
Abstract
Using a recently developed genetically modified microorganisms (GMM) detection strategy, unexpected contaminations of unauthorized GMM in commercialized microbial fermentation products have been reported. A first-line real-time PCR screening analysis was initially performed to determine the presence of key targets frequently found in genetically modified (GM) bacteria. A second-line real-time PCR analysis was subsequently applied to identify specific GMM, including to date a GM Bacillus velezensis producing protease and a GM B. subtilis producing vitamin B2. In this study, an additional real-time PCR method specific to a newly identified GMM producing alpha-amylase was developed to be integrated in such second-line real-time PCR analysis, allowing to strengthen the GMM detection strategy. This method was successfully validated based on the assessment of its specificity and sensitivity performance. In addition, its applicability was confirmed using several food enzyme products commercialized on the market. Finally, via its transfer to an external laboratory, the transferability of the in-house validated method was positively evaluated, allowing its easy implementation in enforcement laboratories.
Similar content being viewed by others


Introduction
In the food and feed industry, genetically modified microorganisms (GMM), harbouring usually antimicrobial resistance (AMR) genes as selection marker, are frequently used to produce microbial fermentation products, including food and feed enzymes, additives and flavourings (Deckers et al. 2020a). According to the European (EU) legislation, the presence of GMM, both viable strains and associated recombinant DNA, is currently unauthorized in the food and feed chain (Regulation (EC) 2003/1829; Regulation (EC) 2003/1830). However, the unauthorized presence of a genetically modified (GM) Bacillus velezensis producing protease (RASFF2019.3332) and a GM Bacillus subtilis producing vitamin B2 (RASFF2014.1249, RASFF2014.1360, RASFF2014.1657, RASFF2018.2755, RASFF2019.0793, RASFF2019.3216) in commercialized microbial fermentation products was observed (Barbau-Piednoir et al. 2015a, b; Berbers et al. 2020; Fraiture et al. 2020d; Paracchini et al. 2017). In addition to the non-compliance with the EU legislation and the failure to respect consumers’ freedom of choice, safety concerns were raised because such GMM were characterized as carrying full-length AMR genes conferring a resistance to ampicillin, chloramphenicol, erythromycin, kanamycin, neomycin or bleomycin antibiotics. The presence of AMR genes in the food and feed chain increases seriously the risk of horizontal AMR gene transfers to environment, pathogens and gut microbiota (Bacanli and Basacan 2019; Barbau-Piednoir et al. 2015a, b; Berbers et al. 2020; Fraiture et al. 2020a, b, c, d, 2021a, b; Munita and Arias 2016; Paracchini et al. 2017; Rozwandowicz et al. 2018; Sharma et al. 2018; von Wrighta and Bruce 2003; Xiong et al. 2018).
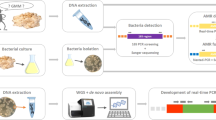
To guarantee the traceability and safety of the food and feed chain, a PCR-based strategy targeting recombinant DNA has therefore recently been proposed to highlight the potential presence of unauthorized GM bacteria carrying AMR genes. A first-line screening analysis is first performed to investigate the presence of key targets by real-time PCR, being frequently found in GM bacteria used by the food and feed industry to produce microbial fermentation products. This set of four key targets is composed of a cat gene (GenBank: NC_002013.1) conferring a resistance to chloramphenicol (CmR), an aadD gene (GenBank: M19465.1) conferring a resistance to both kanamycin and neomycin (KanR), a tet-L gene (GenBank: D00946.1) conferring a resistance to tetracycline (TetR) and the pUB110 shuttle vector (GenBank: M19465.1) carrying the aadD gene (Fraiture et al. 2020a, b, c, 2021b). The detection of at least one of these key targets allowed to strongly suspect GMM contaminations. Consequently, a second-line analysis is subsequently applied in order to identify specific GMM, using real-time PCR methods targeting their unnatural associations of sequences. Currently, this second-line analysis step is only performable for two GMM used for the production of microbial fermentation products, being the GM B. velezensis producing protease and the GM B. subtilis producing vitamin B2 (Barbau-Piednoir et al. 2015b; Fraiture et al. 2020d; Paracchini et al. 2017).
Recently, a new unauthorized GMM producing alpha-amylase was discovered using a PCR-based DNA walking strategy anchored on the pUB110 shuttle vector. In this manner, an unnatural association of sequences between the pUB110 shuttle vector (GenBank: M19465.1) and a Bacillus gene coding for alpha-amylase (GenBank: CP041693.1, J01542.1) was characterized, allowing proving and identifying the new unauthorized GMM producing alpha-amylase (Fraiture et al. 2021b). Based on this characterized sequence, a new real-time PCR method, specific to the GMM producing alpha-amylase, was developed in this study with the aim to be integrated in the second-line analysis of the GMM detection strategy. This novel real-time PCR method was in-house validated, including specificity and sensitivity assessments. Using several commercialized food enzyme products, the applicability of the proposed real-time PCR method was then investigated. Finally, in order to be easily implemented in enforcement laboratories, the transferability of the in-house validated real-time PCR method was tested.
Materials and Methods
Materials
DNA from the control plasmid (Genecust, FRA-GS-200729), artificially synthetized to carry one copy of the targeted sequences to detect the GMM producing alpha-amylase, was used under its intact form (Oldham and Duncan 2012) (Tables 1, 2, 3). DNA from Homo sapiens (G3041 Promega), Zea mays (reference material ERM-BF413ak), wild-type microbial species and GM bacterial strains (B. subtilis RASFF2014.1249 and B. velezensis RASFF2019.333) were obtained as previously described (Table 2) (Fraiture et al. 2020a, b, c, d, 2021b). Commercialized food enzyme preparation products labelled as containing at least alpha-amylase (sample nos. 1–7) or only protease (sample no. 8) were collected from the market (Table 4). These products, in liquid or solid forms, were intended for use in various sectors (brewing, distillery, fruit processing and baking) and were from different brands. DNA from these products was extracted using the NucleoSpin Food kit (Macherrey-Nagel) associated to an initial bead beating step (Precellys® Glass kit 0.5 mm) of 2 × 2 min at 5000 rpm (MiniLys, Bertin Instruments) (Table 4). For all materials, DNA concentration was measured by spectrophotometry using Nanodrop® 2000 (Thermo Fisher) and DNA purity was evaluated using the A260/A280 and A260/A230 ratios (values > 1.8). In addition, for the control plasmid, the concentration was measured by fluorometry using Qubit 3.0 Fluorometer (Thermo Fisher).
Real-time PCR Assays
Each real-time PCR assay was performed in a standard 25 μl reaction volume containing 1X TaqMan® PCR Mastermix (Diagenode), 400 nM of each primer (Eurogentec), 200 nM of the probe (Eurogentec) and 5 μl of DNA. The real-time PCR program consisted of a single cycle of DNA polymerase activation for 10 min at 95 °C followed by 45 amplification cycles of 15 s at 95 °C (denaturing step) and 1 min at 64 °C (for the first-line screening analysis including KanR and pUB110 markers) or 60 °C (for the second-line analysis including GMM protease and GMM alpha-amylase markers) (annealing-extension step) (Table 1, Supplementary file 1) (Fraiture et al. 2020b, d, 2021b). All runs were performed on a CFX96 Touch Real-Time PCR Detection System (BioRad). For each assay, positive controls and NTC (no template control) were included.
Development of the Real-time PCR Method Specific to the GMM Producing Alpha-Amylase
To develop the GMM alpha-amylase marker, a set of primers and probe were designed, using the software Primer3, to cover 139 bp of the transgene flanking region between the pUB110 shuttle vector and a Bacillus gene coding for alpha-amylase (Table 1). This marker was developed to be compatible with the real-time PCR conditions of the second-line analysis of the GMM detection strategy (section “Real-time PCR Assays”).
Validation of the Real-time PCR Method Specific to the GMM Producing Alpha-Amylase
For the specificity assessment, an in silico analysis was first performed where the amplicon sequence was blasted against the NCBI database (Refseq_genomes) from fungi, yeast and bacteria with default parameters (Table 1; Supplementary file 2). Then, the specificity of the method was experimentally assessed using a set of materials from Homo sapiens, Zea mays and 81 wild-type microbial species frequently used by the food and feed industry to produce microbial fermentation products, and GM Bacillus strains (RASFF2014.1249 and RASFF2019.333). For each material, 10 ng of DNA (including at least 5000 genome copies of non-targeted and targeted DNA) was tested in duplicates (Table 2) (ENGL 2015; Li and Du 2014). Such DNA were previously assessed as amplifiable (Deckers et al. 2020b, c; Fraiture et al. 2020d, 2021b). As positive controls, 10 ng of DNA from a commercialized alpha-amylase product (Dextzyme HT, being sample no. 1 in Table 4) and 5 ng of DNA from the control plasmid were tested also in duplicates. The amplicon generated from sample no. 1, previously purified using USB ExoSAP-IT PCR Product Cleanup (Affymetrix), was also sequenced on a Genetic Sequencer 5000 (ThermoFisher) using the Big Dye Terminator Kit v3.1 (Applied Biosystems). The generated sequence was aligned against the targeted reference sequence using MUSCLE (Fraiture et al. 2021b) (Table 1, Supplementary file 3). The generated sequence was considered similar to the reference sequence when a minimum identity and coverage of 99% were observed.
For the sensitivity assessment, serial dilutions of the control plasmid DNA (Genecust, FRA-GS-200729), from 162 to 0.1 estimated target copy number, were prepared (Table 3). The calculation of the estimated target copy number was based on the control plasmid size (2867 bp) and the formula mentioned in Barbau-Piednoir et al. (2015b). Each dilution point was tested in 12 replicates. The limit of detection LOD95%, defined as the number of copies of the target required to ensure a 95% probability of detection (POD), was determined by using Quodata web application (Supplementary file 4A) (Uhlig et al. 2015; Grohmann et al. 2016; https://quodata.de/content/validation-qualitative-pcr-methods-single-laboratory).
Applicability Assessment
For the applicability assessment of the real-time PCR specific to the GMM producing alpha-amylase, 10 ng of DNA from commercialized food enzyme products collected from the market was tested in duplicates (Table 4). To verify their identity, the generated amplicons were purified, sequenced and aligned against the reference targeted sequence as described in the section “Real-time PCR Assays” (Supplementary file 3).
Transferability Assessment
The transferability assessment of the real-time PCR specific to the GMM producing alpha-amylase consisted of a repetition of the sensitivity assessment by an external laboratory, being here the Unità Operativa Semplice a valenza Direzionale - Ricerca e controllo degli organismi geneticamente modificati at the Istituto Zooprofilattico Sperimentale del Lazio e della Toscana “M.Aleandri” (Rome, Italy) (Table 3, Supplementary file 4B). The same serial dilutions of the control plasmid DNA (genecust, FRA-GS-200729), from 162 to 0.1 estimated target copy number, prepared for the in-house validation were also used by the external laboratory. All reagents were ordered by the external laboratory, including oligonucleotides (Metabion International AG) and TaqMan™ Universal PCR Master Mix (Applied Biosystems®). All runs were performed on a QuantStudio™ 7 Flex Real-Time PCR System (Life Technologies).
Conventional PCR and Sequencing
The presence of the full-length aadD gene was assessed using the KanR marker as previously described (Fraiture et al. 2020b). For each sample, 10 ng of DNA was tested in duplicates (Table 4). Each PCR assay was applied on a standard 25 μl reaction volume containing 1X Green DreamTaq PCR Master Mix (ThermoFisher Scientific), 400 nM of each primer (Eurogentec) and 5 μl of DNA. The PCR program consisted of a single cycle of 1 min at 95 °C (initial denaturation) followed by 35 amplification cycles of 30 s at 95 °C (denaturation), 30 s at 60°C (annealing) and 1 min at 72 °C (extension) and finishing by a single cycle of 5 min at 72 °C (final extension). The run was performed on a Swift MaxPro Thermal Cycler (Esco). For each assay, positive controls and NTC (no template control) were included. The final PCR products were visualized by electrophoresis using the Tapestation 4200 device with the associated D1000 Screen Tape and reagents (Agilent) (Supplementary file 5). Final PCR products, earlier purified using USB ExoSAP-IT PCR Product Cleanup (Affymetrix), were sequenced on a Genetic Sequencer 5000 using the Big Dye Terminator Kit v3.1 (Applied Biosystems). The generated sequences were aligned against the reference aadD gene sequence (GenBank: M19465.1) using MUSCLE (Supplementary file 5). The generated sequences were considered as similar to the reference sequence when a minimum identity and coverage of 99% were observed.
Results and Discussion
Development of the Real-time PCR Method
A new real-time PCR method was developed to target specifically the unauthorized GMM producing alpha-amylase based on its previously characterized transgene flanking region between the pUB110 shuttle vector (GenBank: M19465.1) and a Bacillus gene coding for alpha-amylase (GenBank: CP041693.1, J01542.1). The designed set of primers and probe generated an amplicon of 139 bp covering this unnatural association of sequences (Table 1) (Fraiture et al. 2021b). This real-time PCR method was developed to be compatible with the real-time PCR conditions of the second-line analysis of the current GMM detection strategy, allowing strengthening the latter. The developed method was further in-house validated.
Specificity Assessment
The specificity of the generated amplicon was first confirmed in silico as no hits of 100% in terms of identity and recovery were observed following its BLAST against the NCBI database (Supplementary data 2). As expected, one part of the amplicon sequence (72 bp) corresponded to the pUB110 shuttle vector (GenBank: M19465.1), which is naturally carried by Staphylococcus aureus, and the other part of the amplicon sequence (67 bp) corresponded to a Bacillus gene coding for alpha-amylase (GenBank: CP041693.1, J01542.1). Both parts of this amplicon sequence are individually found naturally while their association is considered unnatural. A similar type of GM construct including the pUB110 shuttle vector (GenBank: M19465.1), carrying the aadD gene, flanked to a Bacillus gene coding for a protease (GenBank: WP_032874795.1), was previously found in the GM B. velezensis producing protease (RASFF2019.3332) (Fraiture et al. 2020d).
The specificity of the method was then experimentally tested (Table 2). For positive controls, DNA from a commercialized alpha-amylase product (Dextzyme HT; sample no. 1 in Table 4) and from a control plasmid, artificially synthetized to carry one copy of the targeted sequence, was used. For negative controls, DNA from 91 wild-type microbial strains frequently used to produce microbial fermentation products, the GM B. subtilis producing vitamin B2 (RASFF 2014.1249 strain), the GM B. velezensis producing protease (RASFF2019.3332 strain), plant (Zea mays) and human was used (EFSA 2017; Fraiture et al. 2020a, b, c, d, 2021b). No amplification was observed for all negative controls while each positive control presented an amplification (Table 2). With 100% identity and coverage to the target reference sequence (Fraiture 2020 DNA walking), the identity of the generated amplicon from the commercial alpha-amylase product (Dextzyme HT; sample no. 1 in Table 4) was also confirmed by sequencing, which is particularly important in the context of this validation process as no official reference material was available (Supplementary file 3).
Based on these results, only the targeted sequence was detected with the proposed method as well as no false negative (at levels above LOD) and false positive were observed. Therefore, the proposed method was successfully proved to be specific.
Sensitivity Assessment
The sensitivity of the proposed real-time PCR method was tested using plasmid DNA, from the control plasmid carrying only one copy of the targeted sequence, at different estimated target copy numbers (162, 20, 10, 5, 1, 0.1 and 0) (Table 3). For all 12 replicates, an amplification was observed as low as 20 estimated target copies. LOD95% of the method was determined at 17 estimated target copies per reaction, complying with the “Minimum Performance Requirements for Analytical Methods of GMO Testing” of the European Network of GMO Laboratories (ENGL 2015). On this basis, the proposed method was successfully assessed as sensitive.
Applicability Assessment
The potential presence of the unauthorized GMM producing alpha-amylase was investigated in several commercialized food enzyme preparation products labelled as containing at least alpha-amylase (sample nos. 1–7) or exclusively protease (sample no. 8) (Table 4). These products, in liquid or solid forms, were intended for use in various sectors (brewing, distillery, fruit processing and baking) and were from different brands.
As a first-line real-time PCR screening analysis, the potential presence of GMM was first assessed using two key targets, being the aadD gene (GenBank: AAA88361.1) conferring KanR and the pUB110 shuttle vector (GenBank: M19465.1) carrying the aadD gene. These two key targets were previously identified as being commonly harboured by GM bacteria used by the food and feed industry to produce microbial fermentation products. Moreover, both of these key targets were recently reported as being carried by the GMM producing alpha-amylase (Fraiture et al. 2020b, d, 2021b). For all tested food enzyme preparation samples (nos. 1–7), except sample no. 8, the presence of both the aadD gene in its full-length and the pUB110 shuttle vector was observed, suggesting a potential GMM contamination (Table 4, Supplementary file 5).
A second-line real-time PCR analysis was subsequently performed to determine the presence of the GMM producing alpha-amylase (Fraiture et al. 2020b, d, 2021b). As expected, no signal for the GMM producing alpha-amylase was observed for sample no. 8 for which the presence of the aadD gene and the pUB110 shuttle vector was not detected in the first-line real-time PCR screening analysis (Table 4). For all suspicious samples presenting in the first-line real-time PCR screening analysis a positive signal for both key targets (nos. 1–7), the presence of the GMM producing alpha-amylase was confirmed (Table 4). In addition, the sequence identity of the generated amplicons was confirmed by sequencing, which is especially important in the frame of this validation process as no official reference material was available (Supplementary file 3). These results, leading to several RASFF notifications, allowed pointing out the presence of unauthorized GMM in numerous food products commercialized on the EU market, for which there is zero tolerance in the food and feed chain at any contamination levels (Table 4).
According to these results, the applicability of the in-house validated real-time PCR method targeting the GMM producing alpha-amylase was successfully demonstrated. Moreover, based on the numerous GMM contaminations observed in the various tested commercial samples labelled as containing alpha-amylase, these results suggest a high frequency of GMM contaminations in commercial microbial fermentation product available on the market. Consequently, the importance of using the novel in-house validated method specific to the GMM producing alpha-amylase was highlighted.
Transferability Assessment
In order to evaluate whether the in-house validated method performs correctly in an external laboratory with different operators using different equipment and reagents, a transferability study was carried out. To this end, the same experimental setup as for the in-house sensitivity assessment was used. The results obtained by the external laboratory were comparable with the ones observed during the in-house validation. An amplification was observed as low as 20 estimated target copies for all 12 replicates (Table 3). Moreover, the LOD95% of the transferred real-time PCR method was determined at 14 estimated target copies. This result is highly comparable to the LOD95% of the developed real-time PCR method determined at 17 estimated target copies as observed in the in-house sensitivity assessment (Supplementary file 4). These results demonstrated thus the successful transferability of the novel method.
Conclusion
In the present study, we successfully developed and validated a real-time PCR method targeting specifically an unauthorized GMM producing alpha-amylase. The novel method was designed to be compatible with the PCR conditions of the second-line analysis from the current GMM detection strategy, allowing to strengthen the latter (Fraiture et al. 2020d). The novel method was assessed as being specific, sensitive and applicable in microbial fermentation products and complies with the criteria for GMO detection methods as described in “Minimum Performance Requirements for Analytical Methods of GMO Testing” of the European Network of GMO Laboratories (ENGL 2015). The novel in-house validated method was also positively assessed as transferable to an external enforcement laboratory. This successful transferability to another enforcement laboratory constitutes a necessary prerequisite for a full validation of a method in order to be implemented in all enforcement laboratories (Broeders et al. 2014).
Using the strengthened GMM detection strategy on various commercial products containing alpha-amylase, a high frequency of GMM contaminations on the market with the GMM producing alpha-amylase was observed. Therefore, the present study clearly highlights the importance to pursue and extend the control of GMM in order to guarantee the traceability and the safety of the food and feed chain.
References
Bacanli M, Basacan N (2019) Importance of antibiotic residues in animal food. FCT 125:462–466
Barbau-Piednoir E, De Keersmaecker SCJ, Wuyts V, Gau C, Pirovano W, Costessi A, Philipp P, Roosen NH (2015a) Genome sequence of the EU-unauthorized genetically modified Bacillus subtilis strain 2014–3557 overproducing riboflavin, isolated from an imported lot of Vitamin B2 80 % feed additive. Genome Announc 3:e00214–e00215
Barbau-Piednoir E, De Keersmaecker SCJ, Delvoye M, Gau C, Philipp P, Roosens NHC (2015b) Use of next generation sequencing data to develop a qPCR method for specific detection of EU-unauthorized genetically modified Bacillus subtilis overproducing riboflavin. BMC Biotechnol 15:103
Berbers B, Saltykova A, Garcia-Graells C, Philipp P, Arella F, Marchal K, Winand R, Vanneste K, Roosens NHC, De Keersmaecker SCJ (2020) Combining short and long read sequencing to characterize antimicrobial resistance genes on 2 plasmids applied to an unauthorized genetically modified Bacillus. Sci Rep 10:4310
Broeders S, Huber I, Grohmann L, Berben G, Taverniers I, Mazzara M, Roosens N, Morisset D (2014) Guidelines for validation of qualitative real-time PCR methods. Trends Food Sci 37:115–126
Deckers M, Deforce D, Fraiture MA, Roosens NHC (2020a) Genetically modified micro-organisms for industrial food enzyme. Foods 9:326
Deckers M, Vanneste K, Winand R, De Keersmaecker SCJ, Denayer S, Heyndrickx M, Deforce D, Fraiture MA, Roosens NHC (2020b) Strategy for the identification of micro-organisms producing food and feed products: bacteria producing food enzymes as study case. Food Chem 305:125431
Deckers M, Vanneste K, Winand R, Hendrickx M, Becker P, De Keersmaecker SCJ, Deforce D, Fraiture MA, Roosens NHC (2020c) Screening strategy targeting the presence of food enzyme-producing fungi in food enzyme preparations. Food Control 117:107295
EFSA Panel on Biological Hazards (2017) Scientific Opinion on the update of the list of QPS-recommended biological agents intentionally added to food or feed as notified to EFSA. ej EFSA J 15:4664
ENGL (2015) Definition of minimum performance requirements for analytical methods for GMO testing. http://gmo-crl.jrc.ec.europa.eu/doc/MPR%20Report%20Application%2020_10_2015.pdf
Fraiture MA, Deckers M, Papazova N, Roosens NHC (2020a) Detection strategy targeting a chloramphenicol resistance gene from genetically modified bacteria in food and feed products. Food Control 18:106873
Fraiture MA, Deckers M, Papazova N, Roosens NHC (2020b) Are antimicrobial resistance genes key targets to detect genetically modified microorganisms in fermentation products? Int J Food Microbiol 331:108749
Fraiture MA, Deckers M, Papazova N, Roosens NHC (2020c) Strategy to detect genetically modified bacteria carrying tetracycline resistance gene in fermentation products. Food Anal Methods 13:1929–1937
Fraiture MA, Bogaerts B, Winand R, Deckers M, Papazova N, Vanneste K, De Keersmaecker SCJ, Roosens NHC (2020d) Next-generation sequencing: a key tool to identify unauthorized genetically modified microorganisms in food enzyme preparations. Sci Rep 10:7094
Fraiture MA, Joly L, Vandermassen E, Delvoye M, Van Geel D, Michelet JY, Van Hoeck E, De Jaeger N, Papazova N, Roosens N (2021a) Retrospective survey of unauthorized genetically modified bacteria harbouring antimicrobial resistance genes in feed additive vitamin B2 commercialized in Belgium: Challenges and solutions. Food Control 119:107476
Fraiture MA, Papazova N, Roosens NHC (2021b) DNA walking strategy to identify unauthorized genetically modified bacteria in microbial fermentation products. Int J Food Microbiol 337:108913
Grohmann L, Broll H, Dagand E, Hildebrandt S, Hübert P, Kiesecker H, Lieske K, Mäde D, Mankertz J, Reiting R, Schulze M, Speck B, Uhlig S, Wahler D, Waiblinger HU, Woll K, Zur K (2016) Guidelines for the validation of qualitative real-time PCR methods by means of a collaborative study. Technical report BVL 1
Li XQ, Du D (2014) Variation, evolution, and correlation analysis of C+G content and genome or chromosome size in different kingdoms and phyla. PLoS One 9(2):e88339
Munita JM, Arias CA (2016) Mechanisms of antibiotic resistance. Microbiol Spectr 4:2
Oldham AL, Duncan KE (2012) Similar gene estimates from circular and linear standards in quantitative PCR analyses using the prokaryotic 16S rRNA gene as a model. PLoS One 7(12):e51931
Paracchini V, Petrillo M, Reiting R, Angers-Loustau A, Wahler D, Stolz A, Schönig B, Matthies A, Bendiek J, Meinel DM, Pecoraro S, Busch U, Patak A, Kreysa J, Grohmann L (2017) Molecular characterization of an unauthorized genetically modified Bacillus subtilis production strain identified in a vitamin B2 feed additive. Food Chem 230:681–689
RASFF portal < https://webgate.ec.europa.eu/rasff-window/portal/?event=SearchForm&cleanSearch=1>
Regulation (EC) No 1829/2003 of the European Parliament and of the Council of 22 September 2003 on genetically modified food and feed. Official Journal of the European Union L268:1-23.
Regulation (EC) No 1830/2003 of the European Parliament and of the Council of 22 September 2003 concerning the traceability and labelling of genetically modified organisms and the traceability of food and feed products produced from genetically modified organisms and amending Directive 2001/18/EC. Official Journal of the European Union L268:24-28.
Rozwandowicz M, Brouwer MSM, Fischer J, Wagenaar JA, Gonzalez-Zorn B, Guerra B, Mevius DJ, Hordijk J (2018) Plasmids carrying antimicrobial resistance genes in Enterobacteriaceae. J Antimicrob Chemother 73:1121–1137
Sharma C, Rokana N, Chandra M, Singh BP, Gulhane RD, Gill JPS, Ray P, Puniya AK, Panwar H (2018) Antimicrobial resistance: its surveillance, impact, and alternative management strategies in dairy animals. Front Vet Sci 4:237
Uhlig S, Frost K, Colson B, Simon K, Made D, Reiting R, Gowig P, Grohmann L (2015) Validation of qualitative PCR methods on the basis of mathematical–statistical modelling of the probability of detection. Accred Qual Assur 20:75–83
von Wrighta A, Bruce A (2003) Genetically modified microorganisms and their potential effects on human health and nutrition. Trends Food Sci Technol 14:264–276
Xiong W, Sun Y, Zeng Z (2018) Antimicrobial use and antimicrobial resistance in food animals. Environ Sci Pollut Res 25:18377–18384
Acknowledgements
The Sanger sequencing was performed at the Transversal activities in Applied Genomics Service at Sciensano. The authors would like to thank Patrick Philipp (Service Commun des Laboratoires, France) and Marc Heyndrickx (ILVO, Belgium) for their kindness to provide a commercialized food enzyme preparation (sample no. 6) and bacterial strains, respectively.
Funding
The research that yielded these results was funded by both the Belgian Federal Public Service of Health, Food Chain Safety and Environment through the contract [RT 17/5 SPECENZYM] and the Transversal activities in Applied Genomics (TAG) Service from Sciensano.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Marie-Alice Fraiture declares that she has no conflict of interest. Ugo Marchesi declares that he has no conflict of interest. Daniela Verginelli declares that she has no conflict of interest. Nina Papazova declares that she has no conflict of interest. Nancy Roosens declares that she has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 300 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fraiture, MA., Marchesi, U., Verginelli, D. et al. Development of a Real-time PCR Method Targeting an Unauthorized Genetically Modified Microorganism Producing Alpha-Amylase. Food Anal. Methods 14, 2211–2220 (2021). https://doi.org/10.1007/s12161-021-02044-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-021-02044-x