
Abstract
Structural information pertaining to antigen–antibody interactions is fundamental in immunology, and benefits structure-based vaccine design. Modeling of antigen–antibody immune complexes from co-crystal structures or molecular docking simulations provides an extensive profile of the epitope at the interface; however, the key amino acids involved in the interaction must be further clarified, often through the use of experimental mutagenesis and subsequent binding assays. Here, we describe an in silico mutagenesis method to identify key sites at antigen–antibody interfaces, using significant increase in pH-dependency energy among saturated point mutations. Through a comprehensive analysis of the crystal structures of three antigen–antibody immune complexes, we show that a cutoff value of 1 kcal/mol of increased interaction energy provides good congruency with the experimental non-binding mutations conducted in vitro. This in silico mutagenesis strategy, in association with energy calculations, may provide an efficient tool for antibody–antigen interface analyses, epitope optimization, and/or conformation prediction in structure-based vaccine design.
Similar content being viewed by others


1 Introduction
Most sophisticated biological functions occur through the interaction of two or more components. Consequently, protein–protein interactions play critical roles in such numerous processes, including the immune response, signal transduction and enzyme regulation [1]. Along with the rapid progresses in structural biology in recent years, researchers have been able to expand their interest from the study of single protein structures to that of larger complexes with multiple components. Indeed, the analysis of molecular interactions at the atomic level, such as antigen–antibody interactions in immunity in vivo, is essential for interpreting the structural basis of biological functions. Understanding how an antibody recognizes an antigen and what determines their specificity are crucial in successful vaccine design and continued improvement [2, 3].
The antigenic determinant or ‘epitope’ is presented to the host immune system and subsequently elicits an appropriate antibody reaction. In general, each epitope comprises only a few residues (6–20 residues), which is sufficient to produce an immune response [4, 5]. Structural biological methods and in silico docking prediction methods [6] can model antigen–antibody complexes, and analyses of solvent accessibility surface variation, non-bonding contacts, and the interaction energy can highlight the likely arrangement and potential involvement of specific residues at the interaction interface [7], a process known as epitope mapping. Unfortunately, this relatively static conformation of an antigen–antibody complex is not always sufficient to ascertain the particular residues that will be critical in the binding dynamics and associated energy stability. Thus, epitope residues are often further verified through the use of routine experimental methods, such as site-directed mutagenesis [8] or peptide scanning [9], concomitant with the use of enzyme-linked immunosorbent assay (ELISA), western blotting, Surface Plasmon Resonance (SPR) binding assay and/or analytical ultracentrifugation. However, these strategies are time-consuming and often limited by the creation of alanine point mutations on antigens instead of saturated mutations on the antigen and the antibody. In addition, site-directed mutagenesis of an epitope amino acid might lead to errors in folding of the protein, and thus a disruption of its function or binding capabilities [10], or even a failure in its expression [11]. As such, the peptide scanning approach is only suitable for mapping linear, not conformational, epitopes [12].
Quantitative model for binding energy based on empirical force field had been proposed to determine the real hot spot on protein–protein interface [13]. A knowledge-based hot spot prediction server was also developed using a machine learning approach based on experimental data [14, 15]. These methods provide general protocols to identify hot spot accounting for binding affinity. Here, we describe an in silico saturation mutagenesis method to predict strategic residues on epitope that are energetically critical in mediating the binding of antibody. To this end, we employed a ‘Calculate Mutation Energy (Binding)’ program, using Discovery Studio 4.1 (Accelrys, San Diego, CA) software, to evaluate a series of in silico site-saturated mutations. For these experiments, we employed data from three previously characterized crystal structures with associated alanine substitution mutagenesis results: two crystal structures pertaining to the hepatitis E virus (HEV) and one pertaining to the human immunodeficiency virus (HIV) in combination with their respective antibody binding data [16, 17]. HEV is a small, non-enveloped RNA virus that harbors a protrusion on the basal aspect of the virus, known as the E2s domain. This domain is responsible for host interactions and immunogenicity and is, consequently, the focus of much investigation. The p24 antigen of HIV is a small capsid protein found on the capsule of HIV that is frequently used to detect infection.
Calculations based on saturation mutagenesis predict the key residues required for antigen–antibody interactions, as determined by a significant increase in the free energy in response to a given mutation. These calculations also allow for the identification of potential key epitope sites for further experiments, provide guidance as to the type of amino acid substitution that will be applicable in subsequent experiments, and facilitate antigen design for vaccinations as well as strategies for antibody affinity maturation assays [18, 19]. Fluctuations in the environmental pH may also have a significant effect on binding affinity [20], and, thus, the effects of pH and protein ionization are also considered through energy-term parameterization [21].
Through these in silico interaction and binding energy analyses, we proposed a different cutoff value for predicting key epitope residues in place of that suggested in the guidelines of the software. Furthermore, our findings with this approach show good congruency with the results from our previous in vitro alanine scanning mutagenesis experiments, demonstrating the potential utility of this approach for future rational drug or vaccine design.
2 Materials and Methods
2.1 Preparation of Immune Complex Structures
Three crystal structures of HEV E2s-genotype 1, HEV E2s-genotype 4 and HIV p24 in complex with their respective antibody Fab fragments were determined previously [16, 17, 22]; the PDB IDs for these structures are 3RKD (HEV genotype I E2s with 8C11 Fab, E2s-I:8C11), 4PLK (HEV genotype IV E2s with 8G12 Fab, E2s-IV:8G12) and 3VRL (HIV p24 dimer with A10F9 Fab, p24:A10F9). Using the default CHARMm force field [21], these initial structures were subjected to 2000-step smart minimizer minimization to ensure each of them reached energy minima before subsequent calculations were performed.
2.2 Delineation of the Interface Regions
The interface regions of the antigen–antibody complexes (E2s-I:8C11, E2s-IV:8G12 and p24:A10F9) were defined up to a 6-Å contact distance [23], in combination with a decrease in the solvent accessible surface (SAS) area. Other criteria, including hydrophobic clusters, hydrogen bonding contacts, van der Waals forces, and electrostatic interactions, were also considered for interface definition, which were conducted using the Analyze Protein Interface tool of Accelrys® Discovery Studio 4.1 software. Overall, several residues at the interface were defined as potential key epitope sites for further analysis.
2.3 In Silico Mutagenesis and Energy Evaluation
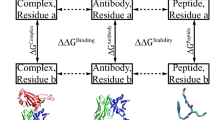
In silico mutagenesis was performed using the Calculate Mutation Energy (Binding) protein design tool embedded in Accelrys® Discovery Studio 4.1. This protocol assesses changes in the binding affinity of protein complexes in response to single-point mutations—in this case, alanine point mutations—among a selected set of amino acid residues. The mutation energy is defined as the free energy shift that occurs upon mutation, and the value is used to hypothesize the effect of a virtual mutation. CHARMm force fields (default settings) were employed to determine the free energy of the complexed and unbound states. A schematic representation delineating the process was showed in Fig. 1.

Schematic representation of the entire process
The pH-dependent mode was set as true to report the binding energy differences. The electrostatic terms were calculated by integration over the proton binding isotherms, derived from fractional protonations of acidic and basic residues which in turn were calculated using the same method as in the Protein Ionization component [24]. The mutation energy was calculated as a function of pH between the mutant and wild type:
where ΔG bind = ΔG AB − ΔG A-B separated. Positive values of ΔΔG mut represent a destabilizing effect of the mutation, whereas negative values represent a stabilizing effect.
The total free energy difference, ΔG tot(pH), between the bound or unbound state in pH-dependent mode is calculated as the following weighted sum of the VDW, electrostatic, entropy and non-polar terms (which includes the effects of both pH and ionic strengths):
In general, proteins were suspended in 1 × phosphate-buffered saline, pH 7.45, with an ionic strength of 0.15 M to simulate a physiological environment.
3 Results
3.1 Preliminary Epitope Residue Identification at the Antigen–Antibody Interface
Antigen–antibody interface residues for the three immune complexes were included in our mutagenesis simulations. In previous studies, we showed that the epitope of 8C11 Fab and 8G12 Fab locates to different positions on the HEV E2s dimer surface: 8G12’s epitope is located at the E2s dimerization region, whereas 8C11 recognizes and binds with the E2s dimer on the opposite side [16, 17, 25]. The binding site of A10F9 Fab is located on one monomer of the shoulder-to-shoulder HIV p24 dimer [22]. Using ΔSAS comparison, hydrogen bonding contact analysis, and energetic calculations, we first omitted any of the residues that showed limited to no involvement in the antigen–antibody interaction.
In the structure of E2s-I:8C11, the antibody recognition sites were identified using eight discontinuous regions on E2s: Thr476–Ala477, Glu479, Thr484–Tyr485, Asp496–Thr499, Val510–Leu514, Lys534, Asn573–Arg578, and Pro592. The relative SAS area (%) for each residue in the complex and on the individual antigen (E2s) was calculated and the ΔSAS values are listed in Table 1. These residues were exclusively distributed on the SAS area, and three major conformational patches (Asp496–Thr499, Val510–Leu514 and Asn573–Arg578) were involved in the center region of the antigen–antibody interface (Fig. 2). Among these major domains, Arg512 showed the strongest interaction with the complementarity determining region (CDR) of the antibody in terms of the abundance of hydrogen bonding contacts formed, which included a hydrogen acceptor from the side chain of AsnL32, and the more electronegative acceptor atoms from the main chains of PheL91 and GlyH107 (Fig. 2; the superscripted “L” denotes the light chain of the mAb and “H” the heavy chain).

Interaction between 8C11 Fab and HEV E2s of genotype I. a Cartoon representation of an overall view of the E2s-I:8C11 Fab complex. The E2s of genotype I is in light pink, the light chain in yellow, and the heavy chain in light blue. Residues involved in the protein–protein interaction are shown in stick mode and colored by element. The carbon element of the key residue, Arg512, is shown in magenta whereas the other residues are shown in green. b Interface residues of E2s of genotype I shown in stick mode overlapping with the transparent surface. Three major conformational patches are shown in cyan, and the other region in yellow. c A close-up view of the interactions. Arg512 forms three hydrogen bonding contacts with GlyH107, AsnL32 and PheL91. Hydrogen bonding contacts are shown as a red dashed line. Key residues on the complementarity determining region (CDR) are shown in line mode. GlyH107 is in blue, and AsnL32 and PheL91 in aqua. d Details of the hydrogen bonding contacts
In the E2s-IV:8G12 complex, an analysis of the crystal structure suggested that the major interactions between the antigen and antibody may be predominantly mediated by fifteen predicted hydrogen bonding contacts. Glu549 and Pro592 were determined as the most energetically critical residues, each observed to be involved in making four hydrogen bonding contacts with the surrounding atoms of the antibody; this also included the hydrogen acceptor atoms from GluL93, ThrL94 and TyrH105 toward Glu549 and GlyH57; and GlnH58 toward Pro592. Furthermore, pi interactions from Lys554 to TrpL92 were observed. Most of the residues with significant interaction energy made close, strong hydrogen bonding contacts between 8G12 and the E2s (Fig. 3).

Interaction between 8G12 Fab and HEV E2s of genotype IV. a Cartoon representation of an overall view of the E2s-IV:8G12 complex. The E2s-IV is colored in light pink, the light chain in pale yellow, and heavy chain in light blue. Residues involved in protein–protein interactions are shown in stick mode. b Interface residues are shown as sticks overlapping with the background of the E2s-IV surface. The major conformation domain is shown in cyan. c A close-up view of the interactions. The key residues—Glu549, Lys554, Gly589, Pro592—are shown with magenta-colored carbons; the others with green-colored carbons. Hydrogen bonding contacts are indicated with red dashed lines. Residues from the antibody are rendered in line mode, with the heavy chain in blue and the light chain in aqua. d Detail interactions of the E2s-IV:8G12 complex
For the P24:A10F9 complex, we previously showed that the A10F9 Fab recognizes a conformational epitope via extensive pi–pi interactions (Pro207 located within an aromatic cage formed by TrpL92, TyrL32 and TyrH105), and a pi interaction between Leu205 and TyrH59 [22]. Residues involved in these aforementioned interactions are listed in Table 2, among which Asp197 forms two hydrogen bonds with SerH52 and SerH56; Arg203 forms three hydrogen bonds with TyrH105, ValH106 and GluH100; and Leu205 forms one hydrogen bond with ThrL94. Furthermore, hydrogen bonds were detected between Pro207 with PheL91 or TrpL92. Besides, Ile201 also maintain indispensable van der Waals interactions with residues on the antibody (Fig. 4; Table 2).

Interaction between A10F9 Fab and HIV p24. a An overall view of dimeric p24:A10F9 in cartoon mode. Structure of p24 dimer is shown in gray, light chain of A10F9 Fab in pale cyan, and heavy chain in light blue. The interface residues are shown in stick mode and colored by element. b A close-up view of the interactions. The interface residues are shown in stick mode and colored by element. The key residues—Asp197, Ile201, Arg203, Leu205 and Pro207—are shown with magenta-colored carbons; the others with green-colored carbons. Hydrogen bonding contacts are marked with red dashed lines. Residues from the A10F9 Fab are rendered in line mode, with the heavy chain in blue and the light chain in aqua
3.2 In Silico Saturation Mutagenesis
In silico, single-point, alanine scanning and saturation mutagenesis was performed on each of the potential key epitope residues defined by the aforementioned interaction analyses. The mutation models were subjected to energy minimization using CHARMm force fields and the difference between the mutation and wild type models was quantified by calculating the energy change. According to the guideline of Discovery studio, if the energy increase upon a mutation reaches a pre-defined cutoff value (0.5 kcal/mol), it could be inferred that the mutation will abrogate the binding or reduce the binding affinity, and the corresponding amino acid should be defined as key site that participates in epitope recognition.
In the E2s-I:8C11 complex, only one mutation (Arg512Ala) was predicted to have a substantially higher free energy (4.84 kcal/mol; Fig. 5), which is consistent with its strategic position at the interface [16]. Replacement of Arg512 with Ala abrogated the formation of three hydrogen bonds with the 8C11 Fab (Fig. 6), and the interaction energy increased dramatically, thereby completely disrupting the binding, as shown by western blotting [16].

Mutation energy landscape of in silico saturation mutation on the E2s-I:8C11 interface. a Mutation energy landscape of in silico single-point, saturation mutations performed on interface residues in the E2s-I:8C11 complex. b Critical residues with alanine mutation energy greater than 1 kcal/mol are shown in the red column. The 1 kcal/mol cutoff line is indicated in red, and alanine mutations are highlighted in red. Congruence between the mutation energy assay and the experimental mutations in our previous study is denoted as follows: “+”, consistent; “N/A” indicates no available experimental data. The cutoff value was reset to 1 kcal/mol, and residue exhibiting greater than 1 kcal/mol of mutation energy were deduced to be strategic for the antigen–antibody interaction

Structure comparison of Arg512 and Ala512 of HEV E2s-I. Arg512 is shown in green, and Ala512 in pink. Residues from the Fab are shown in light blue. Hydrogen bonding contact are displayed as a red dashed line. The Ala512 substitution causes a loss of two hydrogen bonds directed to the 8C11 Fab light chain
Comparatively, alanine scanning mutagenesis simulation of the E2s-IV:8G12 complex showed four main destabilizing effects that boost the interaction energy above 1 kcal/mol: Glu549 (2.03 kcal/mol), Lys554 (1.54 kcal/mol), Gly589 (1.12 kcal/mol) and Pro592 (1.61 kcal/mol). In the saturation mutation simulation, the four sites also showed higher energy changes (>1 kcal/mol) when mutated to any of the other 19 residues (Fig. 7). Except for Gly589, this high level of mutational energy is presumed to be mainly due to the loss of hydrogen bonding contacts and van der Waals interactions. The interaction analysis of the E2s-IV:8G12 interface also helped to elucidate how the epitope will be perturbed by mutations due to energy variation (Fig. 3).

Mutation energy landscape of in silico saturation mutation on the E2s-IV:8G12 interface. a Mutation energy landscape of in silico single-point, saturation mutations performed on interface residues in the E2s-IV:8G12 complex. b Critical residues with alanine mutation energy greater than 1 kcal/mol are shown in the red column. The 1 kcal/mol cutoff line is indicated in red, and alanine mutations are highlighted in red. Congruence between the mutation energy assay and the experimental mutations in our previous study is denoted as follows: “+”, consistent; “–”, inconsistent
In the p24:A10F9 simulation, five mutations showed significant variations in energy: Asp197 (1.24 kcal/mol), Ile201 (2.37 kcal/mol), Arg203 (3.47 kcal/mol), Leu205 (1.36 kcal/mol) and Pro207 (1.93 kcal/mol). Data from saturated mutagenesis confirmed that these five residues are important and irreplaceable, as each mutation was above the 1 kcal/mol cutoff (Fig. 8). Most of these free energy increases above 1 kcal/mol were also induced by saturation mutations at these sites.

Mutation energy landscape of in silico saturation mutation on the HIV p24:A10F9 interface. a Single-point, saturation mutations calculations were performed on interface residues in the p24:A10F9 complex. b Critical residues with alanine mutation energy greater than 1 kcal/mol are shown in the red column. The 1 kcal/mol cutoff line is indicated in red, and alanine mutations are highlighted in red. Congruence between the mutation energy assay and the experimental mutations in our previous study is denoted as follows: “+”, consistent; “N/A” indicates no available experimental data
3.3 Threshold for Key Epitope Residues
The Discovery Studio guideline suggests a threshold of 0.5 kcal/mol for defining the destabilizing or stabilizing effect [26]. In our experience here, a value of 0.5 kcal/mol provides misleading results, and would indicate that most of the residues are critical at the interface. Increasing this threshold to 1.0 kcal/mol gave a more accurate indication of the residues that were significantly contributing to the interaction. Although another physical model for binding energy hot spots was also developed and suggested a similar cutoff value based CHARMM force field [13], the method employ different energy terms while calculation and different test models with respect to ours. In our opinion, a cutoff of 1.0 kcal/mol should be regarded as the energy barrier to hamper an antigen–antibody interaction, and is likely to be applicable for other types of protein–protein interactions. Despite this, residues with minor free energy variations after mutation were considered less-engaged in the epitope recognition. Overall, our findings show that saturation mutations can be used to identify key residues or unknown mutants between specified proteins, quickly and effectively to the same extent or better than the routinely used alanine scanning in vitro mutagenesis assays. Furthermore, this technique is useful for both co-crystal structure and simulated modeling analyses for rational drug design.
4 Discussion
Acute hepatitis E remains a major public health concern, particularly in developing countries, and it appears that prophylactic vaccines are likely to be required to prevent HEV infection [27, 28]. HIV is also an ongoing global concern. In our previous work, we interrogated the binding sites of three immune complexes pertaining to vaccine development for these two viruses using experimental alanine scanning mutagenesis and crystallization [16, 17, 22]. Here, we employed in silico alanine scanning mutagenesis simulations to confirm the essential epitope residues involved in these three complexes to further explore the opportunities available for the timely and cost-effective strategic evaluation of rational drug design.
In the E2s-I:8C11 model, only one candidate residue, Arg512, was identified as a critical site for structural stability by in silico mutation calculations, which agrees with our previous experimental findings [16]. In the E2s-IV:8G12 model, four critical residues—Glu549, Lys554, Gly589 and Pro592—showed elevated levels of free energy. The saturated mutational scanning on the p24:A10F9 complex indicated the importance of five epitope residues, Asp197, Ile201, Arg203, Leu205 and Pro207. Of these, Asp197, Arg203 and Leu205 have been verified previously using experimental EC50 calculations and sigmoidal trend fitting [22]; the additional two residues were not included in the previous binding assay, and further tests are warranted.
The ‘Calculate Mutation Energy (binding)’ protocol integrated in the Discovery Studio software provides a pH-dependent mode and a temperature-dependent mode for different applications, which needs to be set independently. We found that the pH-dependent mode yielded a higher number of residues than the temperature-dependent mode, with the exception of Gly591, which was not indicated to be important in the E2s-IV:8G12 model. Through careful examination of the structure, we found that Gly591 lies in a same loop formed by residues Gly589 to Pro592, and, given its distance from the complementary region of the antibody, would have been predicted to contribute less to binding. In addition, the molecular interaction analysis based on CHARMm force field also suggested fewer substantial interactions between Gly591 and the Fab of 8G12 as compared with other important residues. Comparing the structure of the mutational model at Gly591 with the original wild type model, we note only a slight variation in the local structure. Yet, according to previous reports, a mutation to Gly591 induces significant changes in experimental binding, which is likely to indicate a drastic alteration in the local structure [17]. Indeed, a glycine to alanine mutation might shift the main chain direction and even cause severe changes in local structure. Therefore, we presume that the in silico mutation at Gly591 failed to generate a reasonable local structure and this may have lowered the accuracy of the mutational energy calculation and thus the indicated significance of this residue in the binding. This hypothesis is supported by structure elucidation, as Gly and Pro show differences in flexibility or induce systematic conformational changes when substituted with other types of residues [17].
Despite this discrepancy, overall, the proposed empirical cutoff value successfully predicted most of the key residues of the epitope for antibody recognition in line with those determined using high-resolution complex crystal structures. The only missing site was likely due to insufficient optimization of the mutation structure, which might be overcome by additional energy minimization or dynamic simulation. Our study, thus, presents a reliable computational method for the initial screening of epitope residues before traditional mutation experiments, which would help to facilitate fundamental research on epitopes and vaccine design. Although the proposed cutoff value was in good agreement with the results obtained in previous alanine scanning in vitro experiments, additional studies with other known complexes should be employed in future experiments to confirm the validity of the assay and this proposed cutoff value. In addition to predicting key epitope residues in antigen–antibody interactions, this method could also be extended to other types of protein–protein interactions, with point mutations also aiding in complex determination. Other than focusing on the destabilizing effect induced by a mutation, which abolishes protein–protein binding, the stabilizing effect achieved as a consequence of increasing binding affinity also deserves more attention, particularly in terms of improving the affinity of engineered antibodies [29, 30]. This type of information would provide an alternative computational approach in guided affinity maturation experiments. Prediction of stabilizing mutations was also useful in rational, structure-based design of proteins to generate well-packed conformations [31]. Computational screening of the CDR through mutational trials before in vitro mutations are carried out would also help to circumvent the need for complicated traditional library methods, and one could expect that less time would be required to obtain antibodies with improved affinities. Although the cutoff value used here for the antigen–antibody analysis needs further exploration, this technique seems to provide an effective tool for epitope determination.
References
Almlof M, Aqvist J, Smalas AO, Brandsdal BO (2006) Probing the effect of point mutations at protein-protein interfaces with free energy calculations. Biophys J 90(2):433–442. doi:10.1529/biophysj.105.073239
Haynes BF (2015) New approaches to HIV vaccine development. Curr Opin Immunol 35:39–47. doi:10.1016/j.coi.2015.05.007
Malito E, Carfi A, Bottomley MJ (2015) Protein crystallography in vaccine research and development. Int J Mol Sci 16(6):13106–13140. doi:10.3390/ijms160613106
Bogan AA, Thorn KS (1998) Anatomy of hot spots in protein interfaces. J Mol Biol 280(1):1–9. doi:10.1006/jmbi.1998.1843
Mariuzza RA, Phillips SE, Poljak RJ (1987) The structural basis of antigen-antibody recognition. Annu Rev Biophys Biophys Chem 16:139–159. doi:10.1146/annurev.bb.16.060187.001035
Lengauer T, Rarey M (1996) Computational methods for biomolecular docking. Curr Opin Struct Biol 6(3):402–406
Yunta MaJR (2012) Using molecular modelling to study interactions between molecules with biological activity. Bioinformatics. InTech. doi:10.5772/54007
Benjamin DC, Perdue SS (1996) Site-directed mutagenesis in epitope mapping. Methods 9(3):508–515
Reineke U (2009) Antibody epitope mapping using de novo generated synthetic peptide libraries. Methods Mol Biol 524:203–211. doi:10.1007/978-1-59745-450-6_14
Brinkman A, Kortleve DJ, Schuller AG, Zwarthoff EC, Drop SL (1991) Site-directed mutagenesis of the N-terminal region of IGF binding protein 1; analysis of IGF binding capability. FEBS Lett 291(2):264–268
Antanasijevic A, Basu A, Bowlin TL, Mishra RK, Rong L, Caffrey M (2014) Mutagenesis studies of the H5 influenza hemagglutinin stem loop region. J Biol Chem 289(32):22237–22245. doi:10.1074/jbc.M114.572974
Cretich M, Longhi R, Corti A, Damin F, Di Carlo G, Sedini V, Chiari M (2009) Epitope mapping of human chromogranin A by peptide microarrays. Methods Mol Biol 570:221–232. doi:10.1007/978-1-60327-394-7_10
Kortemme T, Baker D (2002) A simple physical model for binding energy hot spots in protein-protein complexes. Proc Natl Acad Sci USA 99(22):14116–14121
Darnell SJ, LeGault L, Mitchell JC (2008) KFC Server: interactive forecasting of protein interaction hot spots. Nucleic Acids Res 36(Web Server issue):W265–W269. doi:10.1093/nar/gkn346
Zhu X, Mitchell JC (2011) KFC2: a knowledge-based hot spot prediction method based on interface solvation, atomic density, and plasticity features. Proteins 79(9):2671–2683. doi:10.1002/prot.23094
Tang XH, Yang CY, Gu Y, Song CL, Zhang X, Wang YB, Zhang J, Hew CL, Li SW, Xia NS, Sivaraman J (2011) Structural basis for the neutralization and genotype specificity of hepatitis E virus. Proc Natl Acad Sci USA 108(25):10266–10271. doi:10.1073/pnas.1101309108
Gu Y, Tang XH, Zhang X, Song CL, Zheng MH, Wang KH, Zhang J, Ng MH, Hew CL, Li SW, Xia NS, Sivaraman J (2015) Structural basis for the neutralization of hepatitis E virus by a cross-genotype antibody. Cell Res 25(5):604–620. doi:10.1038/cr.2015.34
Ansari HR, Raghava GP (2013) In silico models for B-cell epitope recognition and signaling. Methods Mol Biol 993:129–138. doi:10.1007/978-1-62703-342-8_9
Clementi N, Mancini N, Castelli M, Clementi M, Burioni R (2013) Characterization of epitopes recognized by monoclonal antibodies: experimental approaches supported by freely accessible bioinformatic tools. Drug Discov Today 18(9–10):464–471. doi:10.1016/j.drudis.2012.11.006
Spassov VZ, Yan L (2013) pH-selective mutagenesis of protein-protein interfaces: in silico design of therapeutic antibodies with prolonged half-life. Proteins 81(4):704–714. doi:10.1002/prot.24230
Brooks BR, Brooks CL 3rd, Mackerell AD Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30(10):1545–1614. doi:10.1002/jcc.21287
Gu Y, Cao F, Wang L, Hou WH, Zhang J, Hew CL, Li SW, Yuan YA, Xia NS (2013) Structure of a novel shoulder-to-shoulder p24 dimer in complex with the broad-spectrum antibody A10F9 and its implication in capsid assembly. PLoS ONE. doi:10.1371/journal.pone.0061314
Rao VS, Srinivas K, Sujini GN, Kumar GN (2014) Protein-protein interaction detection: methods and analysis. Int J Proteomics 2014:147648. doi:10.1155/2014/147648
Spassov VZ, Yan L (2008) A fast and accurate computational approach to protein ionization. Protein Sci 17(11):1955–1970
Li SW et al (2009) Dimerization of hepatitis E virus capsid protein E2s domain is essential for virus-host interaction. PLoS Pathog 5(8):e1000537
Feyfant E, Sali A, Fiser A (2007) Modeling mutations in protein structures. Protein Sci 16(9):2030–2041
Zhu FC et al (2010) Efficacy and safety of a recombinant hepatitis E vaccine in healthy adults: a large-scale, randomised, double-blind placebo-controlled, phase 3 trial. Lancet 376(9744):895–902
Zhang J et al (2015) Long-term efficacy of a hepatitis E vaccine. N Engl J Med 372(10):914–922
Choi Y et al (2015) Antibody humanization by structure-based computational protein design. In: MAbs
Magor BG (2015) Antibody affinity maturation in fishes-our current understanding. Biology (Basel) 4(3):512–524
Borgo B, Havranek JJ (2012) Automated selection of stabilizing mutations in designed and natural proteins. Proc Natl Acad Sci USA 109(5):1494–1499
Acknowledgements
This work was supported by the Natural Science Foundation of China under Grants 81571996, 81371818, 31670935; Fujian Provincial Natural Science Foundation under Grant 2013J05053; the Fundamental Research Funds for the Central Universities under Grant 2011121004.
Author contributions
HY and SL designed research; LX, QH, XB, XZ, ZZ, ZK and QZ performed experiments; LX, HY, YG, QZ, JZ, SL, and NX analyzed data; and LX, HY, QZ, and SL wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xin, L., Yu, H., Hong, Q. et al. Identification of Strategic Residues at the Interface of Antigen–Antibody Interactions by In Silico Mutagenesis. Interdiscip Sci Comput Life Sci 10, 438–448 (2018). https://doi.org/10.1007/s12539-017-0242-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12539-017-0242-7