
Abstract
Progesterone is a growth inhibitory hormone in the endometrium. While progestins can be used for the treatment of well-differentiated endometrial cancers, resistance to progestin therapy occurs for reasons that remain unclear. We have previously demonstrated that progesterone receptors (PR) A and B differentially regulate apoptosis in response to overexpression of the forkhead transcription factor, FOXO1. In this study, we further examined the PR-isoform-dependent cellular response to the AKT pathway. Treatment of PRA and PRB-expressing Ishikawa cells (PRA14, PRB23), with an AKT inhibitor API-59CJ-OMe (API-59) promoted apoptosis in the presence and absence of the ligand, R5020 preferentially in PRA14 cells. Upon PR knockdown using small interfering RNA, an increase in apoptosis was observed in PRB23 cells treated with API-59 with or without R5020 while there was no influence in PRA14 cells. Using an apoptosis-focused real-time PCR array, genes regulated by API-59 and R5020 were identified both common and unique to PRA14 and PRB23 cells. BIRC3 was identified as the only gene regulated by R5020 which occurred only in PRB cells. Knockdown of BIRC3 in PRB23 cells promoted a decrease in cell viability in response to API-59 + R5020. Furthermore, the important role of inhibitors of apoptosis (IAPs) in the PRB23 cells to promote cell survival was demonstrated using an antagonist to IAPs, a second mitochondria-derived activator of caspase (Smac also known as DIABLO) mimetic. Treatment of PRB23 cells with Smac mimetic increased apoptosis in response to API-59 + R5020. In summary, our findings indicate a mechanism by which PRB can promote cell survival in the setting of high AKT activity in endometrial cancer cells.
Similar content being viewed by others

Introduction
Progesterone is an essential hormone for the normal functions of the female reproductive tract. In the endometrium, progesterone antagonizes estrogen-mediated cell proliferation and promotes differentiation [1]. Unopposed estrogen action can be the driving force behind such pathologies as endometrial hyperplasia or endometrial adenocarcinoma, which are the most common gynecologic cancers to affect women [2, 3]. Progestins are sometimes used as the primary treatment for endometrial hyperplasia or endometrial carcinoma and are often also used in the setting of recurrent or advanced disease. Response rates to progestin therapy range from 67–82% in atypical hyperplasias to 50–70% in well-differentiated endometrial cancer and to 15–27% in the recurrent setting [4, 5]. In addition, recurrent disease is common among those with atypical hyperplasia or endometrial cancer treated with progestins. The reasons for the declining responsiveness to progesterone/progestins are unclear, and it is essential to understand how the progesterone receptor functions in the setting of endometrial pathologies.
The progesterone receptor is an intracellular steroid receptor that has two isoforms, PRA and PRB. PRA lacks 164 amino acids from the N terminus, and PRA and PRB are translated from individual mRNA species of a single gene under the control of distinct promoters [6]. Although they share many functional domains, they are considered two distinct transcription factors able to regulate different sets of genes [7–10]. The role of PRA and PRB in endometrial tumors remains unclear, with one study suggesting that PRB is predominant in advanced endometrial tumors [11], another study pointing to the loss of both isoforms in advanced endometrial cancer [12], and a third study that indicates only PRA is expressed in poorly differentiated endometrial carcinoma cells [13]. In a recent study, the evaluation of 315 tissue samples from endometrioid endometrial cancer revealed that PRA and PRB were associated with lower-grade tumors and that the ratio of PRA/PRB, if less than 1, was associated with a shorter disease-free survival and a shorter overall survival [14]. In endometrial cancer cell lines expressing either endogenous or recombinant PR, progesterone treatment can inhibit cell growth, invasion, and expression of cellular adhesion molecules, as well as promote differentiation to a secretory phenotype, and induce replicative senescence [8, 15, 16]. Smid-Koopman et al. [8] demonstrated that in the presence of progestins, PRB-expressing Ishikawa cells displayed almost complete inhibition of cell growth, while PRA expressing Ishikawa cells only displayed 50% inhibition of cell growth. In an additional study by Hanekamp et al. [17], it was demonstrated that PRB-expressing Ishikawa cells caused more tumor growth in mice than PRA expressing Ishikawa cells and that tumor growth was inhibited after administration of MPA in only cells expressing PRB. Previously we demonstrated that PRA and PRB differentially influenced the function of the forkhead transcription factor, FOX01, in endometrial cancer cells in that overexpression of the constitutively active FOXO1 (Tm-FOXO1) in PRA- and PRB-expressing Ishikawa cells promoted apoptosis in only PRB cells but not in PRA cells [18]. All together, these studies demonstrate that PRA and PRB have distinct functions in endometrial cancer cells and can differentially regulate the apoptotic response both in a ligand-dependent and ligand-independent manner.
One of the most common mutations that occurs endometrial carcinoma is in the PTEN gene which is seen in more than 50–80% of tumors [19, 20]. PTEN, a tumor suppressor gene, negatively regulates PI3K/Akt-driven cell growth and survival. The loss of PTEN leads to the enhanced activity of the phosphatidylinositol 3′-kinase/Akt signaling pathway which is associated with increased proliferation, survival, and resistance in cancer cells. Endometrial cancer cell lines and cancer specimens have been shown to have increased levels of phosphorylated AKT supporting that the AKT pathway is constitutively active in a subset of endometrial cancers [18, 21–23]. Inhibition of the AKT pathway in endometrial cancer cells that exhibit high AKT activity has been demonstrated to induce apoptosis [22, 24] supporting that increased AKT promotes survival of these cells. Given that PRA and PRB can differentially regulate the apoptotic response when Tm-FOXO1 is overexpressed endometrial cancer cells and given that FOXO1 is a direct target of AKT, we aimed to determine whether inhibition of AKT using a small molecule inhibitor, API-59CJ-OMe (API-59), could induce apoptosis of endometrial cancer cells in a PR-isoform-dependent manner. In this study, we demonstrate that inhibition of AKT using API-59 induces apoptosis preferentially in PRA-specific cells. Apoptotic genes regulated by API-59 in the presence and absence of progestin were identified and found to be both unique and common to PRA and PRB. BIRC3 was highly upregulated by liganded PRB and contributed to the survival of PRB cells. An antagonist to the family of inhibitors of apoptosis (IAP), a Smac mimetic, further promoted apoptosis in PRB cells treated with API-59 and R5020.
Materials and Methods
Endometrial Cancer Tissues and Cell Lines
Endometrial tumors were obtained from women undergoing hysterectomies at Northwestern Memorial Hospital. Patients gave consent before surgery, and these studies were approved by the Human Subject Committee of our institution in accordance with US Department of Health regulations. PRA14 and PRB23 Ishikawa cells were obtained from L. Blok et al. (Erasmus University, The Netherlands) [8]. These cell lines were made by stably transfecting the human PRA and PRB cDNA fragments that were cloned into the expression vector pcDNA3.1 (Invitrogen) into parental Ishikawa cells that were devoid of PR, and thus, PRA or PRB is constitutively expressed. The Ishikawa cells are derived from a well-differentiated human endometrial adenocarcinoma [25] which is PTEN-mutated [22]. HEC1A and HEC1B cells were obtained from American Type Culture Collection (Rockville, MD, USA). HEC1A and HEC1B cells were maintained in MEM (Invitrogen, Carlsbad, CA, USA) supplemented with sodium pyruvate, penicillin/streptomycin, and 10% FBS. Ishikawa cell lines that were stably transfected with PRA or PRB were maintained in DMEM/F12 supplemented with sodium pyruvate, penicillin/streptomycin, neomycin/hygromycin, and 10% FBS. For explant cultures, tumors were cut into small pieces, approximately 2 cm3 in size. Explants were cultured in serum-free DMEM with sodium pyruvate and penicillin/streptomycin with or without 100 nM MPA for 24 h. Tissues were harvested and placed in TRIzol reagent (Invitrogen). For primary endometrial cancer cells, tumors were cut into small pieces and digested in 0.5% collagenase and 0.02% DNase for 30 min in a shaking incubator at 37°C. Cells were centrifuged, resuspended in DMEM/F12 with 10% FBS, and cultured for approximately 3 days. Cells were then serum-starved overnight (DMEM/F12 only), and the next day, media were changed to DMEM/F12 with vehicle or 10 nM R5020 for 24 h. PRA14 and PRB23 cells were cultured in media with 2% charcoal-stripped FBS overnight, followed by treatment with 6-12 μM API-59, 100 nM R5020, or both for 24–48 h in 2% charcoal-stripped FBS.
Real-Time PCR
RNA was isolated from cells using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. Concentration and purity of extracted RNA were determined using the ND-1000 Spectrophotometer (NanoDrop). Total RNA samples were DNase I (Ambion) treated to remove any contaminating DNA. Total RNA was reverse-transcribed with the Smart MMLV reverse transcriptase (Clontech) for the synthesis of cDNA using the manufacturer’s protocol. Real-time PCR was performed using specific primers (Taqman, PE Applied Biosystems) to BIRC3 and the housekeeping gene TBP. The fold change in expression was calculated using the ∆∆Ct method [26], with TBP as an internal control. The focused real-time PCR array used in this study was specifically the Human Apoptosis RT2 Profiler™ PCR Array consisting of 84 key genes involved in apoptosis (SABiosciences). The array includes the TNF ligands and their receptors; members of the bcl-2, caspase, IAP, TRAF, CARD, death domain, death effector domain, and CIDE families; as well as genes involved in the p53 and ATM pathways. Each well in the 96-well plate contained specific primers for each of these genes. Total RNA was reverse-transcribed using SABiosciences RT2 first-strand synthesis kit. The array was performed in triplicate for all samples, and the expression and statistical analyses were done using SABiosciences web-based data analysis program. All PCR reactions were carried out on an ABI PRISM 7000 Sequence Detection System (Applied Biosystems) for 40 cycles (95°C for 15 s, 60°C for 1 min) after 10 min incubation at 95°C.
Western Blot Analysis
Total cell lysates were obtained by lysing cells with M-PER Mammalian lysis solution (Thermo Scientific) supplemented with protease and phosphatase inhibitors (Sigma) on ice. Protein concentration was measured using the Micro BCA kit (Thermo Scientific). Isolated protein samples were run on 7.5% acrylamide gels and transferred onto polyvinylidene difluoride membranes (Whatman). Membranes were blocked in 5% nonfat milk made in TBS-T at room temperature and incubated with primary antibodies, cPARP (Cell Signaling), phosphorylated AKT(Ser 473; Cell Signaling), BIRC3 (BD Pharminogen) either overnight at 4°C (cPARP, pAKT) or overnight at 4°C followed by 1 h at room temperature (BIRC3). After the incubation, membranes were washed three times with TBS-T and then incubated with secondary peroxidase-conjugated goat anti-rabbit or goat anti-mouse antibody for 1 h at room temperature (Bio-Rad). Membranes were then washed three times in TBS-T and developed using the ECL Super Signal West Dura or Femto detection kit (Thermo Scientific). Membranes were stripped using Restore Western Blot Stripping Buffer (Pierce) and reprobed using an antibody to actin (Sigma).
Immunofluorescent Staining
Cells were fixed with 4% paraformaldehyde (Sigma), and coverslips were then washed with PBS and permeabilized with 0.1% Triton–0.1% deoxycholate (Sigma). Cells were blocked with 5% bovine serum albumin (BSA; Sigma) made in PBS. Subsequently, the BIRC3 (Abcam) and phospho-Ser473-AKT (Cell Signaling) antibodies, made in filtered 5% BSA, were added to each sample and incubated overnight at 4°C. A secondary Alexa Fluor® 488 goat anti-rabbit IgG or anti-mouse IgG (Invitrogen) was used. Cells were then mounted with mounting media (Invitrogen) for fluorescence on glass slides and visualized using a fluorescent inverted microscope, Axiovert 200 (Zeiss).
Small Interfering RNA
Cells were grown to 50% confluence. Dharmacon SMARTpool small interfering RNA (siRNA) specific to PR or CIAP2 (BIRC3) were transiently transfected into the cells using Lipofectamine RNAi Max (Invitrogen) according to the manufacturer’s protocol for siRNA. On Target plus Non-targeting pool, siRNA was used as a control (Dharmacon). For silencing PR, cells were transfected for 5 h, and the transfection media was removed and replaced with DMEM/F12 media supplemented with 2% stripped FBS, sodium pyruvate, and penicillin/streptomycin overnight. The following morning cells were treated with API-59 and R5020 for 24–48 h. After incubation, the cells were harvested for Western blot analysis. For silencing BIRC3, cells were transfected for with SMARTpool siRNA specific to BIRC3 for 48 h and treated with API-59 and R5020 for an additional 72 h.
Cell Viability Assay
The cell viability assay used was the Quick Cell Proliferation Assay Kit (BioVision, Mountain View, CA, USA). Cells were plated in a 96-well plate and allowed to attach for 4–6 h. Cells were then treated for 48–72 h. WST-1/ECS (Electro Coupling Solution) solution was added and incubated at 37°C. Samples were read on the Synergy HT from Bio-Tek with the KC4 3.4 software at 420 nm to determine cell viability.
Annexin V Assay
After treatment of cells with vehicle, R5020, API-59, or both for 24 h, cells were trypsinized and resuspended in annexin-binding buffer (10 mM Hepes (Invitrogen), 140 mM NaCl, 2.5 mM CaCl2, pH 7.4) to a concentration of approximately 1 × 106 cells/mL. Annexin V, Alexa Fluor 647 conjugate (Invitrogen), and DAPI (Invitrogen) were added to each cell solution, and samples were analyzed using the CyAn flow cytometer (Dako) for early and late apoptosis.
Smac Mimetic
PRB cells were plated, serum-starved overnight, and then treated with vehicle, R5020, or API-59 with R5020 in the presence or absence of 10 nM Smac mimetic (generously provided by Xiaodong Wang, UT Southwestern [27]) for 48 h. Protein was harvested and Western blots were performed.
Statistical Analysis
Statistical analysis was performed using one-way ANOVA followed by the paired t test.
Results
Inhibition of AKT with API-59 Induces Apoptosis in PR Overexpressing Ishikawa Cells
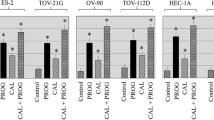
Previously, it was demonstrated that the AKT inhibitor, API-59, inhibited AKT kinase activity without inhibiting phosphorylation of AKT on Ser473 or Thr308 [22]. In addition, ERK, JNK, or PKC pathways were not affected. Treatment of endometrial and ovarian cancer cell lines with this small molecule inhibitor induced apoptosis of several endometrial cancer and ovarian cancer cell lines particularly in cells that expressed elevated levels of phosphorylated AKT indicative of high AKT activity [22, 28, 29]. For these reasons, this AKT inhibitor was used in our study. PRA and PRB-specific Ishikawa cell lines were derived from parental Ishikawa cells that possess a PTEN mutation [22]. PRA14 cells express only PRA while PRB23 cells expressed high levels of PRB with minimal levels of PRA (Fig. 1a). Ishikawa cells (clones from B. Lessey and not the ones used to stably transfect PRA or PRB) also expressed endogenous PRA and PRB protein but at levels much lower than the PR-specific lines. HEC1A and HEC1B did not express PR. Levels of PTEN protein were undetectable in the PRA14and PRB23 cells while p(Ser473)-AKT protein levels were higher in PRA14 and PRB23 than endometrial cancer cells that express wild-type PTEN (HEC1A, HEC1B). Given the high pAKT levels in PRA14 and PRB23 cells, treatment with API-59 promoted apoptosis as expected, as demonstrated by cleaved PARP expression (Fig. 1b) and annexin V staining (Fig. 1c). In addition, a higher percentage of cells underwent apoptosis in PRA14 compared to PRB23 cells treated with API-59 with or without R5020.

The AKT inhibitor, API-59 promotes apoptosis differentially in PRA- and PRB-specific Ishikawa cells. a Five different endometrial cancer cell lines, HEC1A, HEC1B, parental Ishikawa, PRA-specific Ishikawa (PRA14), and PRB-specific Ishikawa (PRB23) cells, were cultured and protein levels of pAKT, AKT, and PTEN were measured by Western blot. b Apoptosis was detected by measuring levels of cleaved PARP. Cells were treated with 12 μM API59, 100 nM R5020, or both for 48 h. Densitometric analysis of cPARP is shown representing the mean ± SEM of three independent experiments. c Early apoptosis was measured by annexin V staining using flow cytometry. Cells were treated with 12 μM API59, 100 nM R5020, or both for 48 h. Data represent the mean ± SEM of six independent experiments. “a” denotes statistical significance compared to non-treated control. p ≤ 0.05
Role of PRA and PRB in API-59-Mediated Apoptosis
In order to determine the role of PRA and PRB in API-59-mediated apoptosis, PR was silenced using siRNA specific to PR. In both PRA14 and PRB23 cells, levels of PR dramatically decreased upon PR knockdown (Fig. 2a, b). Interestingly, PR protein levels increased in response to API-59 in both cell types. Also, while the classic downregulation of PR after R5020 treatment occurred in PRA14 and PRB23 cells, API-59 and R5020 treatment caused PRA levels to remain high in PRA14 cells but not PRB in PRB23 cells. This suggests potential involvement of AKT in specifically PRA protein degradation. Next, apoptosis was measured using cleaved PARP as an indicator. In PRA14 cells, knockdown of PR did not significantly change levels of cPARP observed in response to API-59 with or without R5020 suggesting that PRA does not significantly influence the apoptosis that is observed with API-59. In PRB23 cells, however, silencing PRB increased cPARP levels in all treatments, even at the basal level with no treatment. Thus far, the data suggest that PRB may play a protective role to apoptosis.

Knockdown of PR promotes apoptosis in PRB23 cells. a PRA14 and b PRB23 cells were transiently transfected with a non-specific siRNA (siCont) or siRNA specific to PR (siPR). Cells were then treated with 12 μM API-59, 100 nM R5020, or both for 48 h. PR, cPARP, and actin proteins were measured by Western blot. Densitometric values for cPARP for PRA and PRB cells are represented as the mean ± SEM of three independent experiments
Apoptotic Genes Regulated by API-59 in PRA and PRB Cells
In order to identify the genes that are regulated by API-59 to promote apoptosis and to determine whether genes are differentially regulated depending on the PR isoform, a focused real-time PCR array encompassing 84 genes associated with apoptosis was used. PRA and PRB cells were treated with or without API-59 in the presence or absence of R5020 for 48 h, and the real-time arrays were run in triplicate for each treatment. Table 1 lists the genes that were regulated by 2-fold or greater compared to vehicle-treated control and which also reached statistical significance (p ≤ 0.05). In PRB cells, treatment with R5020 alone regulated only one gene, BIRC3 (Table 1; Supplementary Fig. 1). Treatment with API-59 resulted in eight upregulated and four downregulated genes, and the addition of R5020 to API-59 resulted in 13 upregulated and two downregulated genes. In PRA cells, no genes were regulated by R5020 alone. Treatment with API-59 resulted in 11 upregulated and no downregulated genes, and the addition of R5020 to API-59 resulted in nine upregulated and one downregulated gene. The genes expressed uniquely in PRA or PRB could contribute to the differences observed in the sensitivity to the API-59 compound. The addition of ligand, R5020 to API-59, regulates genes unique to the other treatments. In PRA cells, treatment with API-59 + R5020 regulated two genes unique from API-59 treatment alone (BIRC3, TNFRSF11B; Fig. 3d, Table 1). In PRB cells, API-59 + R5020 regulated five genes distinct from API-59 alone (BCL2L2, BIRC3, CARD6, FASLG, TNFRSF10B; Fig. 3e, Table 1). Taken together, these data demonstrate that API-59 regulates common as well as unique genes in PRA14 and PRB23 cells when ligand is present or absent. Furthermore, R5020 alone regulated only one gene, BIRC3 in PRB cells, suggesting that liganded PRA or PRB alone does not promote apoptosis with the dose and length of treatment used here.

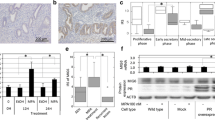
BIRC3 is specifically upregulated by liganded PRB. a PRA14 and PRB23 cells were treated with 100 nM R5020 for 24 h. BIRC3 mRNA and protein were measured using real-time PCR and Western blot. Data represent the mean ± SEM of six independent experiments. b PR was silenced in PRB cells and treated with 100 nM R5020 for 24 h. PRB, BIRC3, and actin proteins were measured by Western blot. c PRB cells were treated with 100 nM R5020, 10 μM RU486, or both for 24 h. BIRC3 protein was measured by Western blot
BIRC3 Is Regulated by PRB
Intriguingly, among the 84 apoptosis-associated genes that were analyzed, BIRC3 was the only gene significantly regulated by more than 2-fold in response to R5020 in PRB cells. Even with API-59 + R5020 treatment, BIRC3 was induced by 7-fold in PRB cells. Confirmatory analysis revealed that BIRC3 mRNA and protein were upregulated significantly in PRB cells but not in PRA cells in response to R5020 (Fig. 3a). In addition, no BIRC3 was detected in the absence of ligand in the PRB cells. To determine whether the progesterone receptor was required for this induction, PR was knocked down in PRB cells using siRNA followed by R5020 treatment. As shown in Fig. 3b, silencing of PRB inhibited the induction of BIRC3 protein by R5020. Furthermore, treatment with the PR antagonist, RU486 also prevented the induction of BIRC3 by R5020 (Fig. 3c), indicating that the induction of BIRC3 requires liganded PRB.
In order to determine whether the induction of BIRC3 by progestins also occurred in primary endometrial tumors, explants from endometrial tumors were treated with the progestin, MPA for 24 h. Among the tumors collected from eight patients, explants from all of them expressed BIRC3 mRNA in response to the progestin medroxyprogesterone acetate, at levels higher than those of vehicle-treated controls (Fig. 4a). The increases, however, ranged from 1.2- to 3.4-fold, and all of these tumors were clinically evaluated to express PR (data not shown). In addition, primary cells from endometrial tumors were cultured and treated with R5020 for 24 h, and immunofluorescent staining for BIRC3 and p(Ser473)-AKT proteins was done (Fig. 4b). These cells exhibited p(Ser473)-AKT staining and treatment with R5020 increased staining for BIRC3 protein in the tumor cells. These data show that the induction of BIRC3 can occur in primary endometrial tumor cultures as well when treated directly with progestins.

a Primary endometrial tumor explants from eight patients were cultured with vehicle or 100 nM MPA for 24 h. BIRC3 mRNA was measured by real-time PCR. Data are presented as fold increase from vehicle-treated samples. b Cells isolated from endometrial tumors were cultured and treated with vehicle (panels 1, 3) or 10 nM R5020 (panels 2, 4) for 24 h. BIRC3 (panels 1 and 2) and p(Ser473)-AKT (panels 3 and 4) proteins were measured using immunofluorescent staining. Magnification ×630
Role of BIRC3 in PRB-Expressing Endometrial Cancer Cells
Thus far, the data show that API-59 promotes apoptosis, more so in PRA14 cells than PRB23 cells and that PRB may be protective against apoptosis. The differential regulation of a unique set of genes by API-59 in these cells in the presence or absence of progestin would explain these differences; however, the underlying mechanisms for these observations remain unclear. In particular, the role of unliganded PRA and PRB in governing the apoptotic responses is unknown. In this study, we chose to study the ligand-dependent role of PR given that the real-time array identified BIRC3 as the only gene regulated by liganded PR. The physiologic relevance of BIRC3 induced by R5020 in PRB23 cells was examined. It was hypothesized that BIRC3 would promote survival of PRB cells that were induced to undergo apoptosis in response to API-59. To demonstrate this, BIRC3 was silenced using siRNA in PRB23 cells followed by treatment with API-59 and R5020. The levels of cPARP increased when BIRC3 was silenced, albeit modestly (Fig. 5a). In addition, annexin V staining using flow cytometry revealed an increase in the percentage of cells in early apoptosis (Fig. 5b). Finally, cell viability assay showed a decrease in the percent of viable cells in siBIRC3 cells compared to siCtrl samples treated with API-59 + R5020 (Fig. 5c). Although modest, the decrease was consistent and statistically significant. It is possible that other IAPs compensated for inhibition of apoptosis when BIRC3 was knocked down and thus BIRC3’s ability to inhibit apoptosis in a progestin-dependent manner may have been masked.

BIRC3 promotes cell survival in PRB cells. PRB23 cells were transfected with a non-specific siRNA (siCont) or siRNA specific to BIRC3 (siBIRC3) and treated with 12 μM API and 100 nM R5020. a Protein levels of BIRC3 were measured to verify efficient knockdown and cPARP levels were measured. b Annexin V staining using flow cytometry was done to measure early apoptosis. c Cell viability was measured with the WST assay. Data are presented as % of viable cells compared to its corresponding siCont and calculated as the means ± SEM of three independent experiments. *p ≤ 0.05. d PRB cells were treated with 100 nM R5020, or 12 μM API-59 + R5020, in the presence and absence of 10 nM Smac mimetic. Levels of cPARP, BIRC3, CIAP1, XIAP, and actin proteins were measured by Western blot
SMAC Mimetics Increase Apoptosis in PRB23 Cells
Next, the efficacy of a small molecule antagonist of IAP, called SMAC mimetics, was examined to inhibit the activity of the IAPs. PRB23 cells were treated with API-59 and R5020, in the presence or absence of a Smac mimetic. The Smac mimetic promoted apoptosis as measured by increased levels of cPARP in response to API-59 and R5020 (Fig. 5d). Interestingly, levels of BIRC3 protein in API59 + R5020-treated cells were lower than that of R5020-treated cells. Furthermore, addition of SMAC mimetic decreased BIRC3 protein levels in R5020 and API59 + R5020-treated cells compared to cell without SMAC mimetic. CIAP1 levels also decreased with SMAC mimetic while XIAP protein levels were similar with all treatments. These data show that SMAC mimetics do potentiate apoptosis triggered by API59 + R5020 through its proposed mechanism of inhibiting IAP activity as well as potentially decreasing expression of BIRC3 and CIAP1. IAPs play an active role in inhibiting apoptosis in PRB23 cells.
Discussion
In this study, several key observations were made. First, the AKT inhibitor, API-59, promoted apoptosis in the PTEN-mutated PRB- and PRA-specific cell lines. PRB cells were less responsive to API-59 compared to PRA cells whether progestin was present or not. Furthermore, knockdown of PR promoted apoptosis in PRB-specific cells but not PRA cells demonstrating a protective effect of PRB to induced apoptosis. The focused real-time array identified genes both common and unique to PRA and PRB. Moreover, PRA and PRB regulated both anti- and pro-apoptotic genes, underlining the complex and intricate nature of PR action. BIRC3 was the only gene that was significantly regulated by R5020 which occurred in PRB cells, not PRA cells. Knockdown of BIRC3 decreased cell viability in response to API-59 and R5020 and treatment with an antagonist to IAPs, Smac mimetic, increased apoptosis further. Based on these findings, we hypothesize that PRB induction of BIRC3 protects endometrial cancer cells from AP1-59-mediated apoptosis.
Differences in responses to API-59 and progestin in PRA and PRB-specific cells were expected. It is well documented that PRA and PRB can regulate a distinct subset of genes in endometrial cancer and breast cancer [7–10]. We previously demonstrated that overexpression of Tm-FOXO1 promoted apoptosis in PRB cells but not in PRA cells [18]. In this study, API-59 induced more apoptosis in PRA compared to PRB cells, which is contrary to the effects observed with Tm-FOXO1 overexpression. While FOXO1 is a direct target of AKT, it is limited to regulating a specific set of genes, while AKT affects a multitude of proteins and pathways that can ultimately result in a different physiological endpoint. It is also plausible that the apoptosis observed with API-59 involves other mechanisms. The AKT inhibitor used in this study, API-59, was initially identified by Jin et al. [22] through a screen 35,000 compounds in the NCI’s anticancer database. While this compound did indeed inhibit AKT activity, without affecting ERK, JNK, or PKC pathways, API-59 belongs to the class of ellipticines, which are known to have several different mechanisms of action. Some of these include binding directly to DNA and intercalating into the DNA strands, stabilizing topoisomerase II–DNA complexes and generating superoxide radicals to promote DNA strand breakage [30]. Thus, the apoptosis that is observed with this compound may not be due solely to selective AKT inhibition, and thus, its use as a specific AKT inhibitor to study the AKT pathway is limited. In this study, we observe that this compound induces apoptosis in the PRB23 and PRA14 cells and have limited its use as such. In order to determine, for example, whether the increase in PR levels that is observed by API-59 is due to specifically inhibition of AKT, further experimentation using more selective inhibitors of AKT will be required. In addition, how AKT inhibition affects the induction of BIRC3 by R5020 in PRB cells cannot be clearly demonstrated with the API-59 compound.
The potential protective effect of PRB was unexpected given the general consensus that progesterone/progestins inhibit growth and decrease tumor integrity both in vitro and in clinical studies [4, 5, 8, 15, 16, 31–33]. It should be noted that the common trend in these studies is that long-term treatment or supraphysiological concentrations of the hormone is required to see an effect in cell growth and apoptosis which makes it challenging to pinpoint specific mechanisms of PR that are associated with these effects. On the other hand, studies have demonstrated a protective effect of progesterone against neuronal cerebral damage [34, 35], in cardiomyocytes treated with doxorubicin [36], spinal cord injury [37, 38], and radiation-induced apoptosis in breast cancer cells [39]. In our study, knockdown of PR in PRB-specific Ishikawa cells for 72 h promoted apoptosis whether ligand was present or not, while knockdown of PR in PRA-specific cells did not significantly affect apoptosis. Whether PRB is indeed protective to other inducers of apoptosis in endometrial cancer cells is under investigation.
The induction of BIRC3 by progestin through PRB could be one mechanism by which PRB is protective against apoptosis in endometrial cancer cells. Few studies have reported an association between progesterone and BIRC3. When human polymorphonuclear leukocytes were treated with progesterone, BIRC3 mRNA levels increased and rates of apoptosis were lower [40]. In breast cancer cell lines overexpressing PRA or PRB, two different microarray analyses identified BIRC3 as one gene upregulated in PRB-specific T47D cells [41, 42]. Estradiol and glucocorticoids have been shown to upregulate BIRC3 as well. In breast cancer cells, estrogen receptor and NF-κB can synergistically induce BIRC3 to promote survival of breast cancer cells [43, 44]. Glucocorticoids increase BIRC3 and play an anti-apoptotic role in ovarian cancer cells [45]. These studies demonstrate that BIRC3 can be hormonally regulated to promote cell survival. To our knowledge, this is the first report to demonstrate upregulation of BIRC3 by liganded PR in endometrial cancer cells.
Studies have demonstrated that BIRC3 promotes resistance of cancer cells to apoptosis [27, 46, 47]. Thus far, its role in endometrial cancer has not been studied. Knockdown of BIRC3 decreased cell viability in this study, albeit modestly. It is possible that the effect of BIRC3 silencing was masked by the compensatory activities of the other IAPs. The robust increase in apoptosis observed with the Smac mimetic supports this. It is likely that the inhibition of all three IAPs with the Smac mimetic contributed to the increase in apoptosis observed with API-59 and R5020. The Smac mimetic mimics the action of naturally occurring inhibitors to the IAP family of proteins. Upon activation of apoptosis, Smac is released from the mitochondria into the cytosol [48]. Smac can bind to the BIR1 or BIR2 domains of IAP proteins to inhibit their anti-apoptotic function and can also cause autoubiquitination of IAPs, thus affecting their function [48]. Most Smac mimetics are still in the preclinical phase of investigation; however, several phase I trials have demonstrated a cytotoxic effect in prostate, pancreatic, colorectal, and breast cancers [48–53]. Smac mimetics can enhance the activity of chemotherapeutics or radiation in cancer cells.
The role of PRA and PRB in endometrial adenocarcinoma remains unclear. This is likely due to multiple factors that are both technical and biological in nature. PRA and PRB expression are usually associated with lower-grade tumors [14]. As tumors become more aggressive, PR expression usually declines. Furthermore, lower-grade tumors tend to respond better to progestin treatment in the clinic, compared to higher-grade tumors, and this has been thought to be due to the expression of PRA and PRB [5]. In our study, we used the Ishikawa cell lines that arise from a well-differentiated tumor, and we have overexpressed PRA and PRB. These cells also carry a PTEN mutation and exhibit high levels of p(Ser473)-AKT. When AKT is inhibited, we see less apoptosis in the PRB cells compared to PRA cells suggesting that PRB promotes the expression of more pro-survival or anti-apoptotic genes compared o PRA genes. This would support observations made by Fujimoto et al. [11] where PRB is predominant in advanced endometrial tumors, as well as the study by Jongen et al. [14] that the ratio of PRA/PRB, if less than 1, was associated with a shorter disease-free survival and a shorter overall survival. From another perspective, we are currently working on a hypothesis that overactive AKT influences PR function due to posttranslational modifications of PR. Previous studies thus far have only correlated PRA and PRB expression with aggressiveness or grade of endometrial carcinoma. It is plausible that PR function is modified in the context of overactive AKT leading to an altered progesterone response. In addition, the dramatic increase in PRA and PRB protein in response to API-59 was unexpected. From this observation, it can be speculated that overactive AKT pathway promotes protein degradation even in the presence of ligand for PRA. Furthermore, although PR levels increased, it is unclear whether PR transcriptional activity increased as well. We are currently investigating the role of AKT in phosphorylation and ubiquitination of PR to influence protein stability and transcriptional function.
In summary, it has been shown here that BIRC3 is induced by progestins through PRB and contributes to the survival of endometrial cancer cells against apoptosis mediated by inhibition of AKT. It is tempting to speculate that the induction of BIRC3 by progestins plays a role in the resistance to progestin therapy observed in some women with endometrial carcinoma. In such instances, inhibition of IAPs using Smac mimetics may prove to be beneficial in combination with hormonal therapy.
References
Clarke CL, Sutherland RL (1990) Progestin regulation of cellular proliferation. Endocr Rev 11(2):266–301
Creasman WT (1997) Endometrial cancer: incidence, prognostic factors, diagnosis, and treatment. Semin Oncol 24(1 Suppl 1):S1-140–S141-150
Munstedt K, Grant P, Woenckhaus J, Roth G, Tinneberg HR (2004) Cancer of the endometrium: current aspects of diagnostics and treatment. World J Surg Oncol 2:24. doi:10.1186/1477-7819-2-24
Chiva L, Lapuente F, Gonzalez-Cortijo L, Carballo N, Garcia JF, Rojo A, Gonzalez-Martin A (2008) Sparing fertility in young patients with endometrial cancer. Gynecol Oncol 111(2 Suppl):S101–S104. doi:10.1016/j.ygyno.2008.07.056
Kim JJ, Chapman-Davis E (2010) Role of progesterone in endometrial cancer. Semin Reprod Med 28(1):81–90. doi:10.1055/s-0029-1242998
Kastner P, Krust A, Turcotte B, Stropp U, Tora L, Gronemeyer H, Chambon P (1990) Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J 9(5):1603–1614
Giangrande PH, Kimbrel EA, Edwards DP, McDonnell DP (2000) The opposing transcriptional activities of the two isoforms of the human progesterone receptor are due to differential cofactor binding. Mol Cell Biol 20(9):3102–3115
Smid-Koopman E, Blok LJ, Kuhne LC, Burger CW, Helmerhorst TJ, Brinkmann AO, Huikeshoven FJ (2003) Distinct functional differences of human progesterone receptors A and B on gene expression and growth regulation in two endometrial carcinoma cell lines. J Soc Gynecol Investig 10(1):49–57
Tung L, Abdel-Hafiz H, Shen T, Harvell DM, Nitao LK, Richer JK, Sartorius CA, Takimoto GS, Horwitz KB (2006) Progesterone receptors (PR)-B and -A regulate transcription by different mechanisms: AF-3 exerts regulatory control over coactivator binding to PR-B. Mol Endocrinol 20(11):2656–2670. doi:10.1210/me.2006-0105
Tung L, Mohamed MK, Hoeffler JP, Takimoto GS, Horwitz KB (1993) Antagonist-occupied human progesterone B-receptors activate transcription without binding to progesterone response elements and are dominantly inhibited by A-receptors. Mol Endocrinol 7(10):1256–1265
Fujimoto J, Ichigo S, Hori M, Nishigaki M, Tamaya T (1995) Expression of progesterone receptor form A and B mRNAs in gynecologic malignant tumors. Tumour Biol 16(4):254–260
Arnett-Mansfield RL, deFazio A, Wain GV, Jaworski RC, Byth K, Mote PA, Clarke CL (2001) Relative expression of progesterone receptors A and B in endometrioid cancers of the endometrium. Cancer Res 61(11):4576–4582
Kumar NS, Richer J, Owen G, Litman E, Horwitz KB, Leslie KK (1998) Selective down-regulation of progesterone receptor isoform B in poorly differentiated human endometrial cancer cells: implications for unopposed estrogen action. Cancer Res 58(9):1860–1865
Jongen V, Briet J, de Jong R, ten Hoor K, Boezen M, van der Zee A, Nijman H, Hollema H (2009) Expression of estrogen receptor-alpha and -beta and progesterone receptor-A and -B in a large cohort of patients with endometrioid endometrial cancer. Gynecol Oncol 112(3):537–542. doi:10.1016/j.ygyno.2008.10.032
Dai D, Wolf DM, Litman ES, White MJ, Leslie KK (2002) Progesterone inhibits human endometrial cancer cell growth and invasiveness: down-regulation of cellular adhesion molecules through progesterone B receptors. Cancer Res 62(3):881–886
Ueda M, Fujii H, Yoshizawa K, Abe F, Ueki M (1996) Effects of sex steroids and growth factors on migration and invasion of endometrial adenocarcinoma SNG-M cells in vitro. Jpn J Cancer Res 87(5):524–533
Hanekamp EE, Kuhne LM, Grootegoed JA, Burger CW, Blok LJ (2004) Progesterone receptor A and B expression and progestagen treatment in growth and spread of endometrial cancer cells in nude mice. Endocr Relat Cancer 11(4):831–841. doi:10.1677/erc.1.00844
Ward EC, Hoekstra AV, Blok LJ, Hanifi-Moghaddam P, Lurain JR, Singh DK, Buttin BM, Schink JC, Kim JJ (2008) The regulation and function of the forkhead transcription factor, Forkhead box O1, is dependent on the progesterone receptor in endometrial carcinoma. Endocrinology 149(4):1942–1950. doi:10.1210/en.2007-0756
Hecht JL, Mutter GL (2006) Molecular and pathologic aspects of endometrial carcinogenesis. J Clin Oncol 24(29):4783–4791. doi:10.1200/JCO.2006.06.7173
Risinger JI, Hayes AK, Berchuck A, Barrett JC (1997) PTEN/MMAC1 mutations in endometrial cancers. Cancer Res 57(21):4736–4738
Gagnon V, Van Themsche C, Turner S, Leblanc V, Asselin E (2008) Akt and XIAP regulate the sensitivity of human uterine cancer cells to cisplatin, doxorubicin and taxol. Apoptosis 13(2):259–271. doi:10.1007/s10495-007-0165-6
Jin X, Gossett DR, Wang S, Yang D, Cao Y, Chen J, Guo R, Reynolds RK, Lin J (2004) Inhibition of AKT survival pathway by a small molecule inhibitor in human endometrial cancer cells. Br J Cancer 91(10):1808–1812. doi:10.1038/sj.bjc.6602214
Kanamori Y, Kigawa J, Itamochi H, Shimada M, Takahashi M, Kamazawa S, Sato S, Akeshima R, Terakawa N (2001) Correlation between loss of PTEN expression and Akt phosphorylation in endometrial carcinoma. Clin Cancer Res 7(4):892–895
Gagnon V, St-Germain ME, Parent S, Asselin E (2003) Akt activity in endometrial cancer cells: regulation of cell survival through cIAP-1. Int J Oncol 23(3):803–810
Nishida M, Kasahara K, Oki A, Satoh T, Arai Y, Kubo T (1996) Establishment of eighteen clones of Ishikawa cells. Hum Cell 9(2):109–116
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25(4):402–408. doi:10.1006/meth.2001.1262
Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J, Harran P, Wang X (2007) Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 12(5):445–456. doi:10.1016/j.ccr.2007.08.029
Hoekstra AV, Ward EC, Hardt JL, Lurain JR, Singh DK, Buttin BM, Schink JC, Kim JJ (2008) Chemosensitization of endometrial cancer cells through AKT inhibition involves FOXO1. Gynecol Oncol 108(3):609–618. doi:10.1016/j.ygyno.2007.11.007
Tang HJ, Jin X, Wang S, Yang D, Cao Y, Chen J, Gossett DR, Lin J (2006) A small molecule compound inhibits AKT pathway in ovarian cancer cell lines. Gynecol Oncol 100(2):308–317. doi:10.1016/j.ygyno.2005.08.044
Harding MM, Grummitt AR (2003) 9-Hydroxyellipticine and derivatives as chemotherapy agents. Mini Rev Med Chem 3(2):67–76
Di Nezza LA, Jobling T, Salamonsen LA (2003) Progestin suppresses matrix metalloproteinase production in endometrial cancer. Gynecol Oncol 89(2):325–333
Saito T, Mizumoto H, Tanaka R, Satohisa S, Adachi K, Horie M, Kudo R (2004) Overexpressed progesterone receptor form B inhibit invasive activity suppressing matrix metalloproteinases in endometrial carcinoma cells. Cancer Lett 209(2):237–243. doi:10.1016/j.canlet.2003.12.017
Moe BG, Vereide AB, Orbo A, Sager G (2009) High concentrations of progesterone and mifepristone mutually reinforce cell cycle retardation and induction of apoptosis. Anticancer Res 29(4):1053–1058
Cervantes M, Gonzalez-Vidal MD, Ruelas R, Escobar A, Morali G (2002) Neuroprotective effects of progesterone on damage elicited by acute global cerebral ischemia in neurons of the caudate nucleus. Arch Med Res 33(1):6–14
Gonzalez-Vidal MD, Cervera-Gaviria M, Ruelas R, Escobar A, Morali G, Cervantes M (1998) Progesterone: protective effects on the cat hippocampal neuronal damage due to acute global cerebral ischemia. Arch Med Res 29(2):117–124
Morrissy S, Xu B, Aguilar D, Zhang J, Chen QM (2010) Inhibition of apoptosis by progesterone in cardiomyocytes. Aging Cell 9(5):799–809. doi:10.1111/j.1474-9726.2010.00619.x
Gonzalez SL, Labombarda F, Deniselle MC, Mougel A, Guennoun R, Schumacher M, De Nicola AF (2005) Progesterone neuroprotection in spinal cord trauma involves up-regulation of brain-derived neurotrophic factor in motoneurons. J Steroid Biochem Mol Biol 94(1–3):143–149. doi:10.1016/j.jsbmb.2005.01.016
Labombarda F, Gonzalez Deniselle MC, De Nicola AF, Gonzalez SL (2010) Progesterone and the spinal cord: good friends in bad times. Neuroimmunomodulation 17(3):146–149. doi:10.1159/000258709
Vares G, Ory K, Lectard B, Levalois C, Altmeyer-Morel S, Chevillard S, Lebeau J (2004) Progesterone prevents radiation-induced apoptosis in breast cancer cells. Oncogene 23(26):4603–4613. doi:10.1038/sj.onc.1207601
Chen H, Wu Z, Li J, Chen R, Yu Y, Xu L, Shuai J, Tu YT (2009) Effect of progesterone on gonococci-induced apoptosis and respiratory burst of human polymorphonuclear leukocytes in vitro. Int J Dermatol 48(9):1011–1016. doi:10.1111/j.1365-4632.2009.04177.x
Quiles I, Millan-Arino L, Subtil-Rodriguez A, Minana B, Spinedi N, Ballare C, Beato M, Jordan A (2009) Mutational analysis of progesterone receptor functional domains in stable cell lines delineates sets of genes regulated by different mechanisms. Mol Endocrinol 23(6):809–826. doi:10.1210/me.2008-0454
Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB (2002) Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem 277(7):5209–5218. doi:10.1074/jbc.M110090200
Frasor J, Weaver A, Pradhan M, Dai Y, Miller LD, Lin CY, Stanculescu A (2009) Positive cross-talk between estrogen receptor and NF-kappaB in breast cancer. Cancer Res 69(23):8918–8925. doi:10.1158/0008-5472.CAN-09-2608
Stanculescu A, Bembinster LA, Borgen K, Bergamaschi A, Wiley E, Frasor J (2010) Estrogen promotes breast cancer cell survival in an inhibitor of apoptosis (IAP)-dependent manner. Horm Canc 1:127–135. doi:10.1007/s12672-010-0018-6
Runnebaum IB, Bruning A (2005) Glucocorticoids inhibit cell death in ovarian cancer and up-regulate caspase inhibitor cIAP2. Clin Cancer Res 11(17):6325–6332. doi:10.1158/1078-0432.CCR-05-0182
Karasawa H, Miura K, Fujibuchi W, Ishida K, Kaneko N, Kinouchi M, Okabe M et al (2009) Down-regulation of cIAP2 enhances 5-FU sensitivity through the apoptotic pathway in human colon cancer cells. Cancer Sci 100(5):903–913. doi:10.1111/j.1349-7006.2009.01112.x
Petersen SL, Peyton M, Minna JD, Wang X (2010) Overcoming cancer cell resistance to Smac mimetic induced apoptosis by modulating cIAP-2 expression. Proc Natl Acad Sci USA 107(26):11936–11941. doi:10.1073/pnas.1005667107
Chen DJ, Huerta S (2009) Smac mimetics as new cancer therapeutics. Anticancer Drugs 20(8):646–658. doi:10.1097/CAD.0b013e32832ced78
Dai Y, Lawrence TS, Xu L (2009) Overcoming cancer therapy resistance by targeting inhibitors of apoptosis proteins and nuclear factor-kappa B. Am J Transl Res 1(1):1–15
Dineen SP, Roland CL, Greer R, Carbon JG, Toombs JE, Gupta P, Bardeesy N et al (2010) Smac mimetic increases chemotherapy response and improves survival in mice with pancreatic cancer. Cancer Res 70(7):2852–2861. doi:10.1158/0008-5472.CAN-09-3892
Lecis D, Drago C, Manzoni L, Seneci P, Scolastico C, Mastrangelo E, Bolognesi M et al (2010) Novel SMAC-mimetics synergistically stimulate melanoma cell death in combination with TRAIL and Bortezomib. Br J Cancer 102(12):1707–1716. doi:10.1038/sj.bjc.6605687
Orzaez M, Gortat A, Mondragon L, Perez-Paya E (2009) Peptides and peptide mimics as modulators of apoptotic pathways. ChemMedChem 4(2):146–160. doi:10.1002/cmdc.200800246
Probst BL, Liu L, Ramesh V, Li L, Sun H, Minna JD, Wang L (2010) Smac mimetics increase cancer cell response to chemotherapeutics in a TNF-alpha-dependent manner. Cell Death Differ 17(10):1645–1654. doi:10.1038/cdd.2010.44
Acknowledgments
We would like to thank Dr. Alok Pant for obtaining endometrial tumors and performing the immunofluorescent experiments, Dr. Jonna Frasor from UIC for her insightful comments, the Robert H. Lurie Comprehensive Cancer Center Flow Cytometry Core Facilities at Northwestern University, and to Dr. Xiaodong Wang from UT Southwestern for providing the Smac mimetic compound. We are especially grateful for the Division of Gynecologic Oncology for their continuous and dedicated efforts in providing endometrial tumors. This work was supported by NIH R21 CA127674-2 (JJK) and Malkins Scholarship (ECW).
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Figure 1
Venn diagram shows the pattern of overlap of genes regulated at least 2-fold in a statistically significant manner. Comparisons were A R5020-treated PRA14 and PRB23 cells, B API-59-treated PRA14 and PRB23 cells, C API-59 + R5020 (A + R)-treated PRA14 and PRB23 cells, D R5020, API-59, and or A + R treatment in PRA14 cells, E R5020, API-59, and/or A + R treatment in PRB23 cells. (PPT 153 kb)
Rights and permissions
About this article
Cite this article
Neubauer, N.L., Ward, E.C., Patel, P. et al. Progesterone Receptor-B Induction of BIRC3 Protects Endometrial Cancer Cells from AP1-59-Mediated Apoptosis. HORM CANC 2, 170–181 (2011). https://doi.org/10.1007/s12672-011-0065-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-011-0065-7