
Abstract
The recent outbreaks of Zika virus and the absence of a specific therapy have necessitated to identify T-cell-stimulating antigenic peptides as potential subunit vaccine candidates. The translated ssRNA (+) genome of Zika virus was explored in EMBOSS antigenic and VaxiJen to predict 63 peptides as potential antigens. Three MHC-II binding peptide prediction tools, viz. NetMHCIIpan, PREDIVAC and immune epitope database (IEDB) were employed in consensus on 63 antigenic peptides to propose 14 T-helper cell epitopes. Similarly, analysis on 63 antigenic peptides through NetMHC, NetCTL and IEDB MHC-I binding peptide prediction tool led to identification of 14 CTL epitopes. Seven T-cell epitopes, C:44-66, M:135-149, NS2A:124-144, NS3:421-453, NS3:540-554, NS4B:90-134 and NS4B:171-188, are observed to share overlapping MHC-I and MHC-II binding motifs and hence, are being proposed to constitute minimum T-cell antigens to elicit protective T-cell immune response against Zika. Three of them, C:44-66, NS3:421-453 and NS3:540-554 are identified to be conserved across all the selected strains of Zika virus. Moreover, the 21 T-cell epitopes are non-self to humans and exhibited good coverage in variable populations of 14 geographical locations. Therefore, 21 T-cell epitopes are proposed as potential subunit vaccines against Zika virus.
Similar content being viewed by others

1 Introduction
The existence of Zika virus is not new, but the recent outbreaks throughout the Americas and in tropical regions have vindicated the urgency to accelerate research both on basic biology as well as vaccine development, to respond to the current and future epidemics [1,2,3,4,5]. Zika, an arbovirus of the flaviviridae family, is transmitted to humans by Aedes aegypti [3]. Zika infections are almost asymptomatic in nature with most common symptoms of infection being rash, fever, and joint pain. The recent Zika outbreaks have caused alarm because of significant increase in Guillain–Barre syndrome and microcephaly in newborns, which led WHO to declare it as global health emergency [3, 4, 6,7,8]. The current Zika epidemics are reported to be part of the Asian lineage of the virus, which have undergone an optimization in codon usage of NS1 gene for expression in humans [9,10,11].
Host–pathogen interaction of Zika virus is reminiscent of other flaviviruses that have been studied [12]. The virus gains access to the host cytoplasm via C-type lectin receptor-mediated endocytosis and infects fibroblast, keratinocytes, and immature dendritic cells which further facilitate viral replication more like dengue virus [12]. Thus, investigational flavivirus vaccine platforms for West Nile and Dengue virus are in primary focus on potential Zika virus vaccines development as instigated by Vaccine Research Centre, NIAID, NIH [13].
Vaccination is considered as an effective and essential strategy for management of viral infections in the absence of a precise antiviral drug. The conventional vaccinology approach to develop new vaccines by cloning and expressing the dominant surface antigen are only effective when given with strong adjuvants [14, 15]. However, this approach is unlikely to work for pathogens that have highly mutable genome such as RNA viruses as in the case of Zika [16]. To resolve such a scenario, integration of computational tools for epitope discovery (immunoinformatics-driven approach) has achieved significant strides through genome-based subunit vaccine design [17, 18]. T-cell epitope can induce pathogen-specific immunogenic memory for a faster and stronger adaptive immune response upon infection, which is hallmark of an efficacious vaccine [16, 19]. As T-cells recognize epitopes that are derived from a broader range of proteins, whole genome can be explored for the selection of T-cell epitopes unlike surface proteins as in case of B-cell epitopes. T-cell subunit vaccines are safer, as they elicit immune responses based on the minimal pathogen-specific antigenic elements, conserved across multiple pathogenic strains and exclude self (host) antigens [16]. Indeed, T-cell epitopes have been shown as potential vaccine candidates in flaviviruses such as Dengue and Yellow fever [20,21,22,23,24,25,26,27]. Therefore, an immunoinformatics approach was implemented in the present study to detect minimal T-cell epitopes from the translated ssRNA (+) genome of Zika virus, capable of inducing cell-mediated immunity in humans.
2 Materials and Methods
2.1 Retrieval of Zika Genome, Proteome and Phylogenetic Analysis
The whole genome and proteome of Zika virus (NC_012532.1) was retrieved from National Centre for Biotechnology Information (NCBI) [28]. A draft homology and phylogenetic analysis were carried out with close homologs: Dengue, Yellow fever, West Nile and Spondweni virus using NCBI BLAST [29] and MEGA 7.0 (Molecular Evolutionary Genetics Analysis) [30]. The mature protein sequences mapped to single polyprotein of Zika virus were retrieved from NCBI RefSeq database.
2.2 Prediction of Antigenic Peptides
Antigenic peptides for Zika virus were predicted from protein sequences using EMBOSS antigenic program, that uses a semi-empirical Kolaskar and Tongaonkar method based on the physico-chemical properties of amino acids and their frequency of occurrence in experimentally known segmental epitopes with about 75% accuracy [31]. The predicted antigenic peptides were validated through VaxiJen v2.0 server with virus as the target organism. The VaxiJen server predicts protective antigens, tumour antigens and subunit vaccines, with prediction accuracy of 70–89% by using an alignment-free algorithm to discriminate between antigens and non-antigens [32].
2.3 Helper T-cell Epitope Prediction
Binding of antigenic peptide to major histocompatibility complex class II (MHC class II) molecules is a key event in T-helper lymphocyte-mediated cellular immunity against infectious pathogens. In this study, MHC-II binding peptide prediction was restricted to HLA-DRB1*0101, HLA-DRB1*0301, HLA-DRB1*0401, HLA-DRB1*0404, HLA-DRB1*0405, HLA-DRB1*0701, HLA-DRB1*0802, HLA-DRB1*0901, HLA-DRB1*1101, HLA-DRB1*1302, HLA-DRB1*1501, HLA-DRB3*0101, HLA-DRB4*0101, HLA-DRB5*0101, due to their inclusion in an HLA-DR supertype or their inclusion in other promiscuous epitope studies [27, 33,34,35,36,37].
NetMHCIIpan is a state-of-the-art method for the quantitative prediction of peptide binding to MHC class II molecule of known sequence [38]. The peptides predicted as antigen in both EMBOSS and VaxiJen were evaluated for their binding ability with the selected predominant HLA-DR alleles using NetMHCIIpan 3.1 server. Intermediate binding affinity (IC50 ≤ 500 nM) with at least one predominant HLA-DR alleles was set as threshold to screen antigenic peptides.
PREDIVAC [39] was employed for evaluation of promiscuous epitopes for selected HLA-DR alleles. It implements a method to predict CD4+ T-cell epitopes that allows coverage of 95% of human protein diversity. Threshold PREDIVAC score of 70 was set to screen MHC-II binding antigenic peptides. The MHC-II binding ability of screened epitopes from PREDIVAC was cross-validated using IEDB consensus method [40]. The CD4+ T-cell epitopes were also validated using PREDIVAC score and IEDB percentile rank of published positive control epitopes from Yellow fever virus [27].
2.4 Cytotoxic T-cell (CTL) Epitopes Prediction
CTLs recognize viral antigens on the surface of virally infected cells in combination with MHC-I molecule and exert their effect by killing the infected cells either by lysis or inducing apoptosis. The antigenic peptides based on EMBOSS and VaxiJen were screened for MHC-I binding propensity using NetMHC 4.0 server [41]. The MHC-I binding was restricted to five predominant HLA-A alleles (HLA-A*0201, HLA-A*0206, HLA-A*2403, HLA-A*3301, HLA-A*6801) and seven predominant HLA-B alleles (HLA-B*0702, HLA-B*1501, HLA-B*3501, HLA-B*4001, HLA-B*5101, HLA-B*5301, HLA-B*5801) with a threshold rank for strong binder 0.5 and weak binder 2.0 [41,42,43,44]. The NetMHC has 75–80% accuracy for peptides binding to HLA class I molecules and is employed widely to predict HLA-binding peptides in several pathogens proteomes including SARS, Influenza and HIV [41].
The antigenic peptides predicted using NetMHC were cross-validated in NetCTL v1.2 by restricting to A2, A24, B7 and B58 supertype representatives [42, 45, 46] with threshold NetCTL score 0.5 [42]. In the NetCTL method, each possible 9mer peptide of a protein was assigned a score based on a combination of proteasomal cleavage, TAP transport efficiency, and HLA-I binding affinity, with the highest weight assigned to HLA-I affinity. The best candidates were further evaluated in IDEB MHC-I binding prediction tool [40].
2.5 Prioritization of Antigenic Peptides as Potential Subunit Vaccine Candidates
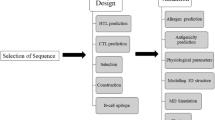
The epitope conservancy analysis tool at the IEDB [47] was applied to identify conservancy of proposed epitopes in all reported strains of Zika virus as well as in its close homologs. The population coverage rates of individual epitopes were calculated using the IEDB population coverage tool [48]. The T-cell epitopes were analysed for the presence of human self-peptides using HLAPred [49]. The immunoinformatics approach used to identify T-cell epitopes as subunit vaccine candidates of Zika virus is schematically represented below (Fig. 1).

Schematic representation of protocol used to identify potential subunit vaccine candidates
3 Results and Discussion
3.1 Zika Genome, Proteome and Its Close Homologs
Identification of potential antigenic peptide from viral proteome is an important strategy for subunit vaccine development. Zika virus has an ssRNA (+) genome (NC_012532.1) of ~ 10 kb length that encodes for single polyprotein which is further cleaved into 14 mature proteins by host and viral proteases (Fig. 2). The BLASTN search revealed that Zika virus genome resembles Spondweni virus with 71% identity and 90% query coverage. The Zika genome was also found to have good homology with the strains of West Nile and Dengue viruses with query coverage and identities of more than 47 and 66%, respectively. Yellow fever virus seemed to have low degree of homology amongst the selected flaviviruses (Table 1).

Genome and proteome of Zika virus
Phylogenetic analysis of genome and proteome sequences of Zika, Dengue, West Nile, Yellow fever and Spondweni viruses revealed four different clades. The Yellow fever virus was found as the common ancestral organism and from which other four flaviviruses diverged. Spondweni virus is the closest relative of Zika virus followed by West Nile and Dengue virus (Fig. 3). Therefore, the disease mechanism and host–pathogen interaction for Zika virus would be similar to the selected flaviviruses and T-cell epitope-based subunit vaccines are likely to be successful against Zika virus as has been for homologous flaviviruses [21, 24,25,26,27]. The close homology also opens up possibility of designing common T-cell epitope-based subunit vaccine candidates for Zika, Dengue, West Nile and Yellow fever viruses.

Phylogeny of Zika virus: a phylogenetic tree based on whole genome alignment, b phylogenetic tree based on whole proteome (translated genome) alignment
3.2 Antigenic Peptide from Mature Proteins of Zika Virus
Detection of antigenic peptides from mature proteins is the first step for epitope-based subunit vaccine design. Analysis of Zika virus proteome using EMBOSS led to the identification of 102 unique antigenic peptides. Sixty-three peptides out of 102 were evaluated as antigens by VaxiJen server. Since, both software recognized 63 antigenic peptides (Suppl. Table 1) as antigens through two independent algorithms, these immunogens are expected to develop adaptive immunity in host through humoral or cellular immune response.
T-cell immunity is crucial for developing immunogenic memory for an effective adaptive immune response against Zika virus. Hence, the predicted immunogens were ultimately directed for both CD4+ and CD8+ T-cell-driven epitope prediction through a consensus of established immunoinformatics tools.
3.3 Potential Helper T-cell Epitopes
Helper T-cell epitopes are critical for generation of strong humoral and cytotoxic T-cell responses. The responses to helper T-cell epitopes, however, are restricted to their affinity and specificity with MHC-II molecules. Thus, MHC-II binding affinity must be considered as major criteria for screening helper T-cell epitopes [50, 51]. In general, an epitope with IC50 ≤ 50 nM towards MHC-II alleles is considered as strong binder, > 50 to ≤ 500 nM as intermediate binder and > 500 to ≤ 5000 nM as weak binder. Thirty-four potential immunogens were found to be intermediate binders of MHC-II alleles in NetMHCIIpan analysis (Suppl. Data 1). Therefore, these immunogens have MHC-II binding motif with reasonable binding affinity to induce T-helper cell-mediated immune response in host.
In order to verify helper T-cell antigenic potential of 34 immunogens, PREDIVAC score was used for screening. The higher PREDIVAC score indicate an immunogen as high-affinity MHC-II binder and, potentially, a CD4+ T-cell epitope [39]. Twelve experimentally validated T-helper cell epitopes of Yellow fever virus [27] were initially assessed to get an idea of a reasonable PREDIVAC score for good CD4+ T-cell epitope. The overall average PREDIVAC score for positive control epitopes towards selected MHC-II alleles was found to be 69.12 (Suppl. Table 2). Consequently, 14 immunogens with threshold average PREDIVAC score ≥ 70 were proposed as potent T-helper cell epitopes (Table 2; Suppl. Table 3).
The inferences from the PREDIVAC assessment were also cross-validated through IEDB consensus method, which reports helper T-cell epitopes based on percentile rank with low percentile rank implying good binding affinity [52, 53]. All the 14 T-cell epitopes have revealed low average percentile rank (Table 2; Suppl. Table 4) compared to positive control epitopes (Suppl. Table 5). Thus, all these evaluations supported the case of 14 immunogens as helper T-cell epitopes of Zika virus.
Conservancy analysis for the helper T-cell epitope have revealed that four peptides, C:44-66, NS3:421-453, NS3540-554, NS4A54-73, were 100% conserved among 100 non-redundant strains of Zika virus isolated from different geographical locations of the world (Suppl. Table 6). Additionally, NS2B4-20, NS5:883-893 and 2K:8-20 showed a conservancy of 99, 98 and 99%, respectively. These seven helper T-cell epitopes are expected to elicit protective immunity against multiple strains of the pathogens.
In our view, the epitope C:44-66 of capsid protein C is the best subunit vaccine candidate to develop helper cell-mediated protective immunity in host across multiple strains of Zika virus as it has showed IC50 < 500 nM with 13 MHC-II alleles in NetMHCIIpan, average PREDIVAC score of 77.93, avg. IEDB percentile rank 3.27 and 100% conservancy in 100 Zika virus strains. Although, the epitopes NS4B:90-134, NS4B:171-188 and NS2A:192-222 showed better binding affinity compared to C:44-66, they would be limited to specific Zika virus stains due to low conservancy of 8, 15 and 7%, respectively. Nevertheless, combinations of multiple antigenic peptides from the 14 helper T-cell epitopes may be tried through a vector to induce optimum helper T-cell cellular response in host.
3.4 Cytotoxic T-lymphocyte (CTL) Epitopes of Zika Virus
MHC-I restricted CTL epitope prediction is vital for designing T-cell epitope-driven subunit vaccine candidates. Forty-three out of 63 immunogens (Suppl. Data 2) have revealed MHC-I binding motifs in NetMHC server [41,42,43,44]. Thirty-eight immunogens were subsequently validated as MHC-I binding epitopes in NetCTL with 0.89 sensitivity and 0.94 specificity with at least one of the A2, A24, B7 and B58 MHC-I supertypes [42, 45, 46]. Fourteen of the 38 epitopes were predicted as CTL epitopes with specificity 0.993 (Table 3; Suppl. Table 7). These epitopes also revealed low percentile rank in IEDB analysis (Table 3; Suppl. Table 8). In fact, 13 of the 14 proposed CTL epitopes had average percentile rank below 5 (Table 3), which justifies high binding affinity of proposed CTL epitopes towards MHC-I alleles. Therefore, the 14 epitopes were proposed as potential CTL epitopes (Table 3).
Five CTL epitopes, C:44-66, NS2B:112-127, NS3:421-453, NS3540-554, NS4A:102-122 were 100% conserved while three epitopes, M:151-165, NS3:236-251, NS3:357-366 showed 96, 97, 91% conservancy, respectively. Hence, these eight epitopes would be useful as subunit vaccines against multiple strains of Zika virus by developing CTL response-based immunity in host.
We have proposed C:44-66 of capsid protein as the best CTL epitope across multiple strains of Zika virus as it showed binding potential towards 10 MHC-I alleles in NetMHC, four supertypes in NetCTL, significantly low average IEDB score of 1.07 and 100% conservancy in 100 non-redundant Zika virus proteome. Nonetheless, NS4B:90-134 and NS2A:124-144 also showed equipotent binding affinity as par with C:44-66 towards MHC-I alleles in all evaluations; however, low conservancy of these two epitopes means, the epitopes would develop immunity against limited number of Zika virus strains.
3.5 Prioritization of T-cell Epitopes
HLA distribution varies among different ethnic groups and geographic regions around the world. Thus, population coverage must be taken into account when designing an effective vaccine to cover as many populations as possible. The proposed T-cell epitopes showed high population coverage for 14 specified geographic regions of the world (Fig. 4). These results suggested that putative helper T-cell epitopes and CTL epitopes can specifically bind with the prevalent MHC molecules in the target population where the vaccine will be employed.

Population coverage of proposed T helper and CTL epitopes
Seven antigenic peptides, C:44-66, M:135-149, NS2A:124-144, NS3:421-453, NS3:540-554, NS4B:90-134, NS4B:171-188 showed shared MHC-I and MHC-II binding motifs (Table 3). Thus, effectively, 21 antigenic peptides are proposed in the present study as potential T-cell epitopes (Tables 2, 3). These 21 epitopes are identified as non-self to host human. The proposed T-cell epitopes represented 12 mature proteins of Zika virus; therefore, could potentially induce T-cell immunity as good as the pathogen. The seven T-cell epitope sharing MHC-I and MHC-II binding motif are anticipated to constitute minimal antigens to elicit both helper T-cell and CTL-mediated immunity against Zika virus. Three of the seven T-cell epitopes, C:44-66, NS3:421-453 and NS3:540-554 were 100% conserved in 100 Zika virus strains. These three epitopes may be tested as subunit vaccine either independently or together with other proposed T-cell epitopes to achieve optimum adaptive immunity in host for all strains of Zika virus. Further, the epitope, NS3:357-366 was found to be conserved in Zika, West Nile and Spondweni virus, hence, may be used for designing common subunit vaccine.
The consensus approach for T-cell epitope prediction is an efficient method in the sense it uses multiple independent algorithms together to propose T-cell epitopes from complete genome sequence by filtering out false positives in the process. Therefore, the 21 T-cell epitopes proposed in the present study are potent T-cell epitopes which may be validated experimentally as subunit vaccine candidates.
Limitations of the approach, however, may be it potentially misses out few good T-cell epitopes due to stringent parameters employed in a single tool. Possibly for this reason, none of the T-cell antigens from E and NS1 protein made it to final list of proposed T helper and CTL epitopes. In case of E protein, nine peptides were predicted as immunogens through EMBOSS and VaxiJen (Suppl. Table 1); while, only two peptides showed MHC-II binding propensities (Suppl. Data 1) and four peptides showed MHC-I binding propensities (Suppl. Data 2). But none of these epitopes could pass through the threshold set at PREDIVAC and NetCTL, respectively. In the case of NS1 protein, although three peptides were predicted as immunogens through EMBOSS and VaxiJen (Suppl. Table 1), none of them showed potential as T-cell antigens in subsequent analysis. Therefore, the antigenic peptides of E and NS1 proteins may be involved in inducing B-cell-mediated humoral response as have been reported for Dengue virus [9, 54,55,56]. Our reasoning also complemented well with Khan et al. [57], wherein through an in vivo T-cell responses assay, authors have demonstrated T-cell epitopes were distributed to a lesser extent in NS1 and E proteins of Dengue virus compared to other proteins.
To summarize, the 21 T-cell epitopes (Tables 2, 3) are proposed as potential subunit vaccines in this study. Seven of them, namely, C:44-66, M:135-149, NS2A:124-144, NS3:421-453, NS3:540-554, NS4B:90-134, NS4B:171-188 possibly hold the clue for both helper T-cell and CTL-based cellular immunity. Epitopes, C:44-66, NS3:421-453 and NS3:540-554 may be used as subunit vaccines for achieving immunity against multiple stains of Zika virus. Our study suggested C:44-66 of capsid protein as the best T-cell-driven subunit vaccine candidate. The epitopes being predicted in silico, these must be carefully validated experimentally to evaluate their ability to elicit cellular immune response. Care must be taken to use different combinations of proposed antigenic peptides along with their MHC-I and MHC-II binding core residues (Suppl. Data 3; Suppl. Data 4) for achieving success in designing subunit-based vaccines against Zika virus.
4 Conclusion
Emergence of Zika virus epidemics exposed limitations of conventional vaccinology in addressing the immunologic issues created by hypervariable flaviviruses. The immunoinformatics approach in this context have enlightened scopes for designing T-cell-driven subunit vaccine from conserved regions of genome sequences. We have used a consensus of multiple immunoinformatics tools for prediction of both helper T-cell and CTL epitopes. Twenty-one T-cell epitopes representing 12 mature proteins of the Zika virus were proposed in the present study with seven of them having shared MHC-I and MHC-II binding motifs. The seven T-cell epitopes, C:44-66, M:135-149, NS2A:124-144, NS3:421-453, NS3:540-554, NS4B:90-134, NS4B:171-188 would be of particular interest to start with experimental evaluations. We specially mention three epitopes, C:44-66, NS3:421-453 and NS3:540-554 as potential subunit vaccines across multiple strains of Zika virus. C:44-66 is the best T-cell epitope among the 21 potential subunit vaccines. Nevertheless, each of the proposed 21 T-cell epitopes are good immunogens to develop cellular response in host against Zika virus as all of them showed better binding affinity in immunoinformatics tools compared to positive control epitopes of Yellow fever virus. These proposed epitopes may be tested experimentally either independently or in combination as subunit vaccine candidates to achieve optimum immunity in host. We anticipate, the in silico subunit vaccines proposed in the present study would provide basis for future development of potent vaccines against Zika virus.
References
Campos GS, Bandeira AC, Sardi SI (2015) Zika Virus Outbreak, Bahia, Brazil. Emerg Infect Dis 21(10):1885–1886
Zanluca C, de Melo VCA, Mosimann ALP, Santos GIVD, Santos CNDD, Luz K (2015) First report of autochthonous transmission of Zika virus in Brazil. Mem Inst Oswaldo Cruz 110(4):569–572
(2016) Zika virus on the move. Cell 164(4):585–587
Musso D, Baud D (2016) Zika virus: time to move from case reports to case control. Lancet Infect Dis
Fauci AS, Morens DM (2016) Zika virus in the Americas—yet another arbovirus threat. N Engl J Med 374(7):601–604
Vogel G (2016) Emerging diseases. A race to explain Brazil’s spike in birth defects. Science 351(6269):110–111
(2016) Zika virus: a new global threat for 2016. Lancet Lond Engl 387(10014):96
WHO (2016) Zika virus and potential complications [Internet]. WHO. http://www.who.int/emergencies/zika-virus/en/
Freire CC de M, Iamarino A, Neto DF de L, Sall AA, Zanotto PM de A (2015) Spread of the pandemic Zika virus lineage is associated with NS1 codon usage adaptation in humans. bioRxiv 25;032839
Enfissi A, Codrington J, Roosblad J, Kazanji M, Rousset D (2016) Zika virus genome from the Americas. Lancet Lond Engl 387(10015):227–228
Zhu Z, Chan JF-W, Tee K-M, Choi GK-Y, Lau SK-P, Woo PC-Y et al (2016) Comparative genomic analysis of pre-epidemic and epidemic Zika virus strains for virological factors potentially associated with the rapidly expanding epidemic. Emerg Microbes Infect 5:e22
Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N et al (2015) Biology of zika virus infection in human skin cells. J Virol 89(17):8880–8896
(2016) Zika Virus Vaccine Research [Internet]. https://www.niaid.nih.gov/topics/Zika/ResearchApproach/Pages/vaccineResearch.aspx
De Groot AS (2003) How the SARS vaccine effort can learn from HIV-speeding towards the future, learning from the past. Vaccine 21(27–30):4095–4104
De Groot AS, Moise L, Liu R, Gutierrez AH, Terry F, Koita OA et al (2014) Cross-conservation of T-cell epitopes: now even more relevant to (H7N9) influenza vaccine design. Hum Vaccines Immunother 10(2):256–262
Terry FE, Moise L, Martin RF, Torres M, Pilotte N, Williams SA et al (2015) Time for T? Immunoinformatics addresses vaccine design for neglected tropical and emerging infectious diseases. Expert Rev Vaccines 14(1):21–35
Mishra S, Losikoff PT, Self AA, Terry F, Ardito MT, Tassone R et al (2014) Peptide-pulsed dendritic cells induce the hepatitis C viral epitope-specific responses of naïve human T cells. Vaccine 32(26):3285–3292
Oyarzun P, Kobe B (2015) Recombinant and epitope-based vaccines on the road to the market and implications for vaccine design and production. Hum Vaccines Immunother
De Groot AS, Sbai H, Aubin CS, McMurry J, Martin W (2002) Immuno-informatics: mining genomes for vaccine components. Immunol Cell Biol 80(3):255–269
Chen H-W, Hu H-M, Wu S-H, Chiang C-Y, Hsiao Y-J, Wu C-K et al (2015) The immunodominance change and protection of CD4+ T-cell responses elicited by an envelope protein domain III-based tetravalent dengue vaccine in mice. PLoS ONE 10(12):e0145717
Shi J, Sun J, Wu M, Hu N, Li J, Li Y et al (2015) Inferring protective CD8+ T-cell epitopes for NS5 protein of four serotypes of dengue virus Chinese isolates based on HLA-A, -B and -C Allelic distribution: implications for epitope-based universal vaccine design. PLoS ONE 10(9):e0138729
Duan Z-L, Liu H-F, Huang X, Wang S-N, Yang J-L, Chen X-Y et al (2015) Identification of conserved and HLA-A*2402-restricted epitopes in Dengue virus serotype 2. Virus Res 22(196):5–12
Duan Z, Guo J, Huang X, Liu H, Chen X, Jiang M et al (2015) Identification of cytotoxic T lymphocyte epitopes in dengue virus serotype 1. J Med Virol 87(7):1077–1089
Comber JD, Karabudak A, Huang X, Piazza PA, Marques ETA, Philip R (2014) Dengue virus specific dual HLA binding T cell epitopes induce CD8 + T cell responses in seropositive individuals. Hum Vaccines Immunother 10(12):3531–3543
Mathew A, Townsley E, Ennis FA (2014) Elucidating the role of T cells in protection against and pathogenesis of dengue virus infections. Future Microbiol 9(3):411–425
James EA, LaFond RE, Gates TJ, Mai DT, Malhotra U, Kwok WW (2013) Yellow fever vaccination elicits broad functional CD4+ T cell responses that recognize structural and nonstructural proteins. J Virol 87(23):12794–12804
de Melo AB, Nascimento EJM, Braga-Neto U, Dhalia R, Silva AM, Oelke M et al (2013) T-cell memory responses elicited by yellow fever vaccine are targeted to overlapping epitopes containing multiple HLA-I and -II binding motifs. PLoS Negl Trop Dis 7(1):e1938
Kuno G (2007) Chang G-JJ. Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch Virol 152(4):687–696
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol
Kolaskar AS, Tongaonkar PC (1990) A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett 276(1–2):172–174
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinf 8:4
Southwood S, Sidney J, Kondo A, del Guercio MF, Appella E, Hoffman S et al (1998) Several common HLA-DR types share largely overlapping peptide binding repertoires. J Immunol Baltim Md 1950 160(7):3363–3373
Priyadarshini V, Pradhan D, Munikumar M, Swargam S, Umamaheswari A, Rajasekhar D (2014) Genome-based approaches to develop epitope-driven subunit vaccines against pathogens of infective endocarditis. J Biomol Struct Dyn 32(6):876–889
Munikumar M, Priyadarshini IV, Pradhan D, Umamaheswari A, Vengamma B (2013) Computational approaches to identify common subunit vaccine candidates against bacterial meningitis. Interdiscip Sci Comput Life Sci 5(2):155–164
Umamaheswari A, Pradhan D, Hemanthkumar M (2012) Computer aided subunit vaccine design against pathogenic Leptospira serovars. Interdiscip Sci Comput Life Sci 4(1):38–45
Rakesh S, Pradhan D, Umamaheswari A (2009) In silico approach for future development of subunit vaccines against Leptospira interrogans serovar Lai. Int J Bioinforma Res 1:85–92
Andreatta M, Karosiene E, Rasmussen M, Stryhn A, Buus S, Nielsen M (2015) Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics 67(11–12):641–650
Oyarzún P, Ellis JJ, Bodén M, Kobe B (2013) PREDIVAC: CD4 + T-cell epitope prediction for vaccine design that covers 95% of HLA class II DR protein diversity. BMC Bioinf 14:52
Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR et al (2015) The immune epitope database (IEDB) 3.0. Nucleic Acids Res 43(Database issue):D405–12
Andreatta M, Nielsen M (2016) Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinforma Oxf Engl 32(4):511–517
Lund O, Nascimento EJM, Maciel M, Nielsen M, Larsen MV, Lundegaard C et al (2011) Human leukocyte antigen (HLA) class I restricted epitope discovery in yellow fewer and dengue viruses: importance of HLA binding strength. PLoS ONE 6(10):e26494
Weiskopf D, Angelo MA, de Azeredo EL, Sidney J, Greenbaum JA, Fernando AN et al (2013) Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8 + T cells. Proc Natl Acad Sci USA 110(22):E2046–E2053
Sette A, Sidney J (1999) Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics 50(3–4):201–212
Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M (2007) Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinf 8:424
Khan MA, Hossain MU, Rakib-Uz-Zaman SM, Morshed MN (2015) Epitope-based peptide vaccine design and target site depiction against Ebola viruses: an immunoinformatics study. Scand J Immunol 82(1):25–34
Bui H-H, Sidney J, Li W, Fusseder N, Sette A (2007) Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinf 8:361
Bui H-H, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A (2006) Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinf 7:153
Adams HP, Koziol JA (1995) Prediction of binding to MHC class I molecules. J Immunol Methods 185(2):181–190
Maenaka K, Jones EY (1999) MHC superfamily structure and the immune system. Curr Opin Struct Biol 9(6):745–753
Stern LJ, Wiley DC (1994) Antigenic peptide binding by class I and class II histocompatibility proteins. Struct Lond Engl 1993 2(4):245–251
Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M et al (2010) Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinf 11:568
Wang P, Sidney J, Dow C, Mothé B, Sette A, Peters B (2008) A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol 4(4):e1000048
da Silva ANMR, Nascimento EJM, Cordeiro MT, Gil LHVG, Abath FGC, Montenegro SML et al (2009) Identification of continuous human B-cell epitopes in the envelope glycoprotein of dengue virus type 3 (DENV-3). PLoS ONE 4(10):e7425
Wu HC, Huang YL, Chao TT, Jan JT, Huang JL, Chiang HY et al (2001) Identification of B-cell epitope of dengue virus type 1 and its application in diagnosis of patients. J Clin Microbiol 39(3):977–982
Jiang L, Zhou J-M, Yin Y, Fang D-Y, Tang Y-X, Jiang L-F (2010) Selection and identification of B-cell epitope on NS1 protein of dengue virus type 2. Virus Res 150(1–2):49–55
Khan AM, Miotto O, Nascimento EJM, Srinivasan KN, Heiny AT, Zhang GL et al (2008) Conservation and variability of dengue virus proteins: implications for vaccine design. PLoS Negl Trop Dis 2(8):e272
Acknowledgements
The authors highly acknowledge the Indian Council of Medical Research for supporting facilities to carry out the research work in Biomedical Informatics Centre, National Institute of Pathology, New Delhi, vide RFC No. BIC/NTF/1/2013–2014 dated 18/02/2013.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pradhan, D., Yadav, M., Verma, R. et al. Discovery of T-cell Driven Subunit Vaccines from Zika Virus Genome: An Immunoinformatics Approach. Interdiscip Sci Comput Life Sci 9, 468–477 (2017). https://doi.org/10.1007/s12539-017-0238-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12539-017-0238-3