
Abstract
Neuropeptides are important signaling molecules used by nervous systems to mediate and fine-tune neuronal communication. They can function as neurotransmitters or neuromodulators in neural circuits, or they can be released as neurohormones to target distant cells and tissues. Neuropeptides are typically cleaved from larger precursor proteins by the action of proteases and can be the subject of post-translational modifications. The short, mature neuropeptide sequences often entail the only evolutionarily reasonably conserved regions in these precursor proteins. Therefore, it is particularly challenging to predict all putative bioactive peptides through in silico mining of neuropeptide precursor sequences. Peptidomics is an approach that allows de novo characterization of peptides extracted from body fluids, cells, tissues, organs, or whole-body preparations. Mass spectrometry, often combined with on-line liquid chromatography, is a hallmark technique used in peptidomics research. Here, we used an acidified methanol extraction procedure and a quadrupole-Orbitrap LC-MS/MS pipeline to analyze the neuropeptidome of Caenorhabditis elegans. We identified an unprecedented number of 203 mature neuropeptides from C. elegans whole-body extracts, including 35 peptides from known, hypothetical, as well as from completely novel neuropeptide precursor proteins that have not been predicted in silico. This set of biochemically verified peptide sequences provides the most elaborate C. elegans reference neurpeptidome so far. To exploit this resource to the fullest, we make our in-house database of known and predicted neuropeptides available to the community as a valuable resource. We are providing these collective data to help the community progress, amongst others, by supporting future differential and/or functional studies.

ᅟ
Similar content being viewed by others


Introduction
Neuropeptides and peptide hormones have been discovered in animals ranging from cnidarians to mammals and are indispensable modulators of virtually all physiological processes in Metazoa [1]. By acting as neurohormones, neurotransmitters, or neuromodulators, neuropeptides fine-tune the communication between specialized cells and tissues in response to an altering environment. Most neuropeptides exert their effects via G protein-coupled receptors (GPCRs), which are prime pharmacological targets, making the study of neuropeptide signaling and their functions of great interest to both biological and medical research [2,3,4,5,6,7].
Neuropeptides are encoded by larger, inactive precursor proteins that are enzymatically processed by prohormone convertases and carboxypeptidases to yield mature, bioactive peptides [8]. In addition, post-translational modifications can be essential to acquire biological activity. These include N-terminal cyclization of glutamine/glutamic acid to pyroglutamate, N-terminal acetylation, C-terminal amidation, phosphorylation, or sulfation [9, 10]. Recognition sites for cleavage of neuropeptide precursors by proprotein convertases typically consist of a dibasic motif involving arginine and/or lysine residues (KR, RR, KK, and RK, in respective order of susceptibility to proprotein convertase cleavage) [11]. However, not all mature peptides are cleaved after dibasic cleavage sites. Frequently, basic residues that are interspersed with an even number of non-basic amino acids ([K/R]Xn[K/R], with n = 2, 4, 6, or 8) will serve as cleavage recognition sequences, or cleavage may take place following but a single basic residue (K, R) [12]. Less commonly so, certain bioactive peptides – such as NPFF – can be produced by processing via non-classic pathways, utilizing other recognition sites [13,14,15]. In mammals, endothelin-converting enzyme-2 (ECE-2) and carboxypeptidases A5 and A6 have been suggested as possible neuropeptide processing enzymes that act on sites other than the classic dibasic residues [13]. The cleavage sites recognized by these proposed peptidases are far less clear-cut than the simple K/R combination used by classic proprotein convertases, and remain to be investigated in detail [16].
Non-standard cleavage sites are an inherent drawback when in silico prediction methods are used for the prediction of bioactive neuropeptides in the proteome. While in silico analyses are invaluable and well-established tools, the algorithms available today are unsuitable to identify non-classically processed neuropeptides. Complementary in vivo techniques involving tissue extraction, chromatography, and Edman degradation can be applied, but are highly inefficient for large-scale identification of mature neuropeptide sequences [17,18,19].
Following developments in mass spectrometry (MS) and high-performance liquid chromatography (HPLC) in the early 21st century, it became possible to analyze integral tissue peptide extracts. The ability of studying the entire peptidome of a single sample introduced the term “peptidomics”, defined as the comprehensive characterization of peptides present in a biological sample [20]. Peptidomics approaches based on liquid chromatography coupled on-line to mass spectrometry (LC-MS) quickly expanded the repertoire of characterized peptides in a multitude of species [5,6,7]. MS and tandem mass spectrometry (MS/MS) enable de novo characterization of peptides and post-translational modifications: each experiment displays a snapshot of the full complexity of peptide profiles as they were present in vivo, without a strict requirement for prior in silico knowledge of the genome or transcriptome of the sampled organism [21]. Since 2001, the number of scientific publications dealing with peptidomics or the peptidome keeps increasing faster than the overall database, highlighting a continuously growing interest [22,23,24,25]. Indeed, peptidomics has become a highly invaluable tool for the analysis of (neuro)peptidomes in biological samples [7].
Recent years brought about major increases in sensitivity of mass spectrometers, which significantly improved the acquired data from mass spectrometric experiments. Recent Orbitrap and Q-TOF MS systems are specifically designed to provide fast scanning in combination with high mass accuracy, dynamic range, and resolving power [26]. When using high pressure liquid-chromatography (HPLC) in combination with MS/MS, not only the mass spectrometer determines identification rate but the ultra (U)HPLC column makes an important contribution to resolution as well. Continuous improvement of (U)HPLC columns leads to increasingly lower dispersion rates, resulting in a higher efficiency [27]. Comparison of platforms performed by parallelized high-resolution and normal-resolution peptidomics show a significant increase in peptide identifications from novel high-resolution set-ups. For instance, a comparative experiment using a nanobore high-resolution C18-LC-ESI-MS/MS and a low-resolution standard bore C18-LC-ESI-MS/MS system revealed a 12-fold increase in unique peptide identifications from gastrointestinal digestions of hemoglobin using the high-resolution system [28].
Peptidomics can provide valuable knowledge on peptidergic effectors involved in physiological and behavioral processes. Neuropeptidomics have revealed thousands of bioactive peptides, guiding functional studies on neuropeptide functions. Often, small invertebrate model organisms have been favored for such studies [5, 29,30,31,32]. Caenorhabditis elegans has emerged as a powerful model to study peptidergic modulation on a cellular and molecular level, facilitated by its completely mapped nervous system constituting 302 neurons [30, 33, 34]. In C. elegans, neuropeptides are classified into three major families based on their mature sequences: insulin-like peptides (INS), FMRFamide-related peptides (FLPs), and non-insulin/non-FMRFamide-related peptides (NLPs) [35]. Wormbase (release WS259) specifies 40 INS precursors, 31 FLP precursors, and 59 NLP precursors (NLP-1 to 57, PDF-1, and NTC-1) encoded by the C. elegans genome. Additional NLP precursors have been suggested in recent phylogenetic studies, expanding the amount of putative NLP precursors to 75 [36, 37]. Previous peptidomic studies in C. elegans reported up to 75 NLP- and FLP-type neuropeptides in a single experiment, processed from 29 distinct precursors [38].
Here, we prepared peptide-enriched extracts from whole-mount C. elegans samples [39] and relied on quadrupole-Orbitrap LC-MS/MS to biochemically identify mature neuropeptides. Using this pipeline, we identified 203 unique C. elegans peptides, including 35 peptide sequences that so far have not been predicted by in silico methods. We discovered 29 novel peptides encoded by known or in silico predicted precursors, as well as eight mature peptides originating from seven prepropeptides that were not yet annotated as neuropeptide precursors. By collecting robust data from mixed-stage cultures reared under standard conditions, this study represents the currently most comprehensive reference neuropeptidome of C. elegans.
Experimental
C. elegans Strains
The wild-type N2 Bristol strain was obtained from the Caenorhabditis Genetics Stock Center (University of Minnesota). Worms were cultivated in standard liquid cultures supplied with flash frozen E. coli K12 as food source as described previously [40, 41]. The concentration of bacteria was verified daily, and new K12 bacteria were added to maintain optimal food levels (OD600 = 1.68).
Extraction of Peptides
Wild-type mixed stage worms from liquid cultures were collected and pooled. This pool was aliquoted into 10 samples, each containing approximately 1 mL of biological material. Peptides were extracted using an acidified methanol extraction solvent as described previously [31]. This precipitates larger proteins, while smaller peptides remain in solution. A size exclusion column (Sephadex PD MiniTrap G-10, GE Healthcare) and a 10 kDa cut-off filter (Amicon Ultra-4, Merck Millipore) were used to enrich the samples for peptide content by isolating the 700–10,000 Da mass fraction. Samples were briefly stored at 4 °C prior to MS analysis.
Quadrupole-Orbitrap LC-MS/MS
Quadrupole-Orbitrap LC-MS/MS experiments were conducted using a Dionex UltiMate 3000 UHPLC coupled on-line to a Thermo Scientific Q Exactive mass spectrometer. The UHPLC is equipped with a guard pre-column (Acclaim PepMap 100, C18, 75 μm × 20 mm, 3 μm, 100 Å; Thermo Scientific) and an analytical column integrated in the nano-electrospray ion source (EASY-Spray, PepMap RSLC, C18, 50 μm × 150 mm, 2 μm, 100 Å; Thermo Scientific). The sample was separated at a flow rate of 300 nL/min, using a 45-min. linear gradient from 3% to 55% acetonitrile containing 0.1% formic acid. MS data were acquired using a data-dependent (dynamic exclusion settings at 15 s) Top10 method, choosing the most abundant precursor ions (charge selection ranging from 2 to 5) from a full MS survey scan for Higher-energy Collisional Dissociation fragmentation (HCD). Full MS scans were acquired at a resolution of 70,000 at m/z 200, with a maximum injection time of 256 ms and scan range of 400 to 1600 m/z. The resolution for MS/MS scans after HCD fragmentation was set at 17,500 at m/z 200, with a maximum injection time of 64 ms.
Mass Spectrometry Data Analysis
To correct for inter-run variation causing retention time shifts between the 10 different runs, all data files were aligned using Progenesis LC-MS software (Nonlinear Dynamics). Peak picking was done in automatic mode, using default sensitivity settings. Results were then filtered on charge state, retaining all features with charges ranging from 1 to 7. All selected features were exported to a .csv file containing the m/z, charge, deconvoluted mass, abundance, and retention times. For peptide annotation, we developed a custom R script [42] that compares all the detected masses in the Progenesis .csv file to an in-house peptide library containing the masses of 354 C. elegans peptides and their post-translational modifications (supplementary Tables S1 and S2). Deconvoluted masses that match within an error margin of 5 ppm were interpreted as a positive hit.
MS/MS fragmentation data were analyzed using PEAKS software (Bioinformatics Solutions) with a custom-made library containing all known C. elegans proteins (Wormbase ver. WS256), E. coli proteins (SwissProt, acquired September 2016), and a list of common protein contaminants (common Repository of Adventitious Proteins, cRAP). Parent mass error was set at 10 ppm, and fragment mass error at 0.02 Da. The following variable modifications were taken into account: oxidation (+15.99 Da), glycine-loss in combination with amidation (–58.01 Da), pyroglutamation from glutamic acid (–18.01 Da), pyroglutamation from glutamine (–17.03 Da), phosphorylation of serine, threonine, or tyrosine (+79.97 Da).
Filtering Parameters for the Discovery of Novel Putative Neuropeptide Precursors
All peptide identifications were exported from PEAKS as a .csv file. A custom R script was devised to cross-reference peptides in this file with the known C. elegans neuropeptides in our in-house database (supplementary Tables S1 and S2), thereby creating a dataset containing peptides hitherto not present in the predicted C. elegans peptidome. In a next filtering step, the location of the peptide within its corresponding precursor was examined, and only peptides flanked by a basic amino acid (K, R, or a combination of both) were retained. Two variations to this rule were allowed: first, if no N-terminal basic residue is present, the peptide must be located 30 amino acids or less from the N-terminus of the precursor protein. This considers that neuropeptides may be located right after the signal peptide cleavage site, typically situated no more than 30 amino acids from the N-terminus of the precursor protein. Second, if no C-terminal basic amino acid is present, the peptide must be located at the C-terminus of the precursor protein. All precursor proteins containing peptides that comply with this set of rules were retained. The resulting list of precursor proteins was subjected to SignalP to look for the presence of a signal peptide sequence, reducing the number of candidate neuropeptide precursors from the initial 20 to 16 [43]. The location of the signal peptide cleavage site was compared with C-termini of peptides that were identified as being located right after the signal peptide sequence, to confirm consistent processing. After manual inspection of MS/MS spectra, seven candidates were withheld for further analysis. All of these candidate precursors were found to be shorter than 300 amino acids, supporting reported observations for neuropeptide precursors to typically not be longer than this length [44, 45]. Indeed, all known FLP and NLP precursors in C. elegans are shorter than 300 amino acids, with the single exception of an NLP-16 splice variant, which is 326 amino acids long. For the prediction of putative proprotein convertase cleavage sites, we used both the NeuroPred and ProP algorithms [46, 47].
Sequence Alignment of Putative Novel Neuropeptide Precursors
Protein BLAST was used to search for protein homologs in all Metazoa of the presumed neuropeptide precursors we found in C. elegans. Potential homologs were selected based on the conservation of the (di)basic cleavage sites as well as the presence of conserved motifs within the putative neuropeptide itself. These hits were used for sequence alignment with the PSI-Coffee algorithm, which is optimized for the alignment of distantly related proteins using homology extension [48]. Shading was added using ver. 3.21 of BOXSHADE [49].
Results
Knowledge of the C. elegans neuropeptidergic repertoire provides invaluable information for further functional and behavioral studies. We ventured to maximize detection of the integral C. elegans peptidome using an in-house developed peptidomics pipeline. In order to include low-abundant and stage-specific neuropeptides as much as possible, we increased sample sizes and made use of mixed-stage cultures. This study focuses on the FLP and NLP families of neuropeptides for which our extraction protocol is optimized.
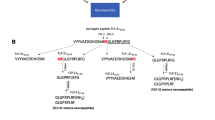
Current predictions estimate that the 31 FLP and 75 NLP precursor proteins in the C. elegans genome could generate up to 354 mature neuropeptides (supplementary Tables S1 and S2). The effectiveness of our peptidomics pipeline was evaluated by assessing how much of the predicted C. elegans peptidome could be observed in each individual sample, compared with the overall success of peptide identification. Peptide mass matching analysis of MS1 data of 10 mixed-stage C. elegans samples identified 203 individual peptides, corresponding to nearly 58% of the predicted peptidome (Figure 1, Supplementary Tables S1–S2). Furthermore, 131 neuropeptide sequences were successfully detected by tandem MS, confirming 37% of the known and predicted C. elegans peptide sequences (Figure 1, Supplementary Tables S1, S2). Robustness of the method was examined by counting the occurrence of individual peptides in the 10 replicates (Supplementary Figure S1). Approximately 80% of the 203 detected peptides were measured in seven or more replicates, indicating a high reproducibility of the method.

Percentage of the predicted C. elegans peptidome (FLP and NLP) observed with LC-MS. In total, 203 neuropeptides were detected in MS mode, together almost 58% of the C. elegans peptidome (containing 354 known and predicted NLP/FLP-type neuropeptide sequences prior to this study). We confirmed 131 neuropeptides – or 37% of this peptidome – with MS/MS
Detection of Novel Peptides Derived from Known C. elegans FLP and NLP Precursors
Next to the identification of already predicted neuropeptides, our peptidomics dataset revealed several peptides that are additionally processed from known C. elegans FLP or NLP precursors. These neuropeptides were identified with MS/MS and retained as targets of interest based on the presence of flanking basic residues. Our analysis identified 29 hitherto unstudied peptide sequences, present in 22 precursor proteins: FLP-1, FLP-3, FLP-5, FLP-6, FLP-9, FLP-10, FLP-15, FLP-18, NLP-3, NLP-6, NLP-8, NLP-9, NLP-10, NLP-12, NLP-17, NLP-42, NLP-43, NLP-49, NLP-50, NLP-51, and NLP-52 (supplementary Table S3). Often, these peptides are located in the precursor between the already known neuropeptides sequences, implying that they may be remnants following proprotein cleavage. However, as we do not detect other remnants of these precursors, nor similar remnants for others, and since all these detected sequences are conserved in Caenorhabditis species, they may be biologically active peptides.
Mass Spectrometric Evidence for the Recently Predicted C. elegans Neuropeptide Precursor Proteins NLP-53, NLP-55, NLP-56, NLP-57, NLP-58, and NLP-65
The C. elegans peptidome has recently been extended with the genes nlp-53 (Y12A6A.2), nlp-54 (C30H6.10), nlp-55 (F14F11.2), nlp-56 (Y57G11C.45), and nlp-57 (F08G12.8). Additionally, we assigned gene names to the novel peptide precursors (supplementary Table 1) suggested by Mirabeau and Joly and Koziol et al., including nlp-58 (T07C12.15) and nlp-65 (R06F6.7) [36, 37]. Based on predictions, these genes are presumed to encode neuropeptide precursor proteins (supplementary Table S2). We here present mass spectrometric evidence that these genes indeed give rise to mature neuropeptides. Using MS/MS, we successfully identified NQGAGSVSLDSLASLPMLRYamide from NLP-53 (supplementary Figure S2), MYINPDYYYVEQLPTM from NLP-55 (supplementary Figure S3), and SSIMTDDVEPPQLLTRQL from NLP-56 (supplementary Figure S4). Three mature neuropeptides were identified for NLP-57: SPIHGIWNNLPAPPQ, VYGFYNYLPKEEDDRD, and NTILLLTPNEDYVE (supplementary Figure S5). In NLP-58 we detected ARIFDGQEEQ as a mature neuropeptide (supplementary Figure S6), but not VPMMSLKGLRamide (present in MS data only, supplementary Table S2), which was predicted by Mirabeau and Joly (2013). Finally, in C. elegans protein NLP-65, we identified the previously in silico predicted neuropeptides DGLPSFYDIR and GLPSAYDIR (supplementary Figure S7) [37].
Discovery of Seven Putative Novel Neuropeptide Precursors in C. elegans
Besides providing evidence for the actual in vivo presence of peptides from the predicted C. elegans peptidome, the MS/MS dataset was mined further for possible neuropeptides encoded by yet to be annotated neuropeptide precursor genes. The dataset was filtered using several neuropeptide characteristics as parameters (Material and Methods: § “Filtering Parameters for the Discovery of Novel Putative Neuropeptide Precursors”). Seven proteins, which we suggest to name NLP-76 to NLP-82, were previously overlooked in bioinformatic surveys and complied with these assumptions. The peptide sequences that we identified for these proteins are: SPIVELYPVVDSGNVEPEAFPAYFRF in C02B4.4 (Figure 2), pQPAGGQDVPPFL in C06A8.3 (Figure 3) GDVDSVFFSPFRIIamide and ALLAGPHDYDLGDFISNPNV in C16D2.2 (Figure 4, LSADFRNEIPPPDYI in F09E8.8 (Figure 5), HSAGSTYPESL in T04C12.3 (Figure 6), NDFFLRSA in Y67D8B.4 (Figure 7), and FILQDLPLERFE in H32K21.1 (Figure 8).

C. elegans protein C02B4.4 encodes the putative neuropeptide SPIVELYPVVDSGNVEPEAFPAYFRF. (a) Precursor sequence with the predicted signal peptide marked in yellow and putative proprotein convertase cleavage sites in green (coloring is based on results from SignalP, NeuroPred, and ProP prediction algorithms). The peptide detected with MS/MS is shown in red. (b) Fragmentation spectrum of the detected neuropeptide SPIVELYPVVDSGNVEPEAFPAYFRF

C. elegans protein C06A8.3 encodes putative neuropeptide pQPAGGQDVPPFL. (a) Precursor sequence with the predicted signal peptide marked in yellow and putative proprotein convertase cleavage sites in green (coloring is based on results from SignalP, NeuroPred, and ProP prediction algorithms). The peptide detected with MS/MS is shown in red. (b) Fragmentation spectrum of the detected neuropeptide pQPAGGQDVPPFL. The N-terminal glutamine has undergone a post-translational modification to pyroglutamic acid

C. elegans protein C16D2.2 encodes putative neuropeptides GDVDSVFFSPFRIIamide and ALLAGPHDYDLGDFISNPNV. (a) Precursor sequence with the predicted signal peptide marked in yellow and putative proprotein convertase cleavage sites in green (coloring is based on results from SignalP, NeuroPred, and ProP prediction algorithms). The peptides detected with MS/MS are shown in red. (b) Fragmentation spectrum of the detected neuropeptide GDVDSVFFSPFRIIamide, with transformation of the C-terminal glycine into an amide as post-translational modification. (c) Fragmentation spectrum of the detected neuropeptide ALLAGPHDYDLGDFISNPNV

C. elegans protein F09E8.8 encodes putative neuropeptide LSADFRNEIPPPDYI. (a) Precursor sequence with the predicted signal peptide marked in yellow and putative proprotein convertase cleavage sites in green (coloring is based on results from SignalP, NeuroPred, and ProP prediction algorithms). The peptide detected with MS/MS is shown in red. (b) Fragmentation spectrum of the detected neuropeptide LSADFRNEIPPPDYI

C. elegans protein T04C12.3 encodes putative neuropeptide HSAGSTYPESL. (a) Precursor sequence with the predicted signal peptide marked in yellow and putative proprotein convertase cleavage sites in green (coloring is based on results from SignalP, NeuroPred, and ProP prediction algorithms). The peptide detected with MS/MS is shown in red. (b) Fragmentation spectrum of the detected neuropeptide HSAGSTYPESL

C. elegans protein Y67D8B.4 encodes putative neuropeptide NDFFLRSA. (a) Precursor sequence with the predicted signal peptide marked in yellow and putative proprotein convertase cleavage sites in green (coloring is based on results from SignalP, NeuroPred, and ProP prediction algorithms). The peptide detected with MS/MS is shown in red. (b) Fragmentation spectrum of the detected neuropeptide NDFFLRSA

C. elegans protein H32K21.1 encodes putative neuropeptide FILQDLPLERFE. (a) Precursor sequence with the predicted signal peptide marked in yellow and putative proprotein convertase cleavage sites in green (coloring is based on results from SignalP, NeuroPred, and ProP prediction algorithms). The peptide detected with MS/MS is shown in red. (b) Fragmentation spectrum of the detected neuropeptide FILQDLPLERFE
We subjected these precursor protein sequences to a BLAST search to look for possible orthologs. All seven precursors – including the peptide sequence and position of the basic residues – are well-conserved in other Caenorhabditis species such as C. brenneri, C. briggsae, and C. remanei (supplementary Figures S7–S14), suggesting a possible biological role. Precursor proteins C02B4.4, C06A8.3, C16D2.2, F09E8.8, and Y67D8B.4 were found to be conserved in several parasitic nematode species as well, including Ancylostoma ceylanicum, Necator americanus, Dictyocaulus viviparus, and Oesophagostomum dentatum (supplementary Figures S7, S8, S9, S10, and S13). Overall, alignments of the seven precursors with their closest homologs in other nematode species display the evolutionary conservation of the detected neuropeptides and the flanked basic cleavage sites. Our BLAST search did not detect any homologs for these new precursor proteins in species other than nematodes.
Discussion
When studying animal physiology and in particular neuronal signaling underlying behavior, aging, and learning and memory formation, an extensive knowledge of the organism’s complete peptidome is highly valuable. Various efforts in creating an integral peptidome library through peptidomics have been made in a wide range of animals, including nematodes (C. elegans, Ascaris suum), arthropods (Tribolium castaneum, Aedes aegypti, Glossina morsitans, Drosophila melanogaster, Apis mellifera, Schistocerca gregaria, Triatoma infestans), molluscs (Aplysia californica, Lymnaea stagnalis), and vertebrates (Rattus norvegicus, Danio rerio) [5, 50,51,52]. Some of these species are considered pests (S. gregaria, A. suum), are known vectors of disease (A. aegypti, G. morsitans, T. infestans) or are invaluable to our ecosystem and to human survival (A. mellifera). Therefore, knowledge of their neuropeptide signaling serves diverse interests, e.g., as an interesting target for pest control, or to aid in ways to promote the survival of biologically valuable species. Since several of these organisms lack a well-annotated reference genome, peptidomics is often the best option to screen for bioactive neuropeptides. On the other hand, knowledge of the integral peptidome of well-established (in)vertebrate model organisms such as C. elegans, D. melanogaster, D. rerio, R. norvegicus, and Mus musculus can provide indispensable information that may be extended to human disease studies. While both the rat and mouse peptidomes have been studied extensively [53,54,55], scientists continuously aim to optimize peptidomics work flows in order to unveil new peptides and advance insights into neuropeptidomes, as exemplified by recent work on dissected rat brains [52].
Increasing the knowledge on the C. elegans peptidome comes with the clear benefits of it being a seasoned model organism, in the sense that an as-complete-as-possible peptidome can quickly integrate with knowledge from diverse biochemical, cellular circuit, and functional studies; hence, it represents a valuable tool for researchers. It should be noted that while mature neuropeptides from both FLP and NLP precursors have been detected in vivo, the complete INS family remains fully based on in silico predictions [56]. Knowledge on how INS protein precursors are expressed and processed into mature INS peptides is thus lacking so far. Previous peptidomics studies in C. elegans have detected up to 75 neuropeptides in a single experiment [57]. Per replicate, we identified on average 170 neuropeptides, with a minimum of 160 and a maximum of 174. When pooling the data from all 10 replicates, we effectively identified 203 out of the 354 known and predicted C. elegans neuropeptides, and confirmed 131 neuropeptides with MS/MS. All of this was done with an optimized peptidomics pipeline, vastly increasing the amount of identifications compared with previous studies. While the identification of 72 neuropeptides, originating from 51 precursors, is based on MS1 data only (supplementary Tables S1 and S2), 10 of these precursors were found to contain multiple MS1 detected neuropeptides (24 in total), which makes it unlikely they were found by random chance.
MS profiling experiments do not allow subjecting the entire predicted C. elegans peptidome to LC-MS/MS analysis. Analyzing whole-mount C. elegans extracts implicates the presence of various metabolites, phospholipids, and proteins in the sample. Their ubiquity may push the lower abundant neuropeptides into the background, resulting in problematic ionization and low-quality fragmentation spectra. To overcome this, we enriched the sample for the 0.7–10 kDa mass range, as the bulk of C. elegans neuropeptides lie within this mass range. There are 33 predicted C. elegans neuropeptides that have a mass lower than 700 Da, which implies they are lost during size exclusion chromatography. Essentially, for discovery (profiling) experiments this is a necessary trade-off with the removal of metabolites from the samples. The maximum number of identifications that could theoretically be achieved with our peptidomics pipeline would therefore encompass 321 (354 minus 33) peptides of the predicted C. elegans peptidome, of which we were able to identify 64%. Altogether, we present an extension of the existing C. elegans peptidome with 35 new mature neuropeptide sequences, including eight neuropeptides originating from seven novel precursors. Thereby, we have expanded the number of C. elegans FLP/NLP neuropeptide precursors to 113, coding for a total of 391 mature neuropeptides. This number may still rise, as the presence of multiple (di)basic cleavage sites and sequence architecture in the seven novel precursors (supplementary Figures S8–S13) suggests the existence of yet to be detected neuropeptides.
Several reasons may underlie the fact that 118 predicted neuropeptides remain undetected in this study. First, the knowledge we have on C. elegans neuropeptides that have not been confirmed by peptidomics consists completely of in silico predictions. All amino acid sequences enclosed by (di)basic cleavage sites in a neuropeptide precursor are considered putative mature neuropeptides. It is plausible that not all sequences encoded in the precursor are processed to mature neuropeptides. In addition, when three adjacent basic amino acids are present, there are two potential cleavage products from proteolytic processing of the precursor that can be predicted. These peptides only differ by one N-terminal basic amino acid residue (K or R) that can be part of the mature peptide if the first two basic residues are used as a cleavage signal. Our peptidomics data reveals a preference for these peptides where the third basic amino acid of the string is part of the mature peptide (e.g., FLP-1, FLP-9, FLP-12, FLP-15, FLP-17), if only one mature peptide is observed. While this may reflect endogenous processing preference, this may also be a technical artefact due to the expectedly more effective ionization of peptides containing basic residues. Individual peptides may deviate from this general trend, and we also observed mature neuropeptides with and without the additional basic residue originating from the tribasic cleavage sites (e.g., FLP-8, FLP-14). Second, as they are modulators of behavior in response to changing environments, peptides may be expressed under specific conditions only. If the cue for their expression – be it internal or external – is absent under standard conditions, these peptides are likely not present in our reference samples. For instance, all NLPs of the nlp-29 gene cluster (nlp-27, nlp-28, nlp-29, nlp-30, nlp-31, nlp-34) are considered antimicrobial peptides that are upregulated after infection [58]. Therefore, it is no surprise that we could detect almost none of those peptides by sampling healthy worms. Finally, it is possible that certain neuropeptides did not reach the detection limit due to highly localized expression and/or low abundance: some peptides are only expressed in one cell and/or at very low levels.
This study presents the first evidence on the in vivo presence of NLP-53-, NLP-55-, NLP-56-, NLP-57-, NLP-58, and NLP-65-derived peptides. We added these putative NLP neuropeptide precursors to Wormbase. NLP-65 was predicted in silico by Koziol and coworkers, although without the C-terminal arginine in both peptides, which they assumed to be part of the cleavage site [37]. Up to now, there was no evidence from peptidomics studies to support their processing to yield mature bioactive peptides. The only recently added NLP-precursor for which we were unable to provide supporting MS-evidence would be NLP-54. Both C. elegans TRH-like peptides that are contained within NLP-54 have, however, a low molecular weight, thereby passing below the 700 Da mass cut-off that is used in our implemented size-exclusion chromatography. Due to their small size they most likely occur as singly charged ions, also escaping MS/MS detection [59, 60]. In addition, no mature peptides processed from NLP-59, -61, -62, -63, -66, -67, -68, -70, and -71 could be detected. The present mass spectrometric detection of the eight peptides contained within the NLP-53, 55-58, and NLP-65 precursors confirms the predicted coding potential of these corresponding peptide encoding genes.
In addition to identifying known or predicted peptides within FLP and NLP precursors, our MS/MS data also revealed 29 novel peptides which are processed from known FLP and NLP precursor proteins (supplementary Table S3). All these peptides are flanked by (di)basic cleavage sites and display conservation in other Caenorhabditis and/or nematode species, with the exception of the newly found NLP-42- and NLP-58-derived peptides. This conservation at least suggests that the peptides could be functionally relevant themselves. We confidently detected eight peptides that are processed from seven distinct proteins previously unannotated as neuropeptide precursors (Figures 2, 3, 4, 5, 6 and 7, supplementary Table S4). All encoded peptides are processed adjacent to (di)basic cleavage sites and the precursor proteins contain a signal peptide, suggesting that they are indeed putative preproproteins harboring mature neuropeptides. Most of these precursors are also enriched in neurons according to Wormbase, which further strengthens a putative function as neuropeptide precursors. The discovery of these novel precursor proteins by our peptidomics study underscores the continued need for wet-lab discovery in peptidomics.
The increased number of predicted and biochemically characterized neuropeptides in C. elegans presents an increasingly complete picture of the reference peptidome of this model organism. The accuracy and completeness of this reference are essential for follow-up (differential) peptidomic studies in C. elegans [61]. This model organism is a powerful model for neurobiological research, furthered even more by the complete mapping of its nervous system [33, 62]. Many neuropeptides are involved in modulating neural circuits, and their functional output often evokes interesting phenotypical changes in C. elegans [34, 63,64,65]. Given a complete picture of the C. elegans peptidome, it will also become possible to map in which neurons specific neuropeptides are present. On top of accumulating expression-based information, recent advances in mass spectrometry suggest that knowledge of peptide content of single neurons in C. elegans will become feasible. Since this may provide additional insights into the regulation of neuronal signaling in this model, we and others are working towards a future in which peptidomics can contribute to the study of neuropeptidergic neuromodulation on a single neuron level, using C. elegans as a research model.
Conclusion
Aiming to expand the C. elegans reference neuropeptidome, we are able to identify 203 neuropeptides based on mass matching, of which 131 were confirmed at the sequence level using LC-MS/MS. This includes neuropeptides in the newly annotated precursors NLP-53, NLP-55, NLP-56, NLP-57, NLP-58 as well as the previously unannotated protein R06F6.7 (now NLP-65), hitherto known by in silico predictions only. Furthermore, we found evidence for 29 putative additional neuropeptides present in known FLP and NLP precursors, as well as eight neuropeptides encoded by seven novel neuropeptide precursors, which were not yet annotated as such. We present both our in-house database of known and predicted peptides, as well as our most complete evidence for in vivo occurrence of these peptides under standard conditions to the scientific community. We hope this resource may be of valuable help in the biochemical and functional studies of C. elegans neurobiology.
References
Schoofs, L., Beets, I.: Neuropeptides control life-phase transitions. Proc. Natl. Acad. Sci. U. S. A. 110, 7973–7974 (2013)
Borbély, E., Scheich, B., Helyes, Z.: Neuropeptides in learning and memory. Neuropeptides. 47, 439–450 (2013)
Hoyer, D., Bartfai, T.: Neuropeptides and neuropeptide receptors: drug targets, and peptide and non-peptide ligands: a tribute to Professor. Dieter Seebach. Chem. Biodivers. 9, 2367–2387 (2012)
Kormos, V., Gaszner, B.: Role of neuropeptides in anxiety, stress, and depression: from animals to humans. Neuropeptides. 47, 401–419 (2013)
De Haes, W., Van Sinay, E., Detienne, G., Temmerman, L., Schoofs, L., Boonen, K.: Functional neuropeptidomics in invertebrates. Biochim. Biophys. Acta Protein Proteomics. 1854, 812–826 (2015)
Bendena, W.G.: Neuropeptide physiology in insects. Adv. Exp. Med. Biol. 692, 166–191 (2010)
Romanova, E.V., Sweedler, J.V.: Peptidomics for the discovery and characterization of neuropeptides and hormones. Trends Pharmacol. Sci. 36, 579–586 (2015)
Rouillé, Y., Duguay, S.J., Lund, K., Furuta, M., Gong, Q., Lipkind, G., Oliva, A.A., Chan, S.J., Steiner, D.F.: Proteolytic processing mechanisms in the biosynthesis of neuroendocrine peptides: the subtilisin-like proprotein convertases. Front. Neuroendocrinol. 16, 322–361 (1995)
Na, S., Paek, E.: Software eyes for protein post-translational modifications. Mass Spectrom. Rev. 34, 133–147
Johnson, H., Eyers, C.E.: Analysis of post-translational modifications by LC-MS/MS. Methods Mol. Biol. 658, 93–108 (2010)
Amare, A., Hummon, A.B., Southey, B.R., Zimmerman, T.A., Rodriguez-Zas, S.L., Sweedler, J.V.: Bridging Neuropeptidomics and Genomics with Bioinformatics: Prediction of Mammalian Neuropeptide Prohormone Processing. J. Proteome Res. 5, 1162–1167 (2006)
Veenstra, J.A.: Mono- and dibasic proteolytic cleavage sites in insect neuroendocrine peptide precursors. Arch. Insect Biochem. Physiol. 43, 49–63 (2000)
Fricker, L.D.: Neuropeptide-processing enzymes: applications for drug discovery. AAPS J. 7, E449–E455 (2005)
Perry, S.J., Yi-Kung Huang, E., Cronk, D., Bagust, J., Sharma, R., Walker, R.J., Wilson, S., Burke, J.F.: A human gene encoding morphine modulating peptides related to NPFF and FMRFamide. FEBS Lett. 409, 426–430 (1997)
Vilim, F.S., Aarnisalo, A.A., Nieminen, M.L., Lintunen, M., Karlstedt, K., Kontinen, V.K., Kalso, E., States, B., Panula, P., Ziff, E.: Gene for pain modulatory neuropeptide NPFF: induction in spinal cord by noxious stimuli. Mol. Pharmacol. 55, 804–811 (1999)
Lyons, P.J., Fricker, L.D.: Substrate specificity of human carboxypeptidase A6. J. Biol. Chem. 285, 38234–38242 (2010)
Kimmel, J.R., Hayden, L.J., Pollock, H.G.: Isolation and characterization of a new pancreatic polypeptide hormone. J. Biol. Chem. 250, 9369–9976 (1975)
Peterson, J.D., Steiner, D.F.: The amino acid sequence of the insulin from a primitive vertebrate, the atlantic hagfish (Myxine glutinosa). J. Biol. Chem. 250, 5183–5191 (1975)
Schoofs, L., Holman, G.M., Hayes, T.K., Nachman, R.J., De Loof, A.: Isolation, primary structure, and synthesis of locustapyrokinin: a myotropic peptide of Locusta migratoria. Gen. Comp. Endocrinol. 81, 97–104 (1991)
Baggerman, G., Clynen, E., Breuer, M., De Loof, A., Tanaka, S., Schoofs, L.: “Peptidomics”, a new way to identify peptides involved in phase polymorphism in locusts. In: The 47th ASMS conference on Mass Spectrometry and Allied Topics (1999)
Boonen, K., Landuyt, B., Baggerman, G., Husson, S.J., Huybrechts, J., Schoofs, L.: Peptidomics: the integrated approach of MS, hyphenated techniques and bioinformatics for neuropeptide analysis. J. Sep. Sci. 31, 427–445 (2008)
Schrader, M., Schulz-Knappe, P., Fricker, L.D.: Historical perspective of peptidomics. EuPA Open Proteomics. 3, 171–182 (2014)
Fricker, L.D., Lim, J., Pan, H., Che, F.-Y.: Peptidomics: identification and quantification of endogenous peptides in neuroendocrine tissues. Mass Spectrom. Rev. 25, 327–344 (2006)
Menschaert, G., Vandekerckhove, T.T.M., Baggerman, G., Schoofs, L., Luyten, W., Van Criekinge, W.: Peptidomics coming of age: a review of contributions from a bioinformatics angle. J. Proteome Res. 9, 2051–2061 (2010)
Baggerman, G., Verleyen, P., Clynen, E., Huybrechts, J., De Loof, A., Schoofs, L.: Peptidomics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 803, 3–16 (2004)
Lesur, A., Domon, B.: Advances in high-resolution accurate mass spectrometry application to targeted proteomics. Proteomics. 15, 880–890
Walter, T.H., Andrews, R.W.: Recent innovations in UHPLC columns and instrumentation. TrAC Trends Anal. Chem. 63, 14–20 (2014)
Caron, J., Chataigné, G., Gimeno, J.-P., Duhal, N., Goossens, J.-F., Dhulster, P., Cudennec, B., Ravallec, R., Flahaut, C.: Food peptidomics of in vitro gastrointestinal digestions of partially purified bovine hemoglobin: low-resolution versus high-resolution LC-MS/MS analyses. Electrophoresis. 37, 1814–1822 (2016)
Hummon, A.B., Amare, A., Sweedler, J.V.: Discovering new invertebrate neuropeptides using mass spectrometry. Mass Spectrom. Rev. 25, 77–98
Taghert, P.H., Nitabach, M.N.: Peptide neuromodulation in invertebrate model systems. Neuron. 76, 82–97 (2012)
Husson, S.J., Reumer, A., Temmerman, L., De Haes, W., Schoofs, L., Mertens, I., Baggerman, G.: Worm peptidomics. EuPA Open Proteomics. 3, 280–290 (2014)
Wegener, C., Neupert, S., Predel, R.: Direct MALDI-TOF mass spectrometric peptide profiling of neuroendocrine tissue of Drosophila. Methods Mol. Biol. 615, 117–127 (2010)
White, J.G., Southgate, E., Thomson, J.N., Brenner, S.: The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. Royal Soc. Lond. B Biol. Sci. 314, 1–340 (1986)
Beets, I., Temmerman, L., Janssen, T., Schoofs, L.: Ancient neuromodulation by vasopressin/oxytocin-related peptides. Worm. 2, e24246 (2013)
Li, C., Kim, K.: Neuropeptides. WormBook 1–36 (2008)
Mirabeau, O., Joly, J.-S.: Molecular evolution of peptidergic signaling systems in bilaterians. Proc. Natl. Acad. Sci. U. S. A. 110, E2028–E2037 (2013)
Koziol, U., Koziol, M., Preza, M., Costábile, A., Brehm, K., Castillo, E.: De novo discovery of neuropeptides in the genomes of parasitic flatworms using a novel comparative approach. Int. J. Parasitol. 46, 709–721 (2016)
Husson, S.J., Landuyt, B., Nys, T., Baggerman, G., Boonen, K., Clynen, E., Lindemans, M., Janssen, T., Schoofs, L.: Comparative peptidomics of Caenorhabditis elegans versus C. briggsae by LC-MALDI-TOF MS. Peptides. 30, 449–457 (2009)
Husson, S.J., Clynen, E., Boonen, K., Janssen, T., Lindemans, M., Baggerman, G., Schoofs, L.: Approaches to identify endogenous peptides in the soil nematode Caenorhabditis elegans. Methods Mol. Biol. 615, 29–47 (2010)
Stiernagle, T.: Maintenance of C. elegans. WormBook 1–11 (2006)
Van Assche, R., Temmerman, L., Dias, D.A., Boughton, B., Boonen, K., Braeckman, B.P., Schoofs, L., Roessner, U.: Metabolic profiling of a transgenic Caenorhabditis elegans Alzheimer model. Metabolomics. 11, 477–486
R Core Team: R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria (2016)
Petersen, T.N., Brunak, S., von Heijne, G., Nielsen, H.: SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods. 8, 785–786 (2011)
Fricker, L.D.: Neuropeptides and other bioactive peptides: from discovery to function. Colloq. Ser. Neuropeptides. 1, 1–122 (2012)
Nishiyama, M., Ishii, M., Hirose, S., Yamazaki, H., Kimura, S.: In silico search for biologically active peptides. In: Kastin, A.J. (ed.) Handbook of Biologically Active Peptides. pp. 1743–1748. Elsevier (2013). https://doi.org/10.1016/B978-0-12-385095-9.00239-6
Southey, B.R., Amare, A., Zimmerman, T.A., Rodriguez-Zas, S.L., Sweedler, J.V.: NeuroPred: a tool to predict cleavage sites in neuropeptide precursors and provide the masses of the resulting peptides. Nucleic Acids Res. 34, W267–W272 (2006)
Duckert, P., Brunak, S., Blom, N.: Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel. 17, 107–112 (2004)
Di Tommaso, P., Moretti, S., Xenarios, I., Orobitg, M., Montanyola, A., Chang, J.-M., Taly, J.-F., Notredame, C.: T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39, W13–W17 (2011)
Hoffman, K., Baron, M.D.: BOXSHADE: Pretty Printing and Shading of Multiple-Alignment files
Traverso, L., Sierra, I., Sterkel, M., Francini, F., Ons, S.: Neuropeptidomics in Triatoma infestans. Comparative transcriptomic analysis among triatomines. J. Physiol. 110, 83–98 (2016)
Van Camp, K.A., Baggerman, G., Blust, R., Husson, S.J.: Peptidomics of the zebrafish Danio rerio: in search for neuropeptides. J. Proteome. 150, 290–296 (2017)
Secher, A., Kelstrup, C.D., Conde-Frieboes, K.W., Pyke, C., Raun, K., Wulff, B.S., Olsen, J.V.: Analytic framework for peptidomics applied to large-scale neuropeptide identification. Nat. Commun. 7, 11436 (2016)
Fälth, M., Sköld, K., Svensson, M., Nilsson, A., Fenyö, D., Andren, P.E.: Neuropeptidomics strategies for specific and sensitive identification of endogenous peptides. Mol. Cell. Proteomics. 6, 1188–1117 (2007)
Van Dijck, A., Hayakawa, E., Landuyt, B., Baggerman, G., Van Dam, D., Luyten, W., Schoofs, L., De Deyn, P.P.: Comparison of extraction methods for peptidomics analysis of mouse brain tissue. J. Neurosci. Methods. 197, 231–237 (2011)
Sturm, R.M., Dowell, J.A., Li, L.: Rat brain neuropeptidomics: tissue collection, protease inhibition, neuropeptide extraction, and mass spectrometric analysis. Methods Mol. Biol. 615, 217–226 (2010)
Pierce, S.B., Costa, M., Wisotzkey, R., Devadhar, S., Homburger, S.A., Buchman, A.R., Ferguson, K.C., Heller, J., Platt, D.M., Pasquinelli, A.A., Liu, L.X., Doberstein, S.K., Ruvkun, G.: Regulation of DAF-2 receptor signaling by human insulin and ins-1, a member of the unusually large and diverse C. elegans insulin gene family. Genes Dev. 15, 672–686 (2001)
Husson, S.J., Clynen, E., Baggerman, G., Janssen, T., Schoofs, L.: Defective processing of neuropeptide precursors in Caenorhabditis elegans lacking proprotein convertase 2 (KPC-2/EGL-3): mutant analysis by mass spectrometry. J. Neurochem. 98, 1999–2012 (2006)
Dierking, K., Yang, W., Schulenburg, H.: Antimicrobial effectors in the nematode Caenorhabditis elegans: an outgroup to the Arthropoda. Philos. Trans. Royal. Soc. Lond. B Biol. Sci. 371, (2016)
Van Sinay, E., Mirabeau, O., Depuydt, G., Van Hiel, M.B., Peymen, K., Watteyne, J., Zels, S., Schoofs, L., Beets, I.: Evolutionarily conserved TRH neuropeptide pathway regulates growth in Caenorhabditis elegans. Proc. Natl. Acad. Sci. (2017). https://doi.org/10.1073/pnas.1617392114
Bauknecht, P., Jékely, G.: Large-scale combinatorial deorphanization of Platynereis neuropeptide GPCRs. Cell Rep. 12, 684–693 (2015)
Verdonck, R., De Haes, W., Cardoen, D., Menschaert, G., Huhn, T., Landuyt, B., Baggerman, G., Boonen, K., Wenseleers, T., Schoofs, L.: Fast and reliable quantitative peptidomics with labelpepmatch. J. Proteome Res. 15, 1080–1089 (2016)
Jarrell, T.A., Wang, Y., Bloniarz, A.E., Brittin, C.A., Xu, M., Thomson, J.N., Albertson, D.G., Hall, D.H., Emmons, S.W.: The connectome of a decision-making neural network. Science. 337, 437–444 (2012)
Bargmann, C.I.: Beyond the connectome: how neuromodulators shape neural circuits. Bioessays. 34, 458–465 (2012)
Beverly, M., Anbil, S., Sengupta, P.: Degeneracy and neuromodulation among thermosensory neurons contribute to robust thermosensory behaviors in Caenorhabditis elegans. J. Neurosci. 31, 11718–11727 (2011)
Bentley, B., Branicky, R., Barnes, C.L., Chew, Y.L., Yemini, E., Bullmore, E.T., Vértes, P.E., Schafer, W.R.: The multilayer connectome of Caenorhabditis elegans. PLoS Comput. Biol. 12, e1005283 (2016)
Acknowledgments
The authors gratefully acknowledge KU Leuven (C14/15/049), the Research Foundation Flanders (FWO G069713 and G052217N), and the European Research Council (ERC-ADG-340318) for financial support. S.Z., L.T. and I.B. are FWO postdoctoral fellows. S.V.B. is supported by the Flemish Institute for Science and Technology (IWT-Flanders). The authors thank Luc Vanden Bosch and Fran Naranjo for technical assistance, Elien Van Sinay for her constructive comments on the final manuscript, and SyBioMa for instrument access.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Van Bael, S., Zels, S., Boonen, K. et al. A Caenorhabditis elegans Mass Spectrometric Resource for Neuropeptidomics. J. Am. Soc. Mass Spectrom. 29, 879–889 (2018). https://doi.org/10.1007/s13361-017-1856-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-017-1856-z