
Abstract
Background and Objective
Abrocitinib is an oral small-molecule Janus kinase (JAK)-1 inhibitor approved for the treatment of moderate-to-severe atopic dermatitis. In vitro studies indicated that abrocitinib is a weak time-dependent inhibitor of cytochrome P450 (CYP) 2C19/3A and a weak inducer of CYP1A2/2B6/2C19/3A. To assess the potential effect of abrocitinib on concomitant medications, drug-drug interaction (DDI) studies were conducted for abrocitinib with sensitive probe substrates of these CYP enzymes. The impact of abrocitinib on hormonal oral contraceptives (ethinyl estradiol and levonorgestrel), as substrates of CYP3A and important concomitant medications for female patients, was also evaluated.
Methods
Three Phase 1 DDI studies were performed to assess the impact of abrocitinib 200 mg once daily (QD) on the probe substrates of: (1) 1A2 (caffeine), 2B6 (efavirenz) and 2C19 (omeprazole) in a cocktail study; (2) 3A (midazolam); and (3) 3A (oral contraceptives).
Results
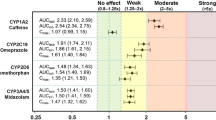
After multiple doses of abrocitinib 200 mg QD, there is a lack of effect on the pharmacokinetics of midazolam, efavirenz and contraceptives. Abrocitinib increased the area under the concentration time curve from 0 to infinity (AUCinf) and the maximum concentration (Cmax) of omeprazole by approximately 189 and 134%, respectively. Abrocitinib increased the AUCinf of caffeine by 40% with lack of effect on Cmax.
Conclusions
Based on the study results, abrocitinib is a moderate inhibitor of CYP2C19. Caution should be exercised when using abrocitinib concomitantly with narrow therapeutic index medicines that are primarily metabolized by CYP2C19 enzyme. Abrocitinib is a mild inhibitor of CYP1A2; however, the impact is not clinically relevant, and no general dose adjustment is recommended for CYP1A2 substrates. Abrocitinib does not inhibit CYP3A or induce CYP1A2/2B6/2C19/3A and does not affect the pharmacokinetics of contraceptives.
Clinical Trials Registration
ClinicalTrials.gov registration IDs: NCT03647670, NCT05067439, NCT03662516.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
To assess the potential effect of abrocitinib on concomitant medications that are substrates of the CYP1A2/2B6/2C19/3A enzymes, drug-drug interaction (DDI) studies were conducted for abrocitinib with sensitive probe substrates, including caffeine, efavirenz, omeprazole and midazolam |
Abrocitinib is a moderate inhibitor of CYP2C19 enzyme. Caution should be exercised when using abrocitinib concomitantly with narrow therapeutic index medicines that are primarily metabolized by CYP2C19 enzyme (e.g., S-mephenytoin, clopidogrel). Abrocitinib is a mild inhibitor of CYP1A2, while the impact is not clinically relevant with no dose adjustment warranted for CYP1A2 substrates |
Hormonal oral contraceptives (a combination of ethinyl estradiol and levonorgestrel) are substrates of CYP3A and important concomitant medications for female AD patients. The oral contraceptive DDI study results indicated that multiple doses of abrocitinib do not reduce the exposure of oral contraceptives |
1 Introduction
Abrocitinib is a small-molecule Janus kinase (JAK)-1 inhibitor [1] that is administered orally once daily and approved for the treatment of moderate-to-severe atopic dermatitis (AD) [2,3,4,5,6]. The recommended starting dose for abrocitinib is 200 mg once daily (QD) with 50 mg and 100 mg QD options available for dose adjustment and special populations. The pharmacokinetic profile of abrocitinib showed a rapid absorption with the median time to reach maximum concentration (Tmax) within 1 h after the administration as tablet. Abrocitinib has a short elimination half-life of approximately 5 h after the administration of 100- or 200-mg doses orally. Both the maximum concentration (Cmax) and area under the concentration time curve (AUC) values of abrocitinib showed a dose-proportional increase within the therapeutic doses. Abrocitinib has two active metabolites, M1 (3-hydroxypropyl) and M2 (2-hydroxypropyl, 12%), attributing ~10% and ~30%, respectively, of the overall pharmacologic activity of abrocitinib. The sum of unbound exposures of abrocitinib, M1 and M2, each in molar units and adjusted for relative potencies, is termed the active moiety of abrocitinib [7, 8].
As assessed in human liver microsomes (HLM) and human hepatocytes, abrocitinib is a weak time-dependent inhibitor (TDI) of cytochrome P450 (CYP) 2C19/3A and a weak inducer of CYP1A2/2B6/2C19/3A enzymes (data on file). TDI studies with abrocitinib (0.1-100 µM) in HLM showed an absence of inhibition of the major CYP enzymes without nicotinamide adenine dinucleotide phosphate (NADPH). In the presence of NADPH, abrocitinib caused relatively weak time-dependent inhibition of CYP2C19 (KI 52 µM; kinact 0.012/min) and CYP3A4/5 (KI 178 µM; kinact 0.032/min). Induction of major CYP enzymes by abrocitinib (1–100 µM) was assessed in human cryopreserved hepatocytes from three donors and compared with the controls. Abrocitinib at ≥ 10 µM caused a concentration-dependent > 2-fold induction of CYP3A4 mRNA (EC50, 61 µM; Emax, 22) in two of the three hepatocyte lots. Abrocitinib also caused > 2-fold induction of CYP2B6 mRNA (EC50, 9.5 µM; Emax, 3.5) at concentrations ≥ 10 µM in all three hepatocyte lots. The CYP2B6 enzyme activity showed > 2-fold induction at ≥ 30 µM in one hepatocyte lot. The treatment of human hepatocytes with abrocitinib resulted in the > 2-fold induction of CYP2C19 mRNA at concentrations ≥ 60 µM in all three lots of human hepatocytes. A concentration-dependent increase occurred in CYP1A2 mRNA in three of three lots of hepatocytes (EC50 19.7 µM; Emax 4.6), while CYP1A2 enzyme activity showed no change in two of three lots and declined in one of three lots.
The relative in vitro TDI or induction risk from each metabolite was less than that of abrocitinib. In HLM, M1 and M2 demonstrated no reversible inhibition of CYP1A2/2B6/2C19/3A and weak TDI of CYP3A at incubation concentration ≥ 99.5 µM after a 30-min preincubation. In the absence of NADPH, M1 and M2 demonstrated weak TDI for CYP3A and 2C19. In the presence of NADPH, M1 and M2 were weak TDIs of CYP3A and CYP2C19.
With the aforementioned in vitro enzyme inhibition and induction profile of abrocitinib, drug-drug interaction (DDI) studies evaluating the effect of abrocitinib on the pharmacokinetics of sensitive CYP1A2, 2B6, 2C19 and 3A substrates were warranted per regulatory guidance [9, 10]. Midazolam, caffeine, efavirenz and omeprazole were used as probe drugs for CYP3A, 1A2, 2B6 and 2C19, respectively. The DDI assessments with caffeine, efavirenz and omeprazole were performed in a single study; they are a subset of the Basel cocktail and have been validated for simultaneous phenotyping of CYP isoforms in the study published by Donzelli et al. [11].
In female patients of childbearing potential, oral hormonal contraceptives (OCs) are anticipated to be administered concomitantly with abrocitinib. The metabolism of such OC steroids such as ethinyl estradiol (EE) and levonorgestrel (LN) is mediated by the CYP3A system and Phase 2 enzymes such as uridine diphospho-glucuronosyltransferase (UGT) and sulfotransferase (SULT) [12, 13]. Induction of the metabolizing systems involved in metabolism of OCs may result in clinically important reduction in the systemic exposure of these hormonal contraceptives, leading to failure of contraception. Abrocitinib showed weak signal for induction via the pregnane X receptor (PXR) pathway (data on file), which is a modulator for the levels of several phase 1 (CYP3A, CYP2B6) and phase 2 (UGTs, SULTs) metabolizing enzymes [14]. Although the risk of induction of metabolic pathways is considered low, clinical data demonstrating the impact of abrocitinib on OCs upon coadministration are desired. Hence, a DDI study estimating the effect of abrocitinib on the pharmacokinetics of two commonly concomitantly administered OCs, EE and LN, was conducted.
The objectives of the DDI studies presented in the current article were to evaluate the effect of abrocitinib on the in vivo pharmacokinetics of midazolam, caffeine, efavirenz and omeprazole, as sensitive substrates of CYP3A, CYP1A2, CYP2B6 and CYP2C19, respectively, and of hormonal OC (EE and LN) in healthy adult participants.
2 Methods
2.1 Study Designs and Participants
The three DDI studies are Phase 1, open-label, multiple dose studies in healthy participants evaluating the effect of multiple doses of abrocitinib 200 mg QD on the pharmacokinetics of midazolam (2 mg single dose [SD]), caffeine (100 mg SD), efavirenz (50 mg SD), omeprazole (10 mg SD) and OC containing ethinyl estradiol (EE) 30 μg and levonorgestrel (LN) 150 μg. The treatment and pharmacokinetic sampling schedules are presented in Table S2–4. DDI assessments with caffeine, efavirenz and omeprazole were conducted under a single protocol as a cocktail DDI study (Table S3).
On pharmacokinetic sampling days, substrate drugs were administered orally with water after overnight fasted conditions. No food was allowed for at least 4 h following dosing. Water is permitted until 1 h prior to study intervention administration. Water may be consumed without restriction beginning 1 h after dosing. Participants were not permitted to lie down during the first 4 h after dosing to standardize the conditions. This research was conducted in accordance with the Helsinki Declaration of 1964 and its later amendments. The final protocol, any amendments and informed consent documentation were reviewed and approved by the Independent Ethics Committee at the investigational center participating in the study. Written informed consent was obtained from all participants prior to enrollment.
Main inclusion criteria were: female or male (female only for the OC DDI study) participants aged 18–55 years; healthy, defined as no clinically relevant abnormalities identified by a detailed medical history, full physical examination, including blood pressure and pulse measurement, 12-lead electrocardiogram or clinical laboratory tests. Main exclusion criteria were: evidence or history of clinically significant dermatological condition or visible rash present during physical examination; any history of chronic infections, any history of recurrent infections, any history of latent infections or any acute infection within 2 weeks of screening; positive urine drug test; use of tobacco or nicotine-containing products within 3 months of screening; use of prescription or non-prescription drugs (except for acetaminophen/paracetamol at doses of ≤1 g/day) and dietary supplements within 7 days or 5 half-lives (whichever was longer) prior to first dose. Hormone replacement therapy was required to be discontinued at least 28 days prior to the first dose of investigational product. For the cocktail DDI study, participants who routinely consumed more than five 8-ounce cups of coffee or other caffeine equivalent beverage per day were excluded.
2.2 Dietary and Activity Restrictions
The daily caloric intake per participant did not exceed 3200 kcal, and the total daily nutritional composition was approximately 55% carbohydrate, 30% fat and 15% protein during confinement at the research center. Participants were not allowed to eat or drink grapefruit or grapefruit-related citrus fruits from 7 days before the first dose of investigational product until collection of the final blood sample. Participants were required to abstain from alcohol products for 24 h before the study and continue to abstain until collection of the final blood sample. Participants were required to abstain from strenuous exercise for at least 48 h before each blood collection.
For the cocktail DDI study, participants were required to abstain from caffeine-containing products for 48 h before the study and continue to abstain until collection of the final blood sample.
2.3 Blood Sample Collection
Blood samples were collected for plasma pharmacokinetic analyses per schedule presented in Table S2–4. In the midazolam DDI study, 4-ml blood samples for midazolam pharmacokinetic analysis were collected up to 24 h after the midazolam doses (Table S2). In the cocktail DDI study, 6-ml blood samples were collected for 48 h (72 h for efavirenz because of its half-life) following dosing on Period 1 Day 1 and Period 2 Day 8. After the omeprazole only dose on Period 2 Day 2, sparse samples up to 8 h were collected to assess the potential inhibition effect of abrocitinib on CYP2C19 (Table S3). In the OC DDI study, serial blood samples (12 ml) were collected for 48 h after the OC dose in Period 1 and 2 (Table S4). Pharmacokinetic sampling schedules were determined based on the pharmacokinetic profiles of the victim drugs and anticipated DDI effect.
In addition, variation in the CYP2C9 and CYP2C19 gene may influence the metabolism and pharmacokinetics of abrocitinib; blood samples were collected on Day 1 pre-dose for genotypic analysis of the CYP2C9 (*2 and *3) and CYP2C19 (*2, *3, *4 and *17). Blood samples were also analyzed for allelic variants of CYP1A2 and 2B6 in the cocktail DDI study, as variation in this gene may influence the metabolism and pharmacokinetics of the probe drugs.
Blood samples were collected into tubes containing dipotassium ethylenediaminetetraacetic acid. The blood collection tube was gently inverted eight to ten times to completely mix the blood and anticoagulant. Within 30 min of collection, centrifugation was carried out at 1700g under 2–8 ℃ until the plasma was separated from the blood cells. Plasma samples for pharmacokinetic analyses were stored at − 80 to −20 ℃ until analysis.
2.4 Pharmacokinetic Sample Analysis
Midazolam and stable isotope-labeled internal standard (SLIS) midazolam-D4 were isolated from K2EDTA human plasma using a liquid-liquid extraction procedure. Following extraction and processing, samples were analyzed by high‐performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS) using a Waters Xbridge HILIC, 2.1 × 50 mm, 3.5-µm column under positive mode with a TurboIonSpray interface.
EE and SLIS EE-D4 were isolated from K2EDTA human plasma using a liquid-liquid extraction and derivatization procedure. After processing, samples were analyzed by high-performance liquid chromatography (UPLC-MS/MS) using a Zorbax SB-C18, 50 × 4.6 mm, 3.5-µm column under positive mode with a TurboIonSpray interface.
Levonorgestrel and its SLIS levonorgestrel-d6 were isolated from K2EDTA human plasma using an automated liquid-liquid extraction procedure. After processing, the samples were injected into LC-MS/MS using a Chromolith/Speed Rod, 50 × 4.6 mm, 2-μm column under positive mode with a TurboIonSpray interface.
Caffeine and its SLIS caffeine-D9 were isolated from K2EDTA human plasma using an automated protein precipitation extraction procedure. After processing, the samples were injected into LC-MS/MS using an ACE 3 C18-PFP, 50 × 4.6 mm, 3-μm column under positive mode with a TurboIonSpray interface.
Omeprazole and its SLIS omeprazole-D3 were isolated from K2EDTA human plasma using an automated protein precipitation extraction procedure. After processing, the samples were injected into LC-MS/MS using an Acquity UPLC BEH C18, 100 × 2.1 mm, 1.7-μm column under positive mode with a TurboIonSpray interface.
Unconjugated efavirenz (referred to as efavirenz from here on) and SLIS efavirenz-D5 were extracted from K2EDTA plasma by an automated liquid-liquid extraction procedure. After processing, the samples were injected into LC-MS/MS using an ACE Excel 2 C18, 50 × 3 mm, 2-μm column under negative mode with a TurboIonSpray interface.
2.5 Mass Spectrometer Calibration Parameters and Statistics of Pharmacokinetic Samples
The calibration range of the HPLC-MS/MS method used to determine the concentration of total midazolam was 0.0500–50.0 ng/ml, and the quality control (QC) concentrations were 0.150 ng/ml, 3.75 ng/ml, 37.5 ng/ml and 375 ng/ml. The inter-day assay accuracy ranged from − 2.67 to 2.00%, and the between-day precision was ≤ 7.06%.
The calibration range of the HPLC-MS/MS method used to determine the concentration of EE in K2EDTA human plasma was 1.00–400 pg/ml, and the QC concentrations were 3.01 pg/ml, 30.0 pg/ml, 200 pg/ml and 300 pg/ml. The inter-day assay accuracy ranged from − 0.7 to 5.0%, and the intra-day precision was ≤ 4.0%.
The calibration range of the HPLC-MS/MS method used to determine the concentration of levonorgestrel in K2EDTA human plasma was 10.0–10,000 pg/ml, and the QC concentrations were 30.0 pg/ml, 500 pg/ml, 5000 pg/ml and 7500 pg/ml. The inter-day assay accuracy ranged from − 4.8 to – 1.0%, and the intra-day precision was ≤ 6.9%.
The calibration range of the HPLC-MS/MS method used to determine the concentration of caffeine in K2EDTA human plasma was 20.0–20,000 ng/ml, and the QC concentrations were 60.0 ng/ml, 1000 ng/ml, 10,000 ng/ml and 15,000 ng/ml. The inter-day assay accuracy ranged from − 4.0 to 3.0%, and the intra-day precision was ≤ 9.5%.
The calibration range of the HPLC-MS/MS method used to determine the concentration of omeprazole in K2EDTA human plasma was 1.00–1250 ng/ml, and the QC concentrations were 3.00 ng/ml, 43.8 ng/ml, 625 ng/ml and 938 ng/ml. The inter-day assay accuracy ranged from − 3.0% to 1.4%, and the intra-day precision was ≤ 6.8%.
The calibration range of the HPLC-MS/MS method used to determine the concentration of efavirenz in K2EDTA human plasma was 1.00–1000 ng/ml, and the QC concentrations were 3.00 ng/ml, 50.0 ng/ml, 500 ng/ml and 750 ng/ml. The inter-day assay accuracy ranged from − 2.1 to 2.3%, and the intra-day precision was ≤ 6.8%.
Full mass spectrometer settings and acquisition parameters for detecting midazolam, EE, Levonorgestrel, caffeine, omeprazole and efavirenz in human plasma are found in Supplementary Table S1.
2.6 Statistical Methods and Pharmacokinetic Parameters
Pharmacokinetic parameters were derived from the concentration-time profiles using standard non-compartmental methods, including AUC from 0 to infinity (AUCinf, if data permitted), AUC from time 0 to the time of last quantifiable concentration (AUClast), Cmax, Tmax and terminal half-life (t½, if data permitted). Parameters noted “if data permitted” were reported only where a well-characterized terminal phase was observed. A well-characterized terminal phase was defined as one with at least three data points, r2 ≤ 0.9, and AUCextrap% ≤ 20. Pharmacokinetic parameter values were calculated using Pfizer’s internally validated software system eNCA (version 2.2.4) for the midazolam and OC DDI studies and oNCA (version 2.5.6) for the cocktail DDI study. The statistical software used was SAS (SAS Institute, Cary, NC, USA). Samples below the lower limit of quantitation (20 ng/ml for caffeine, 1 ng/ml for efavirenz and omeprazole, 1 pg/ml for ethinyl estradiol, 10 pg/ml for levonorgestrel, 0.05 ng/ml for midazolam) were set to 0 ng/ml for the pharmacokinetic analysis. Actual pharmacokinetic sampling times were used in the derivation of pharmacokinetic parameters. The pharmacokinetic parameters were summarized descriptively by treatment. No subgroup analyses by genotype status were performed.
Natural log-transformed parameters were analyzed using a mixed effects model with treatment as a fixed effect and subject as a random effect. Estimates of the adjusted mean differences (test-reference) and corresponding 90% confidence intervals (CIs) were obtained from the model. The adjusted mean differences and 90% CIs for the differences were exponentiated to provide estimates of the ratio of adjusted geometric means (test/reference) and 90% CI for the ratios. The substrates alone was the reference treatment, while the substrate coadministered with abrocitinib was the test treatment.
The safety population was defined as all participants that received at least one dose of study medication. Safety data are summarized descriptively. Adverse events were graded as mild, moderate or severe by the principal investigator based on guidance outlined in the study protocol. Serious adverse events were defined as adverse events that met any of the following criteria: (1) results in death; (2) is life-threatening (immediate risk of death); (3) requires inpatient hospitalization or prolongation of existing hospitalization; (4) results in persistent or significant disability/incapacity (substantial disruption of the ability to conduct normal life functions); (5) results in congenital anomaly/birth defect; (6) is considered an important medical event.
Treatment-emergent adverse events (TEAE) were any events occurring following start of treatment or increasing in severity. Events that occurred in a non-treatment period (for example, washout or follow-up) were counted as treatment emergent and attributed to the previous treatment taken.
3 Results
3.1 Participants Demographics
Demographic characteristics are summarized in Table 1.
The initial number of participants in the cocktail DDI study was 13; however, some of the pharmacokinetic samples from Day 8 Period 2 were lost in transit, and remaining samples were not evaluable. Therefore, the study team decided to repeat the study and enroll 13 replacement participants. The pharmacokinetics collected from the 13 replacement participants were included in the descriptive or statistical summaries. Safety and genotyping results from all 26 participants were reported in this article.
3.2 Midazolam Pharmacokinetics
Median plasma midazolam concentration-time profiles are presented in Fig. 1, and pharmacokinetic parameters are summarized descriptively in Table 2. When midazolam was coadministered with a single oral dose of abrocitinib 200 mg QD on Day 2, median plasma midazolam concentrations were lower than those following administration of midazolam alone with the adjusted geometric mean AUCinf and Cmax values decreased approximately 15.7% and 13.7%, respectively (Table 2). On Day 7, the adjusted geometric mean AUCinf and Cmax values of midazolam decreased approximately 7.7% and 6.5%, respectively, following multiple doses of abrocitinib 200 mg QD compared to midazolam administration alone. The lower bound of corresponding 90% CI for AUCinf ratio was > 80% (Table 3). Following multiple oral doses of abrocitinib 200 mg QD on Day 7, median plasma midazolam concentrations were nearly superimposable with midazolam administered alone. Based on the net effect observed on Day 7 of multiple abrocitinib dosing, a lack of clinically meaningful impact on midazolam from abrocitinib can be concluded.

Median plasma midazolam concentration-time profiles. QD once daily
3.3 Caffeine Pharmacokinetics
Following administration of a single 100 mg oral dose of caffeine administered alone or after 8 days of multiple 200 mg QD doses of abrocitinib in healthy participants, mean Cmax was observed at a median Tmax of approximately 1 h for both treatments (Fig. 2). Terminal elimination t½ values were generally similar for both treatments with mean values of 5.843 h for caffeine alone and 6.830 h when coadministered with multiple doses of abrocitinib (Table 4). Caffeine exposure as assessed by AUC was approximately 40% higher when coadministered with multiple doses of abrocitinib compared to when caffeine was dosed alone, whereas Cmax was similar between both treatments (Table 3).

Median plasma caffeine concentration-time profiles. QD once daily
Pre-dose concentrations were measurable for caffeine (Period 1: 6.83% of Cmax in 1 participant; Period 2: 1.05 - 6.74% of Cmax in 4 participants and 14.87% of Cmax in 1 participant). The quantifiable pre-dose concentrations of caffeine occurred in both Periods 1 and 2 and may be due to dietary consumption of caffeine-related components of a meal by the participants, as the elimination half-life is short (6–7 h) and would have allowed complete washout of dose prior to Period 2. For the calculations of AUC and Cmax for participants with a pre-dose concentration > 5% of the corresponding Cmax, residual concentrations have been corrected assuming first-order elimination. The assumption of first-order elimination is applicable here to calculate the residual exposures, because the terminal phases of the pharmacokinetic profile for caffeine are well characterized. At least three data points are used to describe the terminal phase with high r2 value.
3.4 Efavirenz Pharmacokinetics
Following administration of a single 50 mg oral dose of efavirenz administered alone or after 8 days of multiple 200 mg QD doses of abrocitinib in healthy participants, mean Cmax was observed with a median Tmax of 3.00 h and 3.92 h, respectively (Fig. 3, Table 5). Terminal elimination t½ and AUCinf values for efavirenz could not be reported per the criteria specified in Sect. 2.5. Efavirenz exposures as assessed by AUClast and Cmax were similar when coadministered with multiple doses of abrocitinib compared to when efavirenz was dosed alone (Table 3).

Median plasma efavirenz concentration-time profiles. QD once daily
Pre-dose concentrations were measurable for efavirenz in a total of 13 participants in Period 2 (1.64–8.14% of Cmax in 12 participants and 13.85% of Cmax in 1 participant), likely because of the long elimination half-life of efavirenz in relation to the allowed washout period between treatments in this study. Similar to the calculation of caffeine pharmacokinetic parameters, a correction method for residual concentrations has been applied for AUC and Cmax, assuming first-order elimination, for participants with a pre-dose concentration > 5% of the corresponding Cmax.
3.5 Omeprazole Pharmacokinetics
Following administration of a single 10 mg oral dose of omeprazole alone or after 8 days of multiple 200 mg QD doses of abrocitinib in healthy participants, mean Cmax was observed with a median Tmax of 2.00 h and 2.97 h, respectively (Fig. 4). Terminal elimination t½ values were 0.822 h for omeprazole alone and 1.141 h when coadministrated with multiple doses of abrocitinib (Table 6). Omeprazole exposure as assessed by both AUC and Cmax increased when coadministrated with multiple doses of abrocitinib compared to when omeprazole was dosed alone. Abrocitinib increased omeprazole Cmax by 134% and AUCinf by 189% (Table 3).

Median plasma omeprazole concentration-time profiles. QD once daily
A single dose of 10 mg omeprazole was also administered on Day 2 Period 2 during abrocitinib 200 mg QD doses with sparse pharmacokinetic samples collected. Abrocitinib showed an inhibition effect on CYP2C19 and increased the omeprazole exposure as early as Day 2 of multiple dosing. Comparing the pharmacokinetic concentrations of omeprazole on Day 2 to those on Day 8 that were collected at the same time points, the concentrations on Day 8 were somewhat higher, indicating the lack of induction by abrocitinib.
3.6 Ethinyl Estradiol Pharmacokinetics
Median plasma EE concentration-time profiles with and without multiple doses of abrocitinib are presented in Fig. 5. When EE was coadministered with multiple doses of abrocitinib, median plasma EE concentrations were slightly higher than those following administration of EE alone. The median Tmax of EE was 1.5 h when EE was administered alone and when coadministered with multiple doses of abrocitinib. The mean apparent t½ was 15.64 h for EE alone and 16.70 h for EE with multiple doses of abrocitinib (Table 7). Results of the statistical comparisons are summarized in Table 3. The adjusted geometric mean for AUCinf and Cmax increased by approximately 19% and 7%, respectively, following coadministration with multiple doses of abrocitinib as compared to EE administration alone. The lower bound of corresponding 90% CI for AUCinf ratio was > 80%, indicating a lack of impact on EE from abrocitinib.

Median plasma ethinyl estradiol concentration-time profiles. QD once daily
3.7 Levonorgestrel Pharmacokinetics
Median plasma LN concentration-time profiles with and without multiple doses of abrocitinib are presented in Fig. 6, and pharmacokinetic parameters are summarized descriptively in Table 7. LN t½ and AUCinf were not reported for all participants because of lack of a well-characterized terminal phase (AUCextrap% > 20). When LN was administered with or without multiple doses of abrocitinib, LN exposure was similar for both treatments. The median Tmax of LN was 1 h when LN was administered alone and when coadministered with multiple doses of abrocitinib. The adjusted geometric mean for AUClast did not change. The adjusted geometric mean for Cmax decreased by approximately 14%.

Median plasma levonorgestrel concentration-time profiles. QD once daily
The lower bound of 90% CIs for the adjusted geometric mean ratio of AUClast were > 80% (Table 3). Therefore, the absence of the abrocitinib effect on LN was concluded.
3.8 Pharmacogenomic Evaluations
As variation in the CYP2C19 and CYP2C9 gene may influence the metabolism and pharmacokinetics of abrocitinib, genotyping was performed for CYP2C19 (*2, *3, *4 and *17) and CYP2C9 (*2 and *3). Allelic variants of CYP1A2 and 2B6 were assessed in the cocktail DDI study. The genotyping results are presented in Table 8. No subgroup analyses by genotype status were performed for the current article. The impact of genetic variations on the pharmacokinetics of abrocitinib has been evaluated and will be summarized in a separate publication.
3.9 Safety
No serious or severe adverse events (AEs) were reported. There were no AE-related dose reductions or temporary discontinuations due to AEs. In the midazolam DDI study, one participant permanently discontinued the study because of the AE of first-degree atrioventricular block during treatment with midazolam 2 mg alone, which was considered related to study treatment and moderate in severity. One participant permanently discontinued the study because of an AE of presyncope in the OC DDI study, which was considered treatment related. Incidence of treatment-emergent adverse events (TEAEs) and treatment-related adverse events (TRAEs) are shown in Table 9. In the cocktail DDI study, the most frequently reported treatment-related TEAE was tremor on treatment of caffeine 100 mg + efavirenz 50 mg + omeprazole 10 mg (Days 1–3 of Period 1). In the midazolam DDI study, the most frequently reported all-causality TEAEs were headache, which was experienced by eight participants only during coadministration treatment of abrocitinib 200 mg QD and midazolam 2 mg, and nausea, which was experienced by one and three participants during midazolam alone and coadministration treatments, respectively. Increased plasma exposure of abrocitinib resulted in more TRAEs in the midazolam DDI study, although limited conclusions can be drawn from a single-dose study. In the OC DDI study, the most frequently reported TEAE was headache (6), and all AEs of headache were not considered treatment-related. Overall, abrocitinib, when coadministered with midazolam, caffeine, efavirenz, omeprazole or oral contraceptives, was generally safe and well tolerated in healthy adult participants.
4 Discussion
This article summarized the results of DDI studies in healthy adult participants evaluating the effect of abrocitinib on the in vivo pharmacokinetics of midazolam, caffeine, efavirenz and omeprazole as sensitive substrates of CYP3A, CYP1A2, CYP2B6 and CYP2C19, respectively, as well as hormonal OC (EE and LN). After multiple doses of abrocitinib 200 mg QD, there is a lack of effect on the pharmacokinetics of midazolam, efavirenz or OCs. Therefore, abrocitinib is not an inhibitor of CYP3A or an inducer of CYP2B6/3A. Coadministration of abrocitinib 200 mg QD with omeprazole 10 mg single dose increased the AUCinf and Cmax of omeprazole by approximately 2.9- and 2.3-fold, respectively, indicating that abrocitinib is a moderate inhibitor of CYP2C19 enzyme. Abrocitinib is also a mild inhibitor of CYP1A2 enzyme, which increased the AUCinf of caffeine by 1.4-fold with lack of effect on Cmax.
The DDI studies for CYP3A and 2C19 with midazolam and omeprazole, respectively, were designed to investigate the net inhibition and induction effects of abrocitinib. As assessed in the in vitro assays, abrocitinib showed both weak TDI and weak induction of CYP3A and 2C19. In the DDI evaluations, the pharmacokinetic samples of midazolam and omeprazole were collected after 2 and 7 days (8 days for omeprazole in the cocktail DDI study) of multiple abrocitinib 200 mg QD doses. Multiple dose administration of the investigational drug for a minimum of 7 days is generally recommended to evaluate its induction effect on enzyme activity, as inducers can take several days to exert their effects [15], while the onset of inhibition effect on CYP enzymes by potential inhibitors is relatively quick [16]. Therefore, evaluating the DDI on Day 7 (Day 8 for omeprazole in the cocktail DDI study) informed the net effect of mixed induction and inhibition of abrocitinib. Assessment on Day 2 helps in understanding the inhibition component within the net effect, understanding the mechanism and translation, and eventually informing the label [17]. In the omeprazole study, only sparse pharmacokinetic samples were taken for the Day 2 assessment for future exploration using the physiologically based pharmacokinetic (PBPK) modeling approach to reduce the burden on study participants.
Abrocitinib was demonstrated to be a moderate inhibitor of CYP2C19 based on the omeprazole DDI. Notably, the effect of abrocitinib on omeprazole itself is not considered clinically relevant with the wide safety margin of omeprazole. Adjustment of the omeprazole dose when used concomitantly with a moderate CYP2C19 inhibitor is not generally required [18]. Based on a comprehensive review of recommendations in the labels of known CYP2C19 substrates regarding concomitant use with moderate CYP2C19 inhibitors, dose adjustment is not warranted for the following medications that are primarily metabolized by CYP2C19: cilostazol, lansoprazole, diazepam, esomeprazole, pantoprazole and voriconazole. In contrast, dose reduction is required for citalopram, clobazam, escitalopram and selumetinib; therapeutic dose monitoring is recommended for S-mephenytoin. In addition, clopidogrel is a prodrug, the exposure of which is expected to increase upon coadministration with abrocitinib, while its active metabolite and the clinical effect are expected to decrease. Due to the narrow therapeutic window of clopidogrel, concomitant use of abrocitinib with clopidogrel is discouraged.
CYP2C19 is the main enzyme responsible for approximately 53% of abrocitinib metabolism [8, 19]. The moderate inhibitory effect of abrocitinib on CYP2C19 is consistent with the fact that the observed accumulation ratio of abrocitinib (1.5) is higher than the predicted ratio of 1.1 based on its half-life of approximately 5 h [20]. Omeprazole is a moderate CYP2C19 inhibitor; however, the impact of omeprazole on abrocitinib in the current DDI study is negligible because omeprazole was administered as a single dose at the lowest commercially available dosage. Using omeprazole as the probe of CYP2C19 enzyme maximized the possibility of identifying a DDI.
No subgroup analyses by genotype status were performed for the current manuscript. The impact of genetic variations of CYP2C19 and 2C9 on the pharmacokinetics of abrocitinib has been evaluated and will be summarized in a separate publication. In the omeprazole DDI study, participants who were identified as CYP2C19 ultra-rapid or rapid metabolizers tended to have lower omeprazole exposure compared with CYP2C19 normal, intermediate or poor metabolizers, consistent with expectations. The inhibition effect of abrocitinib on omeprazole in ultra-rapid or rapid metabolizers also tended to be more prominent than that in participants with other metabolism phenotypes. This study enrolled a mixture of various CYP2C19 metabolizer statuses, which enabled a robust assessment of the DDI effect and proper representation of the general population.
The midazolam DDI study was performed first and separate from the cocktail DDI, although midazolam is also part of the Basel cocktail [11]. This is because CYP3A4 is the most sensitive sentinel of risk based on in vitro assessments. The initial thought was to use the midazolam DDI to help to contextualize the potential TDI and/or induction risk of other flagged CYPs. The midazolam and abrocitinib DDI study results turned out to be negative and excluded the DDI potential of abrocitinib on CYP3A enzymes. However, with emerging in vitro data, in vivo evaluations of CYP1A2, 2B6 and 2C19 were warranted. These three DDIs were then conducted within a single cocktail DDI study. The Basel cocktail consists of six commercially available probe drugs, including caffeine 100 mg, efavirenz 50 mg, omeprazole 10 mg, metoprolol 12.5 mg, losartan 12.5 and midazolam 2 mg, to assess the enzyme activity of CYP1A2, 2B6, 2C19, 2D6, 2C9 and 3A, respectively. This combination of probe drugs has been demonstrated to lack mutual interactions and showed sufficient sensitivity to detect inhibition and induction of CYP activities [11, 21].
Not being able to perform the DDI assessment for CYP3A4 and other CYP enzymes within the same phenotyping cocktail study is considered a limitation. Prior to conducting the clinical DDI studies, adequate information from nonclinical or in vitro studies should be collected to inform the clinical plans. Administration of individual probe substrates in separate, stand-alone clinical DDI studies is costly and time consuming and increases unnecessary drug exposure in healthy participants.
The purpose of caffeine DDI assessment was to contextualize the observed induction effect of abrocitinib on CYP1A2 in vitro; however, the total exposure of caffeine was in fact increased by approximately 40%. This observation excluded the induction potential of abrocitinib on CYP1A2. However, the mechanism in this mild inhibition is not clear, although abrocitinib or its metabolites did not show significant competitive or time-dependent inhibition of CYP1A2 in vitro. Overall, the impact on CYP1A2 is not clinically relevant with no dose adjustment warranted for CYP1A2 substrates.
The OC DDI study results demonstrated that multiple doses of abrocitinib 200 mg QD do not reduce the exposures of EE and LN, with the lower bounds of 90% CIs for both EE and LN AUClast ratios above the bioequivalence boundary of 80%. Hormonal OC is the second most commonly used contraceptive method following female permanent contraception [22]. There are no sufficient clinical data to establish an abrocitinib-associated risk regarding female fertility. In addition, AD affects many younger women of childbearing potential. The current study was designed and conducted in accordance with regulatory guidance.
5 Conclusion
Based on the study results, abrocitinib is a moderate inhibitor of CYP2C19 enzyme. Caution should be exercised when using abrocitinib concomitantly with narrow therapeutic index medicines that are primarily metabolized by CYP2C19 enzyme (e.g., S-mephenytoin, clopidogrel). The results of this study suggested that abrocitinib is a mild inhibitor of CYP1A2 enzyme; however, the impact is not clinically relevant, and no general dose adjustment is recommended for CYP1A2 substrates. Abrocitinib does not inhibit CYP3A or induce CYP1A2/2B6/2C19/3A and does not affect the pharmacokinetics of EE or LN.
Availability of data and material
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Code availability
Not applicable.
References
Vazquez ML, Kaila N, Strohbach JW, Trzupek JD, Brown MF, Flanagan ME, et al. Identification of N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfo namide (PF-04965842): a selective JAK1 clinical candidate for the treatment of autoimmune diseases. J Med Chem. 2018;61:1130–52. https://doi.org/10.1021/acs.jmedchem.7b01598.
Simpson EL, Sinclair R, Forman S, Wollenberg A, Aschoff R, Cork M, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;396:255–66. https://doi.org/10.1016/s0140-6736(20)30732-7.
Silverberg JI, Simpson EL, Thyssen JP, Gooderham M, Chan G, Feeney C, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156:863–73. https://doi.org/10.1001/jamadermatol.2020.1406.
Gooderham MJ, Forman SB, Bissonnette R, Beebe JS, Zhang W, Banfield C, et al. Efficacy and safety of oral janus kinase 1 inhibitor abrocitinib for patients with atopic dermatitis: a phase 2 randomized clinical trial. JAMA Dermatol. 2019;155:1371–9. https://doi.org/10.1001/jamadermatol.2019.2855.
Bieber T, Simpson EL, Silverberg JI, Thaci D, Paul C, Pink AE, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med. 2021;384:1101–12. https://doi.org/10.1056/NEJMoa2019380.
Reich K, Thyssen JP, Blauvelt A, Eyerich K, Soong W, Rice KP, et al. Efficacy and safety of abrocitinib versus dupilumab in adults with moderate-to-severe atopic dermatitis: a randomised, double-blind, multicentre phase 3 trial. Lancet. 2022;33:2335–43. https://doi.org/10.1080/09546634.2021.1961997.
Wang EQ, Le V, O’Gorman M, Tripathy S, Dowty ME, Wang L, et al. Effects of hepatic impairment on the pharmacokinetics of abrocitinib and its metabolites. J Clin Pharmacol. 2021;61:1311–23. https://doi.org/10.1002/jcph.1858.
Wang X, Dowty ME, Wouters A, Tatulych S, Connell CA, Le VH, et al. Assessment of the effects of inhibition or induction of CYP2C19 and CYP2C9 enzymes, or inhibition of OAT3, on the pharmacokinetics of abrocitinib and its metabolites in healthy individuals. Eur J Drug Metab Pharmacokinet. 2022;47:419–29. https://doi.org/10.1007/s13318-021-00745-6.
Prueksaritanont T, Chu X, Gibson C, Cui D, Yee KL, Ballard J, et al. Drug-drug interaction studies: regulatory guidance and an industry perspective. AAPS J. 2013;15(3):629–45. https://doi.org/10.1208/s12248-013-9470-x.
Clinical drug interaction studies—cytochrome P450 enzyme- and transporter-mediated drug interactions guidance for industry. FDA Guidance Document January 2020, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions. Accessed Dec 2023.
Donzelli M, Derungs A, Serratore MG, et al. The basel cocktail for simultaneous phenotyping of human cytochrome P450 isoforms in plasma, saliva and dried blood spots. Clin Pharmacokinet. 2014;53(3):271–82. https://doi.org/10.1007/s40262-013-0115-0.
Zhang H, Cui D, Wang B, Han YH, Balimane P, Yang Z, et al. Pharmacokinetic drug interactions involving 17alpha-ethinylestradiol: a new look at an old drug. Clin Pharmacokinet. 2007;46(2):133–57. https://doi.org/10.2165/00003088-200746020-00003.
Lello S, Cavani A. Ethynilestradiol 20 mcg plus levonorgestrel 100 mcg: clinical pharmacology. Int J Endocrinol. 2014;2014: 102184. https://doi.org/10.1155/2014/102184.
Iyer M, Reschly EJ, Krasowski MD. Functional evolution of the pregnane X receptor. Expert Opin Drug Metab Toxicol. 2006;2(3):381–97. https://doi.org/10.1517/17425255.2.3.381.
Galetin A, Burt H, Gibbons L, Houston JB. Prediction of time-dependent CYP3A4 drug–drug interactions: impact of enzyme degradation, parallel elimination pathways, and intestinal inhibition. Drug Metab Dispos. 2006;34:166–75. https://doi.org/10.1124/dmd.105.006874.
Tornio A, Filppula AM, Niemi M, Backman JT. Clinical studies on drug-drug interactions involving metabolism and transport: methodology, pitfalls, and interpretation. Clin Pharmacol Ther. 2019;105(6):1345–61. https://doi.org/10.1002/cpt.1435.
Dutreix C, Munarini F, Lorenzo S, et al. Investigation into CYP3A4-mediated drug–drug interactions on midostaurin in healthy volunteers. Cancer Chemother Pharmacol. 2013;72:1223–34. https://doi.org/10.1007/s00280-013-2287-6.
Omeprazole SmPC, https://www.ema.europa.eu/en/documents/referral/losec-article-30-referrals-annex-iii_en.pdf. Accessed May 2022.
Dowty M, Yang X, Lin J, Bauman J, Doran A, Goosen T, et al. P190 - The effect of CYP2C9 and CYP2C19 genotype on the pharmacokinetics of PF-04965842, A JAK1 inhibitor in clinical development. Drug Metab Pharmacokinet. 2020;35:S80. https://doi.org/10.1016/j.dmpk.2020.04.191.
Peeva E, Hodge MR, Kieras E, Vazquez ML, Goteti K, Tarabar SG, et al. Evaluation of a Janus kinase 1 inhibitor, PF-04965842, in healthy subjects: a phase 1, randomized, placebo-controlled, dose-escalation study. Br J Clin Pharmacol. 2018;84:1776–88. https://doi.org/10.1111/bcp.13612.
Derungs A, Donzelli M, Berger B, Noppen C, Krähenbühl S, Haschke M. Effects of cytochrome P450 inhibition and induction on the phenotyping metrics of the basel cocktail: a randomized crossover study. Clin Pharmacokinet. 2016;55(1):79–91. https://doi.org/10.1007/s40262-015-0294-y.
Kavanaugh ML, Pliskin E. Use of contraception among reproductive-aged women in the United States, 2014 and 2016. F&S Reports. 2020;1(2):83–93. https://doi.org/10.1016/j.xfre.2020.06.006.
Funding
These trials were sponsored by Pfizer Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors are employees and stockholders of Pfizer Inc.
Ethical approval
This research was conducted in accordance with the Helsinki Declaration of 1964 and its later amendments. The final protocol, any amendments and informed consent documentation were reviewed and approved by the Independent Ethics Committee (Comite d'Ethique Hospitalo-Facultaire Erasme-ULB, Brussels, Belgium, for the midazolam DDI study in 2018; IntegReview Ethical Review Board, Austin, TX, USA, for the OC DDI study in 2018; Advarra, Columbia, MD, USA, for the cocktail DDI study in 2021) at the investigational center participating in the study.
Consent to participate
Written informed consent was obtained from all participants prior to enrollment.
Consent for publication
Not applicable.
Author contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by XW, BM, MD, VHL, YH, MC, MO’G, JAW and GC. Bioanalytical data preparation and interpretation were performed by ST. VL: performed all statistical analyses of pharmacokinetic data. MC and GC: contributed to study safety. The first draft of the manuscript was written by XW; all authors critically reviewed the drafts and approved the final manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Wang, X., Dowty, M.E., Tripathy, S. et al. Assessment of the Effects of Abrocitinib on the Pharmacokinetics of Probe Substrates of Cytochrome P450 1A2, 2B6 and 2C19 Enzymes and Hormonal Oral Contraceptives in Healthy Individuals. Eur J Drug Metab Pharmacokinet 49, 367–381 (2024). https://doi.org/10.1007/s13318-024-00893-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-024-00893-5