to interact with several key players of cell cycle progression. These include epidermal growth factor receptor, phosphoinositide 3, and proto-oncogene tyrosine-protein c-Src (SRC) kinases [51,52,53,54,55,56]. It was reported that activation of SRC and SRC-mediated signaling is coordinated by binding of PELP1 and ESR to SRC’s SH3 and SH2 domains, respectively, and is stabilized by the ER-PELP1 interaction through PELP1’s LXXLL motifs [54]. This interaction potentially provides PELP1’s additional level of steroid hormone regulation of cell and/or tissue functions and may affect sperm cell biology.
Our team has already confirmed PELP1 expression in human sperm cells. We established that the elevated expression might be related to deteriorated sperm parameters [57]. Therefore we decided to investigate further whether there is any relationship between estrogen receptors, PELP1, SRC kinase, and sperm parameters, as it may advance our understanding of sperm biology and potentially male fertility.
Materials and methods
Participants
Semen samples were obtained between 2018 and 2020 from 119 participants of Caucasian descent enrolled from the general population with unknown fertility status. They were admitted by the physicians to the Division of Infertility and Reproductive Endocrinology of Poznan University of Medical Sciences (PUMS), due to conception problems, for in-depth andrological consultation. All study participants were of reproductive age. The men who underwent a first-time seminological study and had no knowledge of semen quality (no previous seminological studies) were recruited. Male patients with erectile disorders who could not provide sperm samples during masturbation were excluded from the examinations. To obtain the most homogeneous group, recruited individuals not only complied with the recommended duration of sexual abstinence, from 2 to 7 days but most often, it was a 4-day period. In addition, the men with varicocele, prostate dysfunction, vas deferens obstruction, and metabolic diseases were excluded from further molecular investigations. This study did not analyze the data on having offspring in the participants’ group; therefore, men with reduced semen quality were not necessarily infertile. Patients with fever (≥ 38.5 °C) in the last three months prior to the study were excluded from the study, and if they reported any medical conditions or procedures that might have caused infertility in their history: varicocele, prostate dysfunction, vas deferens obstruction, as well as metabolic diseases, urogenital surgery, scrotal injury, genetic diseases (cystic fibrosis, Kartagener syndrome), cryptorchidism, endocrine hypogonadism, previous radiotherapy or chemotherapy, exposure to diagnostic X-rays, inguinal hernia surgery, suspected urogenital tract infection, chronic parotitis at age 13 and above, chronic diseases or drugs with a gonadotoxic effect, such as psychotropic drugs, antiepileptic drugs or cardiac drugs.
Methods
Semen analysis
Semen analysis was performed manually following “The WHO Laboratory Manual for the Testing and Processing of Human Semen, 5th ed. 2010” [58] under the supervision of an expert in the field of laboratory diagnostics. “Normal” values of semen parameters were referenced as follows: > 32% sperm with progressive motility, > 4% sperm cells with normal morphology, > 15 × 106 sperm per mL, > 58% live spermatozoa, and leukocyte amount < 106 cells per mL. Semen parameters below “normal” WHO reference ranges were defined as “abnormal”.
All analyses were performed using an Axioskop 2 microscope (Zeiss, Carl Zeiss AG, Oberkochen Germany). To determine sperm concentration improved Neubauer hemocytometer chamber (Blaubrand, Brand GmbH, Wertheim, Germany) was used. We focused on the relationship between normal/abnormal parameter values and gene expression; thus, we did not divide patients into groups with normozoospermia, oligozoospermia, asthenozoospermia, etc.
Among 119 participants aged from 23 to 47 years (mean 34 ± 5 years), in 86 patients, at least one semen parameter deviated from the reference range. In 33 participants, all analyzed sperm parameters were within WHO range values [58]. Participants’ clinical characteristics are described in “Results” and Table 1.
Nucleic acid isolation and validation
We aimed to obtain total high molecular weight RNA from the whole fraction of sperm cells, irrespective of their parameters. Thus, we did not use standard isolation techniques (e.g., Percoll, swim-up) from the whole fraction of sperm cells. We did not perform multiple centrifugations to avoid RNA degradation [59]. Semen was washed gently with an equal volume of phosphate-buffered saline (Corning Incorporated, Corning, NY, USA) and pelleted by centrifugation (0.6 × g, 5 min, room temperature). The semen samples were incubated on ice for half an hour in the somatic cell lysis buffer (0.1% SDS, 0.5% Triton X-100 in ddH2O; LabEmpire; Rzeszów, Poland) to lyse the round cells [60, 61] and centrifuged to obtain sperm cells’ fraction. The absence of round cells was established by light-microscopic evaluation. RNA extraction from sperm was performed using 500 μL of acid-guanidinium-phenol reagent (AGP; GenoPlast Biochemicals; Rokocin, Poland). The samples were incubated with gentle shaking in the AGP (250 rpm, 15 min, 55 °C) to separate the proteins and cellular debris. Subsequently, to allow the DNA, RNA, and protein phase-separation, 200 μL of chloroform (Avantor Performance Materials Poland S.A., Gliwice, Poland) were added to the AGP mixture, incubated (3 min, RT), and centrifuged (12,000 × g, 15 min, RT). The aqueous, upper phase was transferred into a new Eppendorf tube, and an equal volume of cold (− 20 °C) absolute ethanol (Avantor Performance Materials Poland S.A., Gliwice, Poland) was then added and thoroughly mixed by inverting the tube a few times. The mixture was transferred into a Direct-zol RNA Miniprep silica matrix column (ZymoResearch; Irvine, CA, USA), and the HMW-RNA was isolated as recommended by the manufacturer. Quantity, quality, and purity were analyzed in compliance with the previously described methodology [62] using a NanoPhotometer® NP-80 UV/VIS Touch (IMPLEN; Munich, Germany). RNA integrity was evaluated using electrophoretic separation under denaturing conditions. The isolated RNA was immediately stored at − 80 °C until further analysis.
Reverse transcription and quantitative PCR
According to the manufacturer’s instructions, a three-step transcriptor reverse transcriptase reaction was performed to synthesize DNA to complementary RNA (cDNA) (Roche; Manheim, Germany). We incubated a mixture of 5 mM oligo(d)T10, 1 mM random hexamer primer (Genomed; Warsaw, Poland), 1 μg HMW-RNA, and RNase-, DNase- and pyrogen-free water (Thermo Fisher Scientific; Waltham, MA, USA) for 10 min at 65 °C. Subsequently, samples were chilled on ice. The HMW-RNA mixture was supplemented with 10 U/rx transcriptor reverse transcriptase, 5 U/rx ribonuclease inhibitor, 1 × reaction buffer (Roche; Manheim, Germany), 0.1 U/μL E. coli poly(A) polymerase, 0.1 mM adenosine triphosphate (New England BioLabs; Ipswich, MA, USA), and 100 mM deoxyribonucleotide triphosphates (Novazym; Poznan, Poland). The total volume of the cDNA reaction mixture was 20 μL. The subsequent steps of cDNA synthesis were followed as described previously [62]. All cDNA samples were synthesized in duplicates and immediately used for qPCR reactions or stored in − 20 °C until further analysis but no longer than seven days.
The quantitative PCR reactions were designed in line with the MIQE guidelines [63] (Online Resource 1, MIQE checklist and data set). The Roche Universal ProbeLibrary (UPL) Assay Design Center (http://qpcr.probefinder.com, last accessed on September 28, 2017) was utilized to determine primer sequences and TaqMan® hydrolysis probe positions for the following genes of interest: ESR1 (F: ccttcttcaagagaagtattcaagg (intron spanning); R: attcccacttcgtagcatttg; probe #69, Roche cat. no.: 04688686001; amplicon length: 160 bp; GenBank sequence accession numbers: NM_001122740.2, NM_001122741.2, NM_001385571.1, NM_001291241.2, NM_001385568.1, NM_001385572.1, NM_001385570.1, NM_001385569.1, NM_000125.4, NM_001328100.2, NM_001291230.2), PELP1 (F: caaggaggagactcacaggag (intron spanning); R: caaggaggagactcacaggag; probe #24, Roche cat. no.: 04686985001; amplicon length: 131 bp; accession numbers: NM_014389.3, NM_001278241.2), and SRC (F: gccatgttcactccggttt; R: cagcgtcctcatctggtttc (intron spanning); probe #21, Roche cat. no.: 04686942001; amplicon length: 100 bp; accession number: NM_005417.5) [64]. The primers were obtained from Genomed (Warsaw, Poland). Regarding ESR2 and HPRT1 reference control gene, we applied ready-to-use assays (PrimePCR, qHsaCEP0052206, BioRad; Hercules, CA, USA and UPL102079, Roche, Manheim, Germany, respectively). The HPRT was used as the most stable of the three reference genes taken under consideration. To assess and evaluate all RNA expression patterns, the LightCycler® 2.0 carousel glass capillary-based system was used (Roche Diagnostics International AG; Rotkreuz, Switzerland).
As described previously, all quantitative polymerase chain reactions (qPCR) were made in a total volume of 20 μL [62] with standard cycling and acquisition steps. Quantitative PCR analysis was performed using the appropriately adjusted and standardized reaction mixtures for Roche UPL probes [62] and 1 × LightCycler® FastStart TaqMan® Probe Master Mix (Roche; Manheim, Germany) according to the manufacturer’s protocol. All reactions were conducted in duplicates. The following actions were performed to avoid contamination with DNA-derived artifacts: non-reverse-transcribed RNA we used as a template instead of cDNA in negative controls and the commercial sets of intron-spanning probes or primers.
For each gene, reaction efficiencies were derived from the relevant standard curves [62]. These values were then compared with the appropriate threshold values and reference gene assays using LC 5.0.0.38 software. All results were presented as concentration ratios (Cr—fluorescence measurement results normalized to standard curves and compared to the HPRT reference gene), corresponding to the relative expression level [65]. Mean values were then evaluated with the use of relevant statistical analyses.
Western blot
For ten randomly selected semen samples, sperm cells were incubated in RIPA Lysis Buffer (Merck KGaA, Darmstadt, Germany). Total extracts were shaken gently (30 min at 4 °C) and centrifuged (10,000 × g, 20 min, 4 °C). The protein-containing supernatant was collected, and protein concentration was assessed using QuickStart Bradford 1 × Dye Reagent (BioRad, USA) and NanoPhotometer® NP80 (IMPLEN, Munich, Germany). Subsequently, 20 µg of protein lysate per lane was diluted with Laemmli buffer (BioRad; Hercules, CA, USA), denatured (80 °C, 10 min), and loaded onto an SDS-polyacrylamide gel (TGX FastCast Acrylamide Kit 10%; BioRad; Hercules, CA, USA). After electrophoretic separation (90 min, 120 V), proteins were transferred (90 min., 0.3 A, 4 °C cold buffer, cooling cartridge) to the activated Immobilon PVDF membranes (Merck KGaA, Darmstadt, Germany) in methanol (Avantor Performance Materials, Poznan, Poland). Subsequently, the membranes were incubated with gentle shaking (200 rpm, 1 h, RT) in blocking buffer (TBS-T; Abcam; Cambridge; UK), supplemented with 5% blotting-grade blocker (BioRad; Hercules, CA, USA). An overnight incubation (4 °C, 200 rpm gentle shaking) was used with primary antibodies against: ESR1 (1:1000, LS-C88420; Lifespan Biosciences; Seattle, WA, USA), ESR2 (1:1000, ab3576; Abcam; Cambridge, UK), PELP1/MNAR (1:1000, A300-180A; Bethyl; Montgomery, TX, USA), SRC (1:1000, orb379229; Biorbyt; Cambridge, UK), p-SRC (phospho-Tyr529 SRC; 1:1000, orb14869; Biorbyt; Cambridge, UK) and GAPDH (1:2500, sc-25778; Santa Cruz; Dallas, TX, USA). All antibodies were diluted in TBS-T buffer with 3% blotting-grade blocker (BioRad; Hercules, CA, USA). The membranes were washed with TBS-T buffer (3 × ; 5 min, RT) and incubated with secondary polyclonal horseradish peroxidase-conjugated goat anti-rabbit antibody (1:1000, Agilent Dako, Santa Clara, USA) on an orbital shaker (1 h, RT with gentle shaking). Blots were visualized by enhanced chemiluminescence using Clarity Western ECL (BioRad, Herkules, CA, USA), and results were documented using G:BOX (Syngene, Cambridge, UK). The 3-color prestained protein marker (Blirt, Gdańsk, Poland) was used as the molecular mass standard.
Immunocytochemistry
Protein localization was assessed using standard immunocytochemical procedures as described previously [57]. Formalin-fixed cell smears were prepared and boiled in a microwave oven twice (2 × 10 min, 200 W, water bath) in citrate buffer (pH 6.0, 0.1 mM citric acid solution, 0.1 mM sodium citrate solution; Avantor Performance Materials; Poznan, Poland). After each step of microwaving antigen retrieval, slides were chilled (20 min, room temperature) and incubated for 3 min in a 3% hydrogen peroxide solution (Avantor Performance Materials, Poznan, Poland). The next step included placing the slides in TBS-T buffer (containing 3% bovine serum albumin, fraction V; Merck KGaA; Darmstadt, Germany) for 1 h at RT. The same primary antibodies were used as described in the western blot section at a 1:100 dilution for all immunocytochemical reactions. Unbounded antibodies were washed in TBS-T buffer (3 × , 10 min, RT). The antigens were visualized using EnVision + /HRP system and 3,3′-diaminobenzidine chromogen (Agilent Dako, Santa Clara, USA). Controls included detection reactions carried out under identical conditions, except that the primary antibodies were replaced by nonimmune serum. The slides were assessed using light microscopy (Olympus CX41 microscope, Olympus Corporation, Tokyo, Japan).
Statistical analyses
Results were evaluated using Statistica® Version 13.5.0 software for Windows (TIBCO Software Inc., Palo Alto, CA, USA). Participants with semen parameters within the WHO 2010 reference range were compared to those with at least one or more parameters outside the range. In addition, subdivisions were created to analyze each semen parameter separately (concentration, progressive motility, vitality, and morphology). All continuous variables were checked for outliers and were winsorized if any were present using the equation (mean ± 2 × standard deviations) [66]. Obtained results were described by the mean ± standard deviation, and median [interquartile range] values. The Shapiro–Wilk test was applied for the normality of continuous variables distribution assessment. The parametric t Student (also with separate variance estimates) and Mann–Whitney U (1- sided and 2-sided) tests were applied for statistical analyses in two groups. The Spearman rank correlation test was applied to evaluate the strength of the correlation coefficient (R). The absolute magnitude of the observed correlation coefficient was interpreted as follows: R < 0.1—negligible, R < 0.4—weak, R < 0.7 moderate, R < 0.9 strong, and R ≥ 0.9 very strong [67]. The Bonferroni–Hochberg correction was applied in the case of multiple testing. Data were considered statistically significant at p < 0.05.
Results
The general approach to data inquiry is shown in Fig. 1. If all analyzed semen parameters were within the WHO reference range values [58], men were assigned to the normal basic parameters subgroup; otherwise, they were assigned to the group with at least one or more parameters outside the range (abnormal values of semen parameters subgroup). We wanted to assess whether the level of expression of the given genes is related to changes in sperm parameters. Thus, participants’ subdivisions were created (Fig. 1).

Patient grouping scheme; n number of participants. The cutoff points for each parameter are defined in square brackets. Participants above the cutoff points were assigned to the subgroup with a normal value of semen parameter and those below to the abnormal semen parameter value subgroup
ESRs, SRC, and PELP1 mRNA expression differences
The purpose of the analysis was to establish the expression pattern of genes of interest in relation to sperm parameters. The normalized quantitative mRNA levels were expressed as concentration ratios (Cr values—see “Materials and Methods”).
The patients differed in all analyzed sperm parameters (p < 0.05). Amplification of ESR1, ESR2, PELP1, and SRC was analyzed in all samples. However, genes of interest expression did not differ significantly between normal and abnormal values of semen parameters. The detailed characteristics of semen parameters and mRNA expression levels are provided in Table 1. It is important to note that the differences were observed for particular sperm parameters when these were analyzed separately, as previously mentioned.
Considering the single sperm parameters, apart from vitality, the normalized expression of ESR1 was higher in subgroups of patients with normal basic parameters, but the differences were insignificant. Regarding ESR2, its expression was significantly higher in patients with abnormal values of semen parameters except for vitality. SRC was highly expressed in the subgroups of patients with normal standard semen parameters, apart from the motility parameter. In contrast, PELP1 was highly expressed in the patients with abnormal values of semen parameters, apart from the motility parameter, where its expression was higher when compared to the subgroup of patients with normal standard semen parameters. The differences were not statistically significant (Online Resource 1, Tables S1–S4).
Concerning sperm concentration, the patients differed in analyzed sperm parameters (p < 0.05). As expected, the observed values were higher in the subgroup with normal values of semen parameters. The total HMW-RNA concentration obtained from the spermatozoa was significantly higher in patients with normal sperm parameters. The normalized expression of all genes of interest did not differ significantly between subgroups with normal and abnormal values of semen parameters (Online Resource 1, Supplementary Table S1).
With regard to progressive motility, the other sperm parameters differed significantly in all cases (p < 0.05) apart from the round cell parameter (p = 0.68). Considering the genes of interest expression level, only the expression of ESR2 differed in patients with abnormal sperm values compared to patients with values within the WHO reference range (p = 0.0008). ESR2 expression was significantly higher in the subgroup with abnormal values of semen parameters (Online Resource 1, Supplementary Table S2).
Regarding vitality, most other sperm parameters differed significantly, apart from morphology and round cell percentage (p > 0.05) (Supplementary Table S3). However, there was no significant difference in the expression level of the analyzed genes. Only ESR2 normalized expression was slightly higher in men with abnormal sperm vitality, but the difference was not statistically significant (p = 0.0852; Supplementary Table S3), However, the number of cases was too low to draw far-reaching conclusions.
For morphology, similarly as in the case of the progressive motility subgrouping, the only difference was observed for the round cell percentage, which was lower in the subgroup with normal values of semen parameters (p > 0.05; Supplementary Table S4). The normalized expression of ESR1 and ESR2 differed between patients with normal and abnormal sperm morphology values (p = 0.006, and p = 0.0158, respectively; Supplementary Table S4).
ESRs, SRC, and PELP1 gene-to-gene ratios’ expression correlations
When analyzed for all participants, gene-to-gene expression Cr ratios were significantly, negatively, and weakly-to-moderately correlated in most cases, apart from SRC&PELP1, which was weak and positive. In the case of ESR1&PELP1 and ESR2&SRC, the correlations were not significant (Fig. 2.; Online Resource 1; Table S5). Considering the Cr ratios in the subgroupings, the gene expression ratios revealed statistically significant, moderate, and negative correlation coefficients for ESR1&ESR2 in both subgroups for all participants (p = 0.016; R = − 0.55 and p < 0.0001; R = − 0.55, respectively). Gene expression ratios were also weakly and negatively but significantly correlated in the case of ESR2&PELP1 in the subgroup of patients with at least one abnormal value of semen parameters (p = 0.0092; R = − 0.33) and moderately and positively correlated for SRC&PELP1 in the subgroup with parameters within the WHO reference range (p = 0.0247; R = 0.43; Fig. 2.; Online Resource 1 Table S5).

Dot plot showing the correlation coefficients 1*log10 p values of all participants (n = 109) and in groups with normal (n = 33) and abnormal (n = 86) values of semen parameters. Reference horizontal lines represent the p values thresholds of p = 0.05 (green line), p = 0.01 (yellow line), and p = 0.001 (red line). Participants above the cutoff points were assigned to the subgroup with a normal value of semen parameter and those below to the abnormal semen parameter value subgroup. The cutoff points were as follows: concentration—15 × 106 sperm/mL, progressive motility—32%, vitality—58%, and morphology—4%
Correlation coefficients for normalized expression were calculated for semen parameters in the subgroups (all parameters within the WHO 2010 reference range vs. at least one or more outside the range) and separately in subdivisions for sperm concentration, progressive motility, vitality, and morphology as previously described.
Correlation coefficients for normalized expression were calculated for semen parameters in the subgroups (all parameters within the WHO 2010 reference range vs. at least one or more outside the range) and separately in subdivisions for concentration, progressive motility, vitality, and morphology as previously described. Regarding ESR1, weak associations were observed between expression level and sperm morphology in participants with all parameters within the WHO 2010 reference range vs. those with at least one or more outside the range. The same tendency applied to ESR1 expression correlation and sperm parameters when analyzed separately for concentration and vitality. The correlation coefficients for the remaining parameters were not significant. Concerning ESR2, associations were observed between the gene of interest and progressive motility and morphology parameters in participants with at least one or more parameters outside the range. A weak negative correlation was observed between the gene of interest and progressive motility in patients with normal concentration and vitality values, and a moderate negative correlation was seen in patients with abnormal sperm concentration and vitality values. In addition, a moderate negative correlation was shown for the gene of interest and round cell percentage and morphology in the normal patient subgroup. Other correlations were not statistically significant. For SRC, only in patients with abnormal sperm motility moderate positive and negative correlations were observed. Those statistically significant correlations are related to the following parameters: total motility (positive), immotile spermatozoa (negative), and vitality (positive). For PELP1, a moderate positive correlation with round cell percentage was found only in patients with abnormal sperm values of motility (Online Resource 1; Supplementary Table S6).
ESRs, SRC, and PELP1 gene-to-gene ratios’ expression correlations with semen parameters
When analyzed separately for each parameter, gene expression ratios significantly correlated in ESR1&ESR2 regardless of subgrouping, except for vitality, where it was not statistically significant in the group with abnormal values of semen parameters. ESR1&SRC Cr values were significantly, moderately, and negatively correlated in the subgroup with the abnormal value of the concentration parameter (p = 0.0117; R = − 0.60) and weakly, negatively correlated in the normal subgroup for the vitality parameter (p = 0.0077; R = − 0.29). Both ESR1&PELP1, as well as ESR2&SRC ratios, showed no significant correlation coefficients. For ESR2&PELP1, the normalized concentration ratios were negatively and significantly correlated in the case of sperm progressive motility and vitality regardless of subgrouping. For sperm concentration and morphology, correlation coefficients were significant in the subgroup with normal value for concentration parameter (p = 0.0158; R = − 0.28), and in the abnormal for morphology (p = 0.0126; R = − 0.9). Interestingly, concerning SRC&PELP1, the Cr values were significantly correlated for all parameters in the “normal” subgroups (Fig. 3.; Online Resource 1; Table S7).

Dot plot showing the correlation coefficients 1*log10 p values. Sperm samples were assigned to subgroups with normal or abnormal values of semen parameters: concentration (n = 89 vs. n = 30), morphology (n = 38 vs. n = 81), progressive motility (n = 84 vs. n = 35), or vitality (n = 110 vs. n = 9). Reference horizontal lines represent the p values thresholds of p = 0.05 (green line), p = 0.01 (yellow line), and p = 0.001 (red line)
ESRs, SRC, and PELP1 protein presence and localization
The identification of immunoreactive bands confirmed the presence of the analyzed proteins in sperm cells at the expected sizes (ESR1: 66 kDa; ESR2: 55 kDa; PELP1: 170 kDa; SRC: 60 kDa and p-SRC: 61 kDa). The GAPDH reference protein bands (37 kDa) were present in all samples (Fig. 4).

Western blot analysis of analyzed proteins in selected samples. Lines: 1—participant no. 12, 2—participant no. 26, 3—participant no. 70. The proteins’ identities are confirmed by their molecular mass in kilo Daltons (kDa)
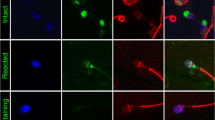
Immunocytochemical staining enabled the identification of all studied proteins in the cells of interest (Fig. 5). The ESR1 protein was primarily localized in the midpiece and postequatorial part of the spermatozoa head, and ESR2 was identified in midpieces. In comparison, PELP1 was expressed not only in midpieces but also in the acrosome region, including peri-acrosomal space. Similarly, SRC was shown in the midpiece and peri-acrosomal space, while p-SRC was mainly expressed in midpieces.

Immunocytochemical staining for analyzed proteins in control samples and analyzed sperm cells. Immunostaining for: a ESR1, b ESR2, c PELP1, d SRC, e p-SRC. The bar represents 100 μm. The arrows indicate a positive reaction
In the case of all samples, the localization of all described proteins did not differ meaningly between normal and abnormal sperm cells.
Discussion
In this paper, we confirmed the presence of ESR1, ESR2, PELP1, and SRC transcripts and proteins in the ejaculated spermatozoa. We analyzed potential relationships between the expression of molecules involved in ESR-mediated signaling pathways, such as ESR1, ESR2, and their coregulators, including PELP1 and SRC kinase.
The role of estrogens is indisputable in regulating spermatogenesis and gamete functional maturation. In ejaculated sperm, estrogens may influence sperm functions such as capacitation, acrosome reaction, motility, and vitality. There are also indications that estrogens take part in the spermatogenic regulation of glucose and lipid metabolism [16, 17, 27,28,29, 42, 68]. As sperm cells are transcriptionally inactive, non-genomic signal transduction via intracellular secondary messengers is the key pathway in these cells. The effects of non-genomic estrogen are mediated via either ESRs or membrane-bound receptors, including GPER, or through interactions with other membrane proteins and/or lipids [27, 69]. PELP1 is considered one of the central modulators of rapid estrogen signaling via estrogen receptors and was reported to interact with several key players of cell cycle progression, i.e., SRC kinases [51,52,53,54,55,56].
In our study, the expression genes of interest did not differ significantly in participants with all sperm parameters within the reference range compared to those with at least one outside the range. Differences were observed, however, for some parameters when analyzed separately. With regard to ESRs, data evaluating their localization in sperm are not entirely consistent. ESR1 is reportedly expressed in the equatorial and upper post-acrosomal regions of the sperm head and the midpiece. Studies suggest that ESR2 is mainly present in the midpiece region and mitochondria but also in the tail region [70,71,72]. The mutual localization of ESRs within the midpiece region and in mitochondria implies an essential role of estrogen in sperm motility and metabolism [14, 42]. Our study confirms these findings, as ESR1 protein was primarily localized in the midpiece and postequatorial part of the spermatozoa head, and ESR2 was identified in midpieces. In addition, ESR1 expression was higher, but not significant, in the participants’ group with the motility parameter values within the WHO range. Only the normalized expression of ESR2 differed significantly in patients with the abnormal motility parameter value compared to those within the WHO reference range [58]. Interestingly, normalized ESR2 expression was also higher for most sperm parameters in men assigned to the abnormal subgroup when the expression pattern was analyzed for each parameter separately, except for concentration.
We established that in the group of men with normal sperm morphology parameters, the ESR1 expression was significantly higher. On the other hand, ESR2 was also higher in normal subgroups apart from the vitality parameter. It may imply the involvement of estrogen receptors in regulating various cell functions. It may also reflect the spermatozoa polarization in the structure and function, expressed in their ability to sectionalize particular signaling pathways to the target regions. Furthermore, due to the presence of ESRs within the equatorial segment or upper post-acrosomal region, it cannot be excluded that estrogens are involved in the activation of the acrosomal reaction and capacitation in cooperation with SRC. Tyrosine kinase SRC has been identified as a crucial molecule that might mediate rapid estrogen action by physical interaction with ESR1 [73,74,75,76,77]. PELP1 could also be involved in this phenomenon as it was reported to control ESR1-induced SRC activation [51, 54]. Our data seem to confirm such a hypothesis, as we observed a positive correlation between SRC and PELP1 expression ratios in normozoospermic patients. In addition, a positive correlation of these genes was observed in patients with normal sperm morphology, progressive motility, and vitality sperm values. In addition, in patients with altered motility, SRC mRNA level was positively associated with sperm progressive motility percentage and negatively with immotile sperm percentage.
Regarding ESR2, its normalized expression differed significantly in patients with abnormal motility values compared to those within the WHO reference range [58]. Interestingly, normalized ESR2 expression was also higher in most subgroups with at least one abnormal value of semen parameters (when the expression pattern was analyzed for each parameter separately), apart from concentration. It seems that estrogen signaling mediated via ESR2 in the presence of PELP1 may contribute to altered sperm characteristics, as we found a significant, weak negative correlation between ESR2 and PELP1 in participants with at least one or more parameters outside the range. It was also seen in the normal subgroups of patients for concentration, progressive motility, and vitality parameters. A negative correlation was also observed in patients with altered morphology and motility parameters (weak and moderate, respectively). A strong, negative correlation was observed in patients with abnormal vitality. This observation suggests that altered expression of not one, but the entire ESR-mediated social network signaling pathway, could influence sperm concentration, motility, vitality, and morphology.
PELP1 expression was higher in participants with at least one or more parameters outside the WHO reference range. This result is consistent with previous investigations aiming to confirm PELP1 presence in human spermatozoa. However, it needs to be mentioned that our previous mRNA analyses showed PELP1 presence in pooled semen samples. We also observed higher protein expression in samples with abnormal values of semen parameters [57]. In this study, we established a method that allowed us to analyze mRNA expression in samples obtained from single patients.
Our immunocytochemical studies revealed that PELP1 is expressed in the midpieces; it cannot be excluded that its expression may be induced by changed cell altered energy metabolism, i.e., due to oxidative stress. Based on the above results, it may be suggested that the molecular mechanism of the ESR2-PELP1 interaction could be a more susceptible target for endocrine disruptors. However, the literature does not unequivocally confirm the negative impact of xenoestrogens on male fertility. Still, in light of numerous studies, xenoestrogens probably have a significant negative effect on fertility. Studies indicate that exposure to certain concentrations of xenoestrogens correlates with lower sperm quality as well as with testicular carcinogenesis and endocrine disorders, including abnormalities in the level of the reproductive hormones [32, 78,79,80,81]. They may increase the number of immature sperm, sperm sex chromosome disomy, and decrease sperm motility. In addition, it has been suggested that estrogen metabolites might increase oxidative stress in human sperm cells [19,20,21]. On the other hand, papers report that some xenoestrogens may have antioxidant and protective properties concerning sperm cells. Under certain circumstances, some xenoestrogens (phytoestrogens, such as genistein) may influence the ability of sperm to fertilize the egg. It should be mentioned that phytoestrogens have incomparably lower biological activity than endocrine disruptors like bisphenol A, DDT, dioxins, or phthalates [22, 23]. However, it seems that the effects of xenoestrogens on sperm biology predominantly depend on substrate type and concentration [24]. All mentioned examples could support our results concerning significant correlations of ESR2 with the percentage of progressive motility and morphology in participants with at least one semen parameter outside the WHO reference range, men with normal/abnormal concentration values, and the normal subgroup of participants when vitality is analyzed.
All the above considerations stay in line with the concept of an exclusive modality of estrogen action in spermatozoa. It can be assumed that estrogen sensitivity depends on the availability of ESRs and their coregulators. Several groups have tried to detect ESRs in sperm to clarify the role of estrogens on spermatozoa. Unfortunately, the results are not always consistent. It may be because several splice variants of the described ESRs, particularly ESR2, are either insufficiently characterized or detected using various methodologies or/and antibodies of different origins. Therefore, it cannot be excluded that an uncharacterized protein sharing homology with ESR2 exists and may cross-react with ESR2 antibodies [27]. Of important note, apart from the well-known classical ESRs and GPER, other novel membrane-associated proteins have been considered estrogen receptors, including estrogen-related receptors, saxiphilin-binding protein, ER-X, ER-x, and putative membrane estrogen receptors. Among many ER-X’s features, those worth mentioning in the context of this discussion are as follows: it has a high affinity for binding estradiol; it has sequence homology to ESR1; it can activate extracellular signal-regulated kinases ERK1/2. ER-x is characterized as a membrane receptor with the ability to mediate estrogen responses in some breast cancer cell lines and, therefore, can influence apoptosis, transcriptional regulation, and growth factor signaling. However, as a consensus has not been reached concerning the definition of an ESR, it is still debatable whether these proteins could be considered estrogen receptors [45,46,47,48,49,50].
Although the key role of estrogens in the male reproductive system has been substantiated, findings regarding estrogens’ impact on sperm biology are inconclusive. It seems that ESR1, ESR2, PELP1, and SRC may be involved in several molecular pathways concerning estrogen signaling in sperm cells by controlling different activities throughout the spermatozoa lifecycle, positively and negatively influencing their biology. However, unveiling all aspects of their interactions and associations with sperm biology requires further assessment. Since male infertility concerns the interplay of thousands of genes, proteins, and metabolites, it is crucial to investigate other molecular targets that could serve as new biomarkers [1,2,3]. Establishing the dependencies within estrogen signaling in sperm cells could help answer several questions concerning idiopathic infertility.
In this paper, we confirmed the presence of ESR1, ESR2, PELP1, and SRC transcripts and proteins in the ejaculated spermatozoa. We analyzed potential mRNA and protein expression relations of molecules involved in ESR-mediated signaling pathways, such as ESR1, ESR2, and their coregulators, including PELP1 and SRC kinase.
Our findings suggest that ESR1, ESR2, PELP1, and SRC may be involved in several molecular pathways concerning estrogen signaling in sperm cells, which can control various activities throughout the spermatozoa life cycle. To a small extent, expression of SRC and PELP1 was linked with the sperm parameters in comparison to ESRs differed levels in subgroups of participants with normal and abnormal values of particular semen parameters. It seems that not the expression of a single gene may affect the sperm quality but more gene-to-gene mutual ratio. The phenomenon of genes relation could be linked with their mutual interactions or other molecules that play important roles in the estrogen-signaling pathways. Characterization of estrogen-signaling pathway-related genes’ modulated expression in sperm cells could aid in better understanding sperm biology and quality. Further colocalization studies would need to be performed to establish the particular relationships between these proteins.
Data availability
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
References
Krzastek SC, Smith RP, Kovac JR. Future diagnostics in male infertility: genomics, epigenetics, metabolomics and proteomics. Transl Androl Urol. 2020;9:S195-205.
Cannarella R, Condorelli RA, Mongioì LM, La Vignera S, Calogero AE, La VS, et al. Molecular biology of spermatogenesis: novel targets of apparently idiopathic male infertility. Int J Mol Sci. 2020;21:1728.
Guerri G, Maniscalchi T, Barati S, Busetto GM, Del Giudice F, De Berardinis E, et al. Non-syndromic monogenic male infertility. Acta Biomed. 2019;90:62–7.
Kothandaraman N, Agarwal A, Abu-Elmagd M, Al-Qahtani MH. Pathogenic landscape of idiopathic male infertility: new insight towards its regulatory networks. NPJ Genomic Med. 2016;1:16023.
Agarwal A, Parekh N, Selvam MKP, Henkel R, Shah R, Homa ST, et al. Male oxidative stress infertility (MOSI): proposed terminology and clinical practice guidelines for management of idiopathic male infertility. World J Mens Health. 2019;37:296–312.
Duffy JMN, Adamson GD, Benson E, Bhattacharya S, Bhattacharya S, Bofill M, et al. Top 10 priorities for future infertility research: an international consensus development study† ‡. Hum Reprod. 2020;35:2715–24.
Bracke A, Peeters K, Punjabi U, Hoogewijs D, Dewilde S. A search for molecular mechanisms underlying male idiopathic infertility. Reprod Biomed. 2018;36:327–39.
Oehninger S, Franken DR, Ombelet W. Sperm functional tests. Fertil Steril. 2014;102:1528–33.
Rex AS, Aagaard J, Fedder J. DNA fragmentation in spermatozoa: a historical review. Androl. 2017;5:622–30.
He B-M, Chen R, Sun T-Q, Yang Y, Zhang C-L, Ren S-C, et al. Prostate cancer risk prediction models in Eastern Asian populations: current status, racial difference, and future directions. Asian J Androl. 2020;22:158.
Gabrielsen JS, Tanrikut C. Chronic exposures and male fertility: the impacts of environment, diet, and drug use on spermatogenesis. Andrology. 2016;4:648–61.
Wijesekara GUS, Fernando DMS, Wijerathna S, Bandara N. Environmental and occupational exposures as a cause of male infertility. Ceylon Med J Ceylon Med J. 2015;60:52–6.
Condorelli RA, Calogero AE, Russo GI, La Vignera S. From spermiogram to bio-functional sperm parameters: when and why request them? J Clin Med. 2020;9:406.
Aquila S, De Amicis F. Steroid receptors and their ligands: effects on male gamete functions. Exp Cell Res. 2014;328:303–13.
Kotwicka M, Skibinska I, Jendraszak M, Jedrzejczak P. 17β-estradiol modifies human spermatozoa mitochondrial function in vitro. Reprod Biol Endocrinol. 2016;14:50.
Adeoya-Osiguwa SA, Markoulaki S, Pocock V, Milligan SR, Fraser LR. 17beta-estradiol and environmental estrogens significantly affect mammalian sperm function. Hum Reprod. 2003;18:100–7.
Carreau S, Delalande C, Silandre D, Bourguiba S, Lambard S. Aromatase and estrogen receptors in male reproduction. Mol Cell Endocrinol. 2006;246:65–8.
Aquila S, Sisci D, Gentile M, Middea E, Catalano S, Carpino A, et al. Estrogen receptor (ER)α and ERβ are both expressed in human ejaculated spermatozoa: evidence of their direct interaction with phosphatidylinositol-3-OH kinase/Akt pathway. J Clin Endocrinol Metab. 2004;89:1443–51.
Li DK, Zhou Z, Miao M, He Y, Wang J, Ferber J, et al. Urine bisphenol-A (BPA) level in relation to semen quality. Fertil Steril Elsevier Ltd. 2011;95:625-630.e4.
Bennetts LE, De Iuliis GN, Nixon B, Kime M, Zelski K, McVicar CM, et al. Impact of estrogenic compounds on DNA integrity in human spermatozoa: evidence for cross-linking and redox cycling activities. Mutat Res Fundam Mol Mech Mutagen. 2008;641:1–11.
Radwan M, Wielgomas B, Dziewirska E, Radwan P, Kałużny P, Klimowska A, et al. Urinary bisphenol A levels and male fertility. Am J Mens Health. 2018;12:2144–51.
Martinez-Soto JC, De Dioshourcade J, Gutiérrez-Adán A, Landeras JL, Gadea J. Effect of genistein supplementation of thawing medium on characteristics of frozen human spermatozoa. Asian J Androl. 2010;12:431–41.
Hinsch KD, Aires V, Hägele W, Hinsch E. In vitro tests for essential sperm functions using the phyto-oestrogen genistein as a test substance. Andrologia. 2000;32:225–31.
Skibińska I, Jendraszak M, Borysiak K, Jędrzejczak P, Kotwicka M. 17β-estradiol and xenoestrogens reveal synergistic effect on mitochondria of human sperm. Ginekol Pol. 2016;87:360–6.
Sikka SC, Wang R. Endocrine disruptors and estrogenic effects on male reproductive axis. Asian J Androl. 2008;10:134–45.
Sifakis S, Androutsopoulos VP, Tsatsakis AM, Spandidos DA. Human exposure to endocrine disrupting chemicals: effects on the male and female reproductive systems. Environ Toxicol Pharmacol Environ Toxicol Pharmacol. 2017;51:56–70.
Dostalova P, Zatecka E, Dvorakova-Hortova K. Of oestrogens and sperm: a review of the roles of oestrogens and oestrogen receptors in male reproduction. Int J Mol Sci. 2017;18:904–27.
Li X, Li H, Jia L, Li X, Rahman N. Oestrogen action and male fertility: experimental and clinical findings. Cell Mol Life Sci Springer Basel. 2015;72:3915–30.
Carreau S, Bouraima-Lelong H, Delalande C. Estrogens: new players in spermatogenesis. Reprod Biol. 2011;11:174–93.
Carreau S, Hess RA. Oestrogens and spermatogenesis. Philos Trans R Soc. 2010;365:1517–35.
Hess RA, Cooke PS. Estrogen in the male: a historical perspective. Biol Reprod. 2018;99:27–44.
Adegoke EO, Rahman MS, Pang MG. Bisphenols threaten male reproductive health via testicular cells. Front Endocrinol (Lausanne). 2020;11:624.
Adegoke EO, Rahman MS, Amjad S, Pang WK, Ryu DY, Park YJ, et al. Bisphenol A damages testicular junctional proteins transgenerationally in mice. Environ Pollut. 2022;302:119067.
Kiwitt-Cárdenas J, Adoamnei E, Arense-Gonzalo JJ, Sarabia-Cos L, Vela-Soria F, Fernández MF, et al. Associations between urinary concentrations of bisphenol A and sperm DNA fragmentation in young men. Environ Res. 2021;199:111289.
Jeminiwa BO, Knight RC, Abbot KL, Pondugula SR, Akingbemi BT. Gonadal sex steroid hormone secretion after exposure of male rats to estrogenic chemicals and their combinations. Mol Cell Endocrinol. 2021;533:111332.
Nudmamud-Thanoi S, Sueudom W, Tangsrisakda N, Thanoi S. Changes of sperm quality and hormone receptors in the rat testis after exposure to methamphetamine. Drug Chem Toxicol. 2016;39:432–8. https://doi.org/10.3109/01480545.2016.1141421.
Mäkinen S, Mäkelä S, Weihua Z, Warner M, Rosenlund B, Salmi S, et al. Localization of oestrogen receptors alpha and beta in human testis. Mol Hum Reprod Oxford Academic. 2001;7:497–503.
Barut O, Seyithanoglu M, Kucukdurmaz F, Demir BT, Olmez C, Dogan NT, et al. Relationship between the G protein-coupled oestrogen receptor and spermatogenesis, and its correlation with male infertility. Andrologia. 2020. https://doi.org/10.1111/and.13779.
Fietz D, Ratzenböck C, Hartmann K, Raabe O, Kliesch S, Weidner W, et al. Expression pattern of estrogen receptors α and β and G-protein-coupled estrogen receptor 1 in the human testis. Histochem Cell Biol Histochem Cell Biol. 2014;142:421–32.
Chimento A, De LA, Nocito MC, Avena P, La PD, Zavaglia L, et al. Role of GPER-mediated signaling in testicular functions and tumorigenesis. Cells. 2020;9:2115 (Multidisciplinary Digital Publishing Institute).
Carreau S, Bois C, Zanatta L, Silva FRMB, Bouraima-Lelong H, Delalande C. Estrogen signaling in testicular cells. Life Sci. 2011;89:584–7.
Guido C, Perrotta I, Panza S, Middea E, Avena P, Santoro M, et al. Human sperm physiology: estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ) influence sperm metabolism and may be involved in the pathophysiology of varicocele-associated male infertility. J Cell Physiol. 2011;226:3403–12.
Cooke PS, Nanjappa MK, Ko C, Prins GS, Hess RA. Estrogens in male physiology. Physiol Rev Am Physiol Soc. 2017;97:995–1043.
Guercio G, Saraco N, Costanzo M, Marino R, Ramirez P, Berensztein E, et al. Estrogens in human male gonadotropin secretion and testicular physiology from infancy to late puberty. Front Endocrinol (Lausanne). 2020;11:1–14.
Micevych PE, Dewing P. Membrane-initiated estradiol signaling regulating sexual receptivity. Front Endocrinol (Lausanne). 2011;2:1–9.
Toran-Allerand CD, Guan X, MacLusky NJ, Horvath TL, Diano S, Singh M, et al. ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J Neurosci. 2002;22:8391–401.
Kampa M, Notas G, Pelekanou V, Troullinaki M, Andrianaki M, Azariadis K, et al. Early membrane initiated transcriptional effects of estrogens in breast cancer cells: first pharmacological evidence for a novel membrane estrogen receptor element (ERx). Steroids. 2012;77:959–67.
Qiu J, Bosch MA, Tobias SC, Grandy DK, Scanlan TS, Rønnekleiv OK, et al. Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J Neurosci. 2003;23:9529–40.
Duliban M, Gurgul A, Szmatola T, Pawlicki P, Milon A, Arent ZJ, et al. Mouse testicular transcriptome after modulation of non-canonical oestrogen receptor activity. Reprod Fertil Dev CSIRO PUBLISHING. 2020;32:903–13.
Misra S, Pandey AK, Gupta S, Kumar A, Khanna P, Shankar J, et al. Estrogen related receptor is required for the testicular development and for the normal sperm axoneme/mitochondrial derivatives in Drosophila males. Sci Rep. 2017. https://doi.org/10.1038/srep40372.
Cheskis BJ, Greger J, Cooch N, McNally C, Mclarney S, Lam HS, et al. MNAR plays an important role in ERa activation of Src/MAPK and PI3K/Akt signaling pathways. Steroids. 2008;73:901–5.
Vadlamudi RK, Kumar R. Functional and biological properties of the nuclear receptor coregulator PELP1/MNAR. Nucl Recept Signal. 2007;5:e004.
Sareddy GR, Vadlamudi RK. PELP1: Structure, biological function and clinical significance. Gene. 2016;585:128–34.
Barletta F, Wong C-W, McNally C, Komm BS, Katzenellenbogen B, Cheskis BJ. Characterization of the interactions of estrogen receptor and MNAR in the activation of cSrc. Mol Endocrinol. 2004;18:1096–108.
Brann DW, Zhang QG, Wang RM, Mahesh VB, Vadlamudi RK. PELP1-A novel estrogen receptor-interacting protein. Mol Cell Endocrinol. 2008;290:2–7.
Vadlamudi RK, Wang RA, Mazumdar A, Kim Y, Shin J, Sahin A, et al. Molecular cloning and characterization of PELP1, a novel human coregulator of estrogen receptor alpha. J Biol Chem. 2001;276:38272–9.
Skibińska I, Andrusiewicz M, Soin M, Jendraszak M, Urbaniak P, Jedrzejczak P, et al. Increased expression of PELP1 in human sperm is correlated with decreased semen quality. Asian J Androl. 2018;20:425.
World Health Organization. WHO laboratory manual for the examination and processing of human semen. 5th ed. Geneva: World Health Organization; 2010.
Georgiadis AP, Kishore A, Zorrilla M, Jaffe TM, Sanfilippo JS, Volk E, et al. High quality RNA in semen and sperm: isolation, analysis and potential application in clinical testing. J Urol. 2015;193:352–9.
Lalancette C, Platts AE, Johnson GD, Emery BR, Carrell DT, Krawetz SA. Identification of human sperm transcripts as candidate markers of male fertility. J Mol Med. 2009;87:735–48.
Ostermeier GC, Goodrich RJ, Moldenhauer JS, Diamond MP, Krawetz SA. A suite of novel human spermatozoal RNAs. J Androl. 2005;26:70–4.
Andrusiewicz M, Słowikowski B, Skibińska I, Wołuń-Cholewa M, Dera-Szymanowska A. Selection of reliable reference genes in eutopic and ectopic endometrium for quantitative expression studies. Biomed Pharmacother. 2016;78:66–73.
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–22.
Englert-Golon M, Andrusiewicz M, Żbikowska A, Chmielewska M, Sajdak S, Kotwicka M. Altered expression of ESR1, ESR2, PELP1 and c-SRC genes is associated with ovarian cancer manifestation. Int J Mol Sci. 2021;22:6216.
Englert-Golon M, Tokłowicz M, Zbikowska A, Sajdak S, Kotwicka M, Andrusiewicz M. Differential Expression of HIF1A EPAS1 and VEGF genes in Benign and malignant ovarian neoplasia. Cancers. 2022;14:4899.
Field A. Discovering Statistics using SPSS. 5th ed. New York: Sage Publications Ltd; 2017.
Schober P, Schwarte LA. Correlation coefficients: appropriate use and interpretation. Anesth Analg Lippincott Williams and Wilkins. 2018;126:1763–8.
Aquila S, Sisci D, Gentile M, Carpino A, Middea E, Catalano S, et al. Towards a physiological role for cytochrome P450 aromatase in ejaculated human sperm. Hum Reprod. 2003;18:1650–9.
Levin ER. Plasma membrane estrogen receptors. Trends Endocrinol Metab. 2009;20:477–82.
Rago V, Siciliano L, Aquila S, Carpino A. Detection of estrogen receptors ER-alpha and ER-beta in human ejaculated immature spermatozoa with excess residual cytoplasm. Reprod Biol Endocrinol. 2006;4:1–6.
Solakidi S, Psarra A-MG, Nikolaropoulos S, Sekeris CE. Estrogen receptors α and β (ERα and ERβ) and androgen receptor (AR) in human sperm localization of ERβ and AR in mitochondria of the midpiece. Hum Reprod. 2005;20:3481–7.
Luconi M, Francavilla F, Porazzi I, Macerola B, Forti G, Baldi E. Human spermatozoa as a model for studying membrane receptors mediating rapid nongenomic effects of progesterone and estrogens. Steroids. 2004;69:553–9.
Bragado MJ, Gil MC, Martin-Hidalgo D, De Llera AH, Bravo N, Moreno AD, et al. Src family tyrosine kinase regulates acrosome reaction but not motility in porcine spermatozoa. Reproduction. 2012;144:67–75.
Varano G, Lombardi A, Cantini G, Forti G, Baldi E, Luconi M. Src activation triggers capacitation and acrosome reaction but not motility in human spermatozoa. Hum Reprod. 2008;23:2652–62.
Breitbart H, Rotman T, Rubinstein S, Etkovitz N. Role and regulation of PI3K in sperm capacitation and the acrosome reaction. Mol Cell Endocrinol. 2010;314:234–8.
Song RXD, Zhang Z, Santen RJ. Estrogen rapid action via protein complex formation involving ERalpha and Src. Trends Endocrinol Metab. 2005;16:347–53.
De LA, Avena P, Sirianni R, Chimento A, Fallo F, Pilon C, et al. Role of scaffold protein proline-, glutamic acid-, and leucine-rich protein 1 (PELP1) in the modulation of adrenocortical cancer cell growth. Cells. 2017;6:42.
Amjad S, Rahman MS, Pang WK, Ryu DY, Adegoke EO, Park YJ, et al. Effects of phthalates on the functions and fertility of mouse spermatozoa. Toxicology. 2021;454:152746.
Gorowska-Wojtowicz E, Duliban M, Kudrycka M, Dutka P, Pawlicki P, Milon A, et al. Leydig cell tumorigenesis–implication of G-protein coupled membrane estrogen receptor, peroxisome proliferator-activated receptor and xenoestrogen exposure. In vivo and in vitro appraisal. Tissue Cell. 2019;61:51–60.
Wang YQ, Li YW, Chen QL, Liu ZH. Long-term exposure of xenoestrogens with environmental relevant concentrations disrupted spermatogenesis of zebrafish through altering sex hormone balance, stimulating germ cell proliferation, meiosis and enhancing apoptosis. Environ Pollut Environ Pollut. 2019;244:486–94.
Hutz RJ, Carvan MJ III, Larson JK, Liu Q, Stelzer RV, King-Heiden TC, et al. Familiar and novel reproductive endocrine disruptors: xenoestrogens, dioxins and nanoparticles. Curr trends Endocrinol. 2014;7:111.
Acknowledgements
We would like to thank Róża Czarnecka-Cierniewska, M.Sc. and Agnieszka Sadowska, M.Sc. for technical support.
Funding
Grant support: This research was funded by National Science Centre Poland [Grant number UMO-2016/23/D/NZ5/02604], and The APC was funded by PUMS. Narodowe Centrum Nauki, UMO-2016/23/D/NZ5/02604, Izabela Skibińska
Author information
Authors and Affiliations
Contributions
Study design: IS, MA, and MK. Data collection: IS, MA, AZ, MJ, and PJ. Statistical analysis: IS, MA, and MK. Data interpretation: IS, MA, MK, MJ, and PJ. Manuscript preparation: IS and MA. Literature search: IS. Funds collection: IS.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of Poznan University of Medical Sciences (PUMS protocol code 698/18, date of approval: 14 June 2018).
Informed consent
Written informed consent was obtained from each participant.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Skibińska, I., Andrusiewicz, M., Jendraszak, M. et al. Expression of estrogen receptors, PELP1, and SRC in human spermatozoa and their associations with semen quality. Human Cell 36, 554–567 (2023). https://doi.org/10.1007/s13577-022-00847-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-022-00847-6