
Abstract
This preliminary study describes the use of high resolution and accuracy mass spectrometry techniques combined with new generation chemical software products for detecting and identifying contaminants in food commodities. As a first step, the extracts of routine target analysis samples (obtained in our official laboratory responsible for food residues control) were acquired and processed with this method in order to search unknown and non-targeted contaminants in food. In order to verify the feasibility of the presented method, the research has been firstly addressed to untargeted pesticides and their metabolites in stone fruits commodities and tomatoes. The differential analysis carried with Compound Discoverer 2.0 between the investigated unknown sample and the blank matrix sample allowed to remove all the matrix molecular components; Aggregated Computational Toxicology Resource (ACToR) helped to understand and predict chemical interpretation of substances. The acquisition in FullScan-AIF and FullScan-ddMS2 allowed the clear detection and identification of isobaric compounds such as quinalphos and phoxim. In order to verify that the proposed method is suitable to the scope of application, the main points of SANTE/11813/2017 Document have been followed. The results demonstrate that no false positives and no false negatives have been detected from the analysis of samples spiked with 55 pesticides at 0.010 and 0.10 mg kg−1. This preliminary study has been also tested with a Proficiency Test (EUPT-FV-SM08) and, according to EUPT-FV-SM08 Final Report, our laboratory has been included in the 67% (56) that clearly detected over 70% pesticides. Finally, this method has been extended to other matrices and contaminants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The market globalization is brought to global hazards, in particular for chemicals; furthermore, international legislation is not harmonized, so some compounds are allowed in some countries and not in others. This leads to consistent risk to introduce in European foods and feeds containing banned compounds or concentrations above the maximum residue levels and a wide number of alert notifications in the Rapid Alert System for Food and Feed (RASFF) for chemical substances and microorganisms (EC Regulation No. 178 /2002).
In order to cope with this new reality, laboratories need to adapt their technologies and analytical approaches to guarantee a suitable degree of safety for consumers. It involves many aspects: legislation enforcement regarding the presence of selected compounds below maximum residue limits (MRLs), the determination of allergens, and the detection of environmental contaminants or natural toxins.
The classic analytical approach for the determination of contaminants in foods involves target analysis based on chromatographic techniques (GC or LC) coupled with tandem mass spectrometry (MS) detection. These procedures allow the quantification of known compounds with great selectivity and sensitivity (Castro-Puyana et al. 2017).
In this regard, the use of non-target MS approaches allowed significant improvements of the analytical determinations. Due to the increasing number of unexpected compounds in food matrices, the untargeted approach by means of high-resolution accurate MS (HRAMS) techniques, combined with new generation software, appears suitable to satisfy the international legislation on food safety, and therefore to enhance the safety level for consumers (Kaufmann 2012; Kaufmann 2014; Rajski et al. 2014; Bletsou et al. 2015; Lehotay et al. 2015; Zomer and Mol 2015; Knolhoff and Croley 2016).
Nevertheless, up to now, the achievement of reliable data using this approach has often been a difficult task, considering that for each molecular formula, a wide range of compounds can be generated, involving, as a consequence, laborious data spectra interpretations and limiting the applicability of this approach to routine analysis (Weissberg and Dagan 2011). According to Kaufmann (2010), the capability for interpreting the elemental composition of a specific ion decreases when the molecular weight and the contained atoms increase. For instance, ions of m/z 250.050 and m/z 500.100, with a mass measurement error of up to 5 m/z units (and assumed to contain C, H, N, O, S atoms), show 36 and 167 possible compositions, respectively. Lehotay et al. (2015) reported some possible molecular formula for exact mass of aflatoxin B1 ± 2 mDa and the number of structures in ChemSpider for each molecular formula.
However, thanks to the combined development of instruments with higher accuracy and resolving power (Gómez-Ramos et al. 2013) and new generation and dedicated software (Milman 2015), the number of possible compositions is strongly reduced (Herrera-Lopez et al. 2014; Fu et al. 2016; Knolhoff et al. 2016; Fu et al. 2017).
The aim of this study is to prove the efficacy of non-targeted approach for detecting and identifying undesirable xenobiotics in food matrices, in order to satisfy the recent requirements set in the programs of food safety. In this first phase of study, stone fruits and tomatoes have been analysed.
For this purpose, we present the development of high-resolution accurate mass spectrometry (HRAMS) approach to enhance the number of contaminants that can be detected by means of untargeted screening methods accomplished by UHPLC-Orbitrap (Q Exactive™) followed by data processing analysis with dedicated compound databases. Empirical formulas of detected molecules were, then, generated through a combination of parameters such as mass accuracy, isotopic clusters, and ion fragments in order to be used for the online research database.
Materials and Methods
Reagents and Materials
Acetonitrile (ACN) LC-MS grade, methanol (MeOH) LC-MS grade, water LC-MS grade, acetic acid (AAc), and formic acid (FAc) for mass spectrometry were obtained from Sigma Aldrich (Steinheim, Germany).
QuEChERS Extraction Packets 5982-7650 (4 g of magnesium sulphate, 1 g of sodium citrate, 0.5 g sodium hydrogen citrate sesquihydrate, 1 g of sodium chloride) and Dispersive SPE 15 mL fruit and vegetable 5982-5056 (150 mg of PSA and 900 mg magnesium sulphate), used in the clean-up procedure, were obtained from Agilent Technologies (Santa Clara, USA).
Individual standards were obtained from Dr. Ehrenstorfer (Augsburg, Germany), Sigma Aldrich (Steinheim, Germany), ORSELL (Modena, Italy), Cerilliant (Darmstadt, Germany), LGC Standards (Wesel, Germany), Australian Government National Measurement Institute (Sydney, Australia), and Cayman Chemical (Michigan, USA), and the respective individual stock standard solutions (1000–2000 mg L−1) were prepared in appropriate solvent (see Supplementary Information Table S1).
Finally, Pierce LTQ Velos ESI Positive Ion Calibration Solution was provided by Thermo Fisher Scientific (Waltham, MA, USA).
UHPLC-HRAMS Analysis
The UHPLC chromatographic run was carried out using Dionex™ Ultimate 3000 (Thermo Scientific™, San Jose, USA) equipped with an analytical column Thermo Scientific™ Accucore™ aQ C18 column (100 × 2.1 mm with particle diameter of 2.6 μm). The oven temperature was set at 40 °C, and the injected sample volume was 10 μL. The autosampler temperature was set at 15 °C. The mobile phases were: (A) water and (B) methanol both containing 5 mm ammonium formate and 0.1% of FAc; the flow rate was 0.4 mL min−1.
The following gradient elution scheme was used: at the beginning, 20% phase B was constant for 0.5 min, and it was increased up to 98% in 10 min. The latter was maintained for 4 min, and then switched back to the initial 20% in 0.5 min and kept constant for 4 min.
The UHPLC system was connected to the single stage Orbitrap mass spectrometer Q Exactive™ from Thermo Fisher Scientific (Bremen, Germany) through a heated electrospray interface (HESI-II) operating in positive ionization (Makarov and Scigelova 2010). The HESI parameters in positive polarity were the following: electrospray voltage of 3.2 kV; sheath gas of 40 arbitrary units; and auxiliary gas of 25 arbitrary units.
The analysis was performed with 4 consecutive UHPLC-HRAMS runs: 3 with a FullScan-All Ion Fragmentation (FullScan-AIF); 1 in Data Dependent Scan Mode (ddMS2).
The FullScan-AIF runs were acquired with resolving power of 140,000 FWHM for parental ions and mass range of 110–950 m/z, AGC target of 3e6, max IT 500 ms; the FullScan-AIF acquisition of all fragments was set with a resolving power of 35,000 FWHM and 63.3–700 m/z as mass range, AGC target of 3e6, max IT 150 ms.
The ddMS2 run was set with a “Homemade Exclusion list” of 70 compounds (see Supplementary Information Table S2) in a mass range from 110 to 950 m/z and carried out in data dependent scan mode (ddMS2) with resolving power of 70,000 FWHM for parental ions and 17,500 FWHM for all fragmentation products, using a mass accuracy ≤ 2 ppm. The exclusion list includes all the ions that are present in the “environmental-laboratory system”, and it was generated by performing the blank reagent chromatographic run.
All the chromatographic runs were carried out using a stepped energy collision of 20, 35, and 60 eV.
Sample Preparation and Comparison of Extraction Protocols
Four extraction protocols were compared: (a) “Dilute-and-shoot” methanol extraction (MeOH); (b) “Dilute-and-shoot” acetonitrile extraction (ACN); (c) QuEChERS (EN 15662: 2008) without clean-up; and (d) QuEChERS (EN 15662: 2008) with clean-up.
The following procedure was applied for all the extraction protocols: 10 g of homogenized sample was weighed in a 50 mL PTFE centrifuge tube; then, 10 mL of the solvent related to the specified protocol were added to the sample that was shaken in an automatic axial extractor (AGYTAX®, Cirta Lab. S.L., Spain) for 15 min.
Then, for (a) and (b) protocols, the sample was centrifuged at 4500 rpm for 10 min at 4 °C, and the recovered supernatant was injected in UHPLC-HRAMS system after diluting it 1:10 in water.
For (c) and (d) protocols, the sample was added with QuEChERS Extraction Packets and then shaken in the automatic axial extractor for 10 min. The sample was centrifuged at 4500 rpm for 10 min at 4 °C, and the recovered supernatant, diluted 1:10 in water, was injected in LC-MS for protocol (c). Alternatively, for protocol (d), 7 mL of the supernatant was transferred to a 15-mL PTFE centrifuge tube containing 900 mg of MgSO4 and 150 mg of PSA. Firstly shaken in a vortex for 60 s, the extract was then centrifuged at 4500 rpm for 10 min, and the recovered supernatant was injected in LC-MS after 1:10 dilution in water.
The extraction protocol was selected in order to have not only the best recovery of analytes but also the best mapping of the matrix components (Gómez-Ramos et al. 2016).
Matrix Blank and Blind Samples
In the first step of this work, the number of contaminants and the kind of matrices have been limited to analytes and food commodities that are routinely analysed in our laboratory, in order to verify if this approach fit for purpose.
For these reasons, 4 reagent blanks and 6 matrix blank samples, belonging to stone fruit commodities, were extracted with the mentioned extraction protocols. Assessment of the samples as “blank” was previously carried out using the multiresidue targeted approach by means of UHPLC-HRAMS (EN 15662:2008). Each matrix blank sample was weighted in order to have 4 portions that were processed by the four different extraction protocols (6 × 4 = 24 portions).
After this step, because the mapping of the matrix components has not shown great differences, 6 matrix blank samples were mixed together in order to have a representative matrix. The samples were spiked at 0.1 mg kg−1 with a standard mix of 14 pesticides having a wide range of chemical-physical properties (see Supplementary Information Table S1). These samples were divided in 4 groups constituted by 2 samples each, in order to be processed by the four extraction protocols. This step was performed in order to obtain a blind sample.
Statistical Analysis and Database Searching
In this work, the following software and databases were used: Thermo Xcalibur™ 3.1 Software, Trace Finder 3.3, Compound Discoverer 2.0 and Mass Frontier 7.0 from Thermo Fisher Scientific (Houston Texas); m/z Cloud Advanced Mass Spectral Database from High Chem LLC, Slovakia; Aggregated Computational Toxicology Resource (ACToR); DrugBank; EPA ToxCast; FDA UNII - NLM; FooDB; Pesticide Common Names; PubChem and Chemistry World from the Royal Society of Chemistry (Judson et al. 2008).
Compound Discoverer 2.0 Workflow
In order to start the statistical analysis of the chromatograms through Compound Discoverer 2.0, a workflow has been defined starting from the definition of parameters, called “nodes” (Thermo Scientific 2015).
The workflow tree is reported in supplementary information (Fig. S1).
The first step for the building of the workflow is the choice of “Input File” that is the raw file generated during the acquisition of the sample through Thermo Xcalibur™ 3.1 Software and that can be imported and processed in Compound Discoverer 2.0.
Our workflow starts with the first node called “Spectrum Files” that reads all the spectrum information contained in the raw data file.
The third node is “Retention Time Aligner” that provides the workflow with an alignment between the sample and the matrix blank chromatograms.
The “Unknown Detector” node identifies unknown compounds by means of different parameters such as mass and intensity tolerance, signal/noise threshold, minimum peak intensity, ions, minimum and maximum element counts, filter peaks, maximum peak width, minimum scans per peak, and minimum isotopes. This node allows the extraction of extracting ion current (XIC) from the Full Scans acquisitions and has been connected with the other nodes with similar functions (“Group Unknown Compounds” and “Merge Features”).
At the same time, this node is also connected with “Search ChemSpider” node that is useful to search mass spectral databases for matching compounds within a specified mass tolerance range or with a certain elemental composition. As set of databases and software application, ACToR has been used.
In this work, the “Differential Analysis” node has been used as post-processing node. It calculates the statistics for performing a differential analysis (fold change, ratio, p values, and so on), stores this data in the result file, and creates a volcano plot by using the data stored in the compounds.
In this workflow, all parameters were set according to the default parameters of Compound Discoverer 2.0, except for mass tolerance that has been set at 5 ppm for all nodes in which it was required. In addition for the elements counts, C, H, Br, Cl, K, N, Na, O, P, and S have been set; regarding the S/N threshold, it was set at 3; the minimum peak intensity has been set at 1,000,000; ACToR has been used as set of databases and software applications to help chemical structure interpretation. In particular, DrugBank and pesticide common names are search oriented to the specific group of analytes and can also be used at a later time for retrospective analysis. In the Differential Analysis node, p value has been set at 2.5E-06 and log2 fold change set at 2.
Validation Approach-Quality Control
In order to verify the suitability of the proposed method for the scope of application (detect and identify xenobiotics), the main points of SANTE/11813/2017 Document (SANTE 2017) have been followed, taking into consideration that for the qualitative screening methods, the acceptable false-negative rate is 5%. To this aim, 14 samples were weighted, and each one spiked at 0.01 and 0.10 mg kg−1 (14 × 2 = 28 aliquots) with a standard mix of 55 pesticides of different chemical-physical properties (see Supplementary Information Table S1) and processed with the above reported procedure.
This protocol was also applied to EUPT-FV-18 Proficiency Test sample (test item spinach) (European Union Proficiency Test in Fruits and Vegetables 18 2016) and compared with the targeted approach.
Proficiency Test
This approach was also proved with the EUPT-FV-SM08 Screening Proficiency Test whose item was spinach (European Union Proficiency Test in Fruits and Vegetables Screening Methods 08 2017). The unknown approach was compared with a targeted screening method in which the compound database used was EFS_HRAM_Compound_Database.cdb (Thermo Fisher Scientific) and contains 1024 compounds.
Method Upgrade to Other Matrices and Contaminants
Furthermore, this method has also been tested on different matrices and on different potential contaminants belonging to different classes in order to assess the performances for non-target applications. With this purpose, 2 tomato samples were spiked at 0.02 mg/kg with a mix containing 3 drugs (methylone, heroin, oxycodone) and 3 aflatoxins (B1, B2, G2).
The samples were processed with the same workflow, using ACToR implemented with DrugBank and EPA ToxCast.
Results and Discussion
HRAMS Set-up
Generally, routine methods focus on the detection and identification of a particular compound or class of compounds; however, there are still problems when other hazardous compounds are present within a sample (Rajski et al. 2014).
Nowadays, the use of high-resolution techniques combined with cutting-edge software for the development of new data analysis workflows can be used to solve these problems.
The elucidation of elemental compositions can be performed by evaluating the high-resolution mass spectra of unknown unfragmented analyte ions. HRAMS instruments allow mass resolutions up to 100,000 FWHM enabling the determination of relative isotopic abundances and the extraction of diagnostic spectra information (Kaufmann 2010).
In targeted analysis using high-resolution techniques, for each compound, it is necessary to have other adducts, besides H+, and also a fragmentation pattern in order to avoid to bump into isobaric matrix components (Marczak et al. 2016; Perez-Ortega et al. 2016).
To this aim, HRAMS in combination with innovative statistical software and dedicated databases represents a strong discriminating factor enabling an innovative differential analysis and the elimination of the majority of matrix components.
The goal of this work was the study of the implementation of a new approach for the analysis of food commodities in order to detect a wider range of potential contaminants as possible, such as pesticides and related metabolites and mycotoxins and drugs that are investigated in targeted routine analysis in our laboratory.
For this study, a series of working steps has been implemented: firstly, the setting of a “Homemade Exclusion list” (for details, see Supplementary Information Table S2) in the LC-MS method to eliminate all the ions belonging to the reagent blank. The second step is the selection of the extraction and, eventual, purification system based on their ability to extract as many compounds as possible. In this context, the software and databases allow the mapping of molecular compounds of the sample in order to evaluate the ability of the extraction procedure, to highlight the potential number of interfering compounds in the sample and their distributions in the investigated mass range.
The acquisition method has been performed by using 4 consecutive injections for each extract (blank reagent, blank matrix sample, quality control sample, and unknown sample), of which 3 with a FullScan-All Ion Fragmentation (FullScan-AIF) as reported above. The 3 chromatograms are processed with online databases in order to detect and identify the parental ion using the high resolution (140,000 FWHM) that allows the reduction of possible isobaric compounds and at the same time, setting a AGC target of 3e6. In particular, the FullScan acquisition parameters allowed a finest detection and identification of parental ions of low molecular weight xenobiotic molecules; AIF is set with a quadrupole opening to collect all fragments of all parental ions in the defined mass range. Three injections of the same sample were run to ensure a significant average of all m/z signals. The obtained data have been stored as it can be used in the future laboratory activities for retrospective analysis.
The resolving power of 140,000 FWHM, applied to the FullScan-AIF runs, is necessary to the collection and the identification of parental ions that are present in the injected extract. Instead, a lower resolving power of 35,000 FWHM is a good compromise between the high resolution used to extract the parental ion and respective adducts and isotopes, for which a high scan rate is required, and the relative lower resolution is used to extract all the fragments.
In the following step, the last run was performed in ddMS2 acquisition. This step allowed to collect fragments belonging to peaks going beyond a defined threshold (i.e. AGC Target 1e6) and provides the exact identification of an unknown compound after its theoretical fragmentation study performed with Mass Frontier 7.0 (Kellmann et al. 2009; Thermo Scientific 2011). The resolving power of 70,000 FWHM applied for the run performed in ddMS2 is enough to identify signals that have been previously identified with the FullScan-AIF runs.
Choice of the Extraction Protocol and Matrix Blank/Blind Samples Results
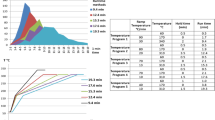
Four extraction protocols were compared in terms of matrix and xenobiotic compounds extracted from the matrix and the samples, respectively. The selection has been performed by means of Compound Discoverer 2.0; firstly for the matrix blank that gives the results as mapping plots (Fig. 1) and, then, as differential analysis between the reagent blank and matrix.

Mapping plot for peach (merendella) as matrix blank
According to the data obtained in this step (Table 1), the number of detected compounds in matrix blanks is comparable among the four extraction protocols. Our choice was addressed to the use of the “Dilute-and-shoot” acetonitrile protocol that showed high number of recovered analytes (validation approach-quality control and proficiency test approaches) with the advantage to be the cheapest extraction method. Fig. 2

Differential analysis and results table for penthiopyrad by means of Compound Discoverer 2.0
It should be noted that, in this step, the differential analysis carried out with Compound Discoverer 2.0, besides the 14 spiked pesticides (Table 2), allowed the detection and identification of myclobutanil alcohol metabolite, which was not determined by targeted analysis.
Validation Approach-Quality Control
Compound ID
This validation protocol was carried following SANTE/11813/2017 Document (SANTE 2017) in order to assess the suitability of the presented approach, and it was divided in 2 steps.
The first part has been performed by spiking 14 stone fruit samples with a standard mix of 55 pesticides of different chemical-physical properties. The spiking levels were at 0.010 mg kg−1 (resulting as LOQ for the most part of the pesticides used) and 0.10 mg kg−1. The choice of using pesticides with different characteristics, starting from the classical to the new generation ones, allowed covering different molecules characterized by specific fingerprints. Particular attention has been paid to new generation pesticides, such as penflufen, penthiopyrad, prosulfocarb, pyrethrins, and spirotetramat, introduced in the European market in the last years and for which the European Commission has expressly requested their inclusion in the national control programmes (SANCO/12745/2013 rev. 9(1) 2013).
Analysing the results obtained in this step (reported in Supplementary Information Table S3), it can be claimed that the differential analysis, performed with Compound Discoverer 2.0, has correctly detected and identified 55 compounds on a total of 55 spiked pesticides. Two peculiarities should be reported: Phorate, a pesticide present in the spiking mix, was not detected and identified as parental ion but as phorate sulfoxide and phorate oxon sulfoxide. Moreover, it was possible to detect and identify phoxim (m/z = 299.06138 as precursor ion [M + H]+) that has been by far distinguished from quinalphos (m/z = 299.06138 as precursor ion [M + H]+), thanks to ddMS2 acquisition. The latter, due to the identification of fragment ions, allowed the clear identification of isobaric compounds by means of differential analysis performed by Compound Discoverer 2.0 (Figs. 3 and 4). Furthermore, eight compounds, not belonging to the pesticide class, were also identified on all the spiked samples and not in the matrix blank. One of these compounds is the diethyl dithiophosphoric acid (C4H11O2PS2) that is probably a product of degradation of thiophosphate pesticides, included in the spiking mix. This was demonstrated by the fragmentation mechanism and the differential analysis performed by means of Mass Frontier 7.0 and Compound Discoverer 2.0, respectively (Figs. 5, 6 and 7) (Weissberg and Dagan 2011).

Identification of phoxim and quinalphos through Compound Discoverer 2.0 after 3 chromatographic runs acquisitions in FullScan-AIF

a Case of isobaric compounds phoxim and quinalphos. b Fingerprint of fragments from phoxim

Identification of diethyl dithiophosphoric acid (C4H11O2PS2) by means of Compound Discoverer 2.0 after 3 chromatographic runs acquisitions in FullScan-AIF

Fragmentation mechanism of diethyl dithiophosphoric acid (C4H11O2PS2) by means of Mass Frontier 7.0

Identification of diethyl dithiophosphoric acid (C4H11O2PS2) by means of Thermo Xcalibur™ 3.1 Software. a) Accurate mass (187.00134 m/z); b) Exact mass (187.00108 m/z); c-d) ddMS2 acquisition: parental ion and fragmentation products.
In this context, it should be highlighted that even if it would be necessary to have a reference standard to the analyte confirmation, however, good confidence in identification can be possible by obtaining the molecule fingerprint and to overlap it with the fingerprint available on databases, such as m/z Cloud, that are continuously updated.
As a result, 198 compounds have been identified by means of ChemSpider referring to online-dedicated databases such as pesticides common names, EPA ToxCast, and FDA implemented in it; 95 compounds have also been identified and reported in the “Mass List search Result”.
In addition, with this untargeted approach, each chromatogram can also be reprocessed in order to perform broad-spectrum retrospective analyses allowing studies with different goals and perspectives.
Proficiency Tests
The second part of the validation protocol was carried with the EUPT-FV-18 Proficiency Test in which the sample and the matrix blank of spinach were analysed both with the targeted and untargeted methods. In particular, the results of both methods clearly gave an unambiguous identification and structural characterization of the compounds, based on accurate mass measurements and informative fragmentation spectra, for all the pesticides spiked by EURL-FV, except for cymoxanil, dimethoate, and omethoate; the latter, although well detected and identified with the targeted method, was not detected by the untargeted method. The failure in cymoxanil detection was probably due to the low concentration of this analyte in the sample; instead, for dimethoate and omethoate, the probable cause was the low recovery obtained with “Dilute-and-shoot” acetonitrile extraction procedure. On the other hand, while imidacloprid desnitro was not identified by multiresidue targeted analysis, it was instead detected and identified by the untargeted approach being its parental ion imidacloprid present in the spiked proficiency test item (Fig. S2) (Gbylik-Sikorska et al. 2015).
All the data results for EUPT-FV-18 (European Union Proficiency Test in Fruits and Vegetables 18, 2016) for the unknown non-targeted method are reported in Supplementary Information Table S4.
During the validation of EUPT-FV-18 Proficiency Test, the samples of EUPT-FV-16 (pepper) and EUPT-FV-17 (broccoli) Proficiency Tests were also tested as quality control in both target and unknown method of analysis. In both cases, all the pesticides were correctly detected and identified.
Finally, in the Screening 2016 Proficiency Test EUPT-FV-SM08 (European Union Proficiency Test in Fruits and Vegetables Screening Methods 08 2017), among 15 pesticides, 10 were correctly detected and identified with the untargeted method; the other 5 pesticides are suitable for the GC-MS determination and not detectable with LC-MS (see Supplementary Information Table S5). Among these 5 pesticides, quintozene, heptachlor, and chlozolinate are more easily detectable with a GC-MS. Tetramethrin determination is carried out both with LC-MS and GC-MS; however, much higher sensitivity is ensured by the latter. In addition, in this study, performed with untargeted method supported by UHPLC-HRAMS, false positives and false negatives have been avoided.
Using the target screening method, similar results have been obtained except for prothiofos that was not included in the EFS_HRAM_Compound_Database.cdb (Thermo Fisher Scientific).
According to the EUPT-FV-SM08 Final Report (European Union Proficiency Test in Fruits and Vegetables Screening Methods 08 2017), our laboratory has been included in the 67% (56) that detected over 70% pesticides.
This particular proficiency test was selected as it involves peculiar pesticides that are not frequently found in food and feed or not monitored by the laboratories because they are not part of EU coordinated programmes. Consequently, the use of screening methods is overwhelmingly increasing the chance of detecting less commonly found pesticides. This expands not only the laboratory scope regarding pesticide residue analysis but also improves the reliability of the screening methods by verification through validation approaches.
Method Upgrade to Other Matrices and Contaminants
All the contaminants spiked in the tomato samples were clearly detected and identified with this method. In particular, Fig. 8, the correct identification of the heroin has been shown, reporting the chromatographic peak of the extracted ion, the isotopic pattern and 4 characteristic adducts. The last chromatogram, not reported in the figure, confirms the identity through the correct fragmentation. The same parameters are shown for the aflatoxin B2 (Fig. 9).

Identification of heroin through Compound Discoverer 2.0

Identification of aflatoxin B2 through Compound Discoverer 2.0
In this context, it has to be highlighted that it is only a very starting limited example of method upgrade to other matrices and contaminants, aimed to only emphasize the method potentiality.
Conclusions and Perspectives
In the context of food safety, the determination of xenobiotics by means of targeted methods is not straightforward due to the application of different analytical methods involving higher costs and time-consuming analyses.
In order to guarantee a suitable degree of safety for consumers, it is essential to identify wider ranges of xenobiotics that can be potentially found in food. Among these, not only environmental, process and food-contact contaminants but also compounds produced by microorganisms can be pointed out.
In the last years, great studies have been addressed to the combination of HRAMS and software to develop targeted analysis.
The novelty introduced by this study largely overcomes targeted method schemes through an innovative untargeted approach based on differential analysis giving the chance of searching unexpected substances in food commodities ensuring higher levels for food safety. The leading-edge workflow based on HRAMS combined with innovative software and databases allows not only the possibility to detect substances not previously pre-selected but also to reprocess raw data files for performing chemometric, differential, and retrospective analyses.
Furthermore, future perspectives will be focused on the monitoring of small molecules that are degradation products of organic compounds inside food samples, and even if this approach is valid for all the analytes which are prone to electrospray ionization, in order to have a complete contaminant analysis, this method will also be extended to the monitoring in GC-HRAMS.
The forthcoming development of this innovative approach would be intensified in order to firstly open new horizons to the research of a wider range of xenobiotic and unexpected compounds and related metabolites in food matrices and secondly to lay the foundation for its applicability to routine analysis in the near future.
References
Bletsou AA, Jeon J, Hollender J, Archontaki E, Thomaidis NS (2015) Targeted and non-targeted liquid chromatography-mass spectrometric workflows for identification of transformation products of emerging pollutants in the aquatic environment. Trends Anal Chem 66:32–44. https://doi.org/10.1016/j.trac.2014.11.009
Castro-Puyana M, Perez-Míguez R, Montero L, Herrero M (2017) Reprint of: application of mass spectrometry-based metabolomics approaches for food safety, quality and traceability. Trends Anal Chem 96:62–78. https://doi.org/10.1016/j.trac.2017.08.007
EC (2002) Regulation No 178/2002 of the European Parliament and of the Council of 28 January 2002 laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety. Off J Eur Union L 31/1, 01.02.
EN 15662:2008 (2008) European Committee for Standardization (2008) CEN EN 15662: Foods of plant origin-determination of pesticide residues using GC-MS and/or LC-MS/MS following acetonitrile extraction/partition and clean-up by dispersive SPE-QuEChERS method. https://www.en-standard.eu/csn-en-15662-foods-of-plant-origin-multimethod-for-the-determination-of-pesticide-residues-using-gc-and-lc-based-analysis-following-acetonitrile-extraction-partitioning-and-clean-up-by-dispersive-spe-modular-quechers-method/
European Union Proficiency Test in Fruit and Vegetables 18 (2016) Pesticide Residues in Spinach Homogenate-Final Report. http://www.eurl-pesticides.eu/userfiles/file//FinalReport_EUPT_FV18.pdf Accessed 24/06/2017
European Union Proficiency Test for Pesticides in Fruit and Vegetables Screening Methods 08 (2017) Pesticide Residues in Spinach Homogenate-Final Report. http://www.eurl-pesticides.eu/userfiles/file//EUPT-FV-SM08-Final-Report.pdf Accessed 24/06/2017
Fu Y, Zhou Z, Kong H, Lu X, Zhao X, Chen Y, Chen J, Wu Z, Xu Z, Zhao C, Xu G (2016) Nontargeted screening method for illegal additives based on ultrahigh-performance liquid chromatography-high-resolution mass spectrometry. Anal Chem 88:8870–8877. https://doi.org/10.1021/acs.analchem.6b02482
Fu Y, Zhao C, Lu X, Xu G (2017) Nontargeted screening of chemical contaminants and illegal additives in food based on liquid chromatography-high resolution mass spectrometry. Trends Anal Chem 96:89–98. https://doi.org/10.1016/j.trac.2017.07.014
Gbylik-Sikorska M, Sniegocki T, Posyniak A (2015) Determination of neonicotinoid insecticides and their metabolites in honey bee and honey by liquid chromatography tandem mass spectrometry. J Chromatogr B 990:132–140. https://doi.org/10.1016/j.jchromb.2015.03.016
Gómez-Ramos MM, Ferrer C, Malato O, Agüera A, Fernandez-Alba AR (2013) Liquid chromatography-high-resolution mass spectrometry for pesticide residue analysis in fruit and vegetables: screening and quantitative studies. J Chromatogr A 1287:24–37. https://doi.org/10.1016/j.chroma.2013.02.065
Gómez-Ramos MDM, Rajski Ł, Lozano A, Fernández-Alba AR (2016) The evaluation of matrix effects in pesticide multi-residue methods via matrix fingerprinting using liquid chromatography electrospray high-resolution mass spectrometry. Anal Methods 23:4664–4673. https://doi.org/10.1039/C6AY00436A
Herrera-Lopez S, Hernando MD, Garcia-Calvo E, Fernandez-Alba AR, Ulaszewska MM (2014) Simultaneous screening of targeted and non-targeted contaminants using an LC-QTOF-MS system and automated MS/MS library searching. J Mass Spectrom 49:878–893. https://doi.org/10.1002/jms.3428
Judson R, Richard A, Dix D, Houch K, Elloumi F, Martin M, Cathey T, Transue TR, Spencer R, Wolf M (2008) ACToR-aggregated computational toxicology resource. Toxicol Appl Pharmacol 233:7–13. https://doi.org/10.1016/j.taap.2007.12.037
Kaufmann A (2010) Strategy for the elucidation of elemental compositions of trace analytes based on a mass resolution of 100 000 full width at half maximum. Rapid Commun Mass Spectrom 24:2035–2045. https://doi.org/10.1002/rcm.4612
Kaufmann A (2012), Chapter 4 - high mass resolution versus MS/MS. In: Compr Anal Chem, 58:169–215
Kaufmann A (2014) Combining UHPLC and high-resolution MS: a viable approach for the analysis of complex samples. Trends Anal Chem 63:113–128. https://doi.org/10.1016/j.trac.2014.06.025
Kellmann M, Muenster H, Zomer P, Mol H (2009) Full scan MS in comprehensive qualitative and quantitative residue analysis in food and feed matrices: how much resolving power is required. J Am Soc Mass Spectrom 20:1464–1476. https://doi.org/10.1016/j.jasms.2009.05.010
Knolhoff AM, Croley TR (2016) Non-targeted screening approaches for contaminants and adulterants in food using liquid chromatography hyphenated to high resolution mass spectrometry. J Chromatogr A 1428:86–96. https://doi.org/10.1016/j.chroma.2015.08.059
Knolhoff AM, Zweigenbaum JA, Croley TR (2016) Non-targeted screening of food matrices: development of a chemometric software strategy to identify unknowns in liquid chromatography-mass spectrometry data. Anal Chem 88:3617–3623. https://doi.org/10.1021/acs.analchem.5b04208
Lehotay SJ, Sapozhnikova Y, Mol HGJ (2015) Current issues involving screening and identification of chemical contaminants in foods by mass spectrometry. Trends Anal Chem 69:62–75. https://doi.org/10.1016/j.trac.2015.02.012
Makarov A, Scigelova M (2010) Coupling liquid chromatography to Orbitrap mass spectrometry. J Chromatogr A 1217:3938–3945. https://doi.org/10.1016/j.chroma.2010.02.022
Marczak Ł, Znajdek-Awizen P, Bylka W (2016) The use of mass spectrometric techniques to differentiate isobaric and isomeric flavonoid conjugates from Axyris amaranthoides. Molecules 21:E1229. https://doi.org/10.3390/molecules21091229
Milman BL (2015) General principles of identification by mass spectrometry. Trends Anal Chem 69:24–33. https://doi.org/10.1016/j.trac.2014.12.009
Perez-Ortega P, Lara-Ortega FJ, Garcia-Reyes JF, Gilbert-Lopez B, Trojanowicz M, Molina-Diaz A (2016) A feasibility study of UHPLC-HRMS accurate-mass screening methods for multiclass testing of organic contaminants in food. Talanta 160:704–712. https://doi.org/10.1016/j.talanta.2016.08.002
Rajski Ł, Gómez-Ramos MDM, Fernández-Alba AR (2014) Large pesticide multiresidue screening method by liquid chromatography-Orbitrap mass spectrometry in full scan mode applied to fruit and vegetables. J Chromatogr A 1360:119–127. https://doi.org/10.1016/j.chroma.2014.07.061
SANCO/12745/2013 rev. 9(1). (2013) Working document on pesticides to be considered for inclusion in the national control programmes to ensure compliance with maximum residue levels of pesticides residues in and on food of plant and animal origin.
SANTE (2017) Document no. SANTE/11813/2017. Guidance document on analytical quality control and method validation procedures for pesticides residues analysis in food and feed
Thermo Scientific (2011) Mass Frontier Version 7.0 User Guide. XCALI-97349 Revision A. https://tools.thermofisher.com/content/sfs/manuals/Man-XCALI-97349-Mass-Frontier-70-User-ManXCALI97349-A-EN.pdf Accessed 26/05/2017
Thermo Scientific (2015) Compound Discoverer User Guide Software Version 2.0 XCALI-97805 Revision A
Weissberg A, Dagan S (2011) Interpretation of ESI(+)-MS-MS spectra - towards the identification of “unknowns”. Int J Mass Spectrom 299:158–168. https://doi.org/10.1016/j.ijms.2010.10.024
Zomer P, Mol HGJ (2015) Simultaneous quantitative determination, identification and qualitative screening of pesticides in fruits and vegetables using LC-Q-Orbitrap-MS. Food Addit Contam Part A 32:1628–1636. https://doi.org/10.1080/19440049.2015.1085652
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Rossana Scarpone, Roberta Rosato, Francesco Chiumiento, Chiara Cipolletti, Manuel Sergi and Dario Compagnone declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
Scarpone, R., Rosato, R., Chiumiento, F. et al. Preliminary Study to Develop an Alternative Method for the Non-targeted Determination of Xenobiotics in Food by Means of Ultra High Performance Liquid Chromatography Coupled to High Resolution and Accuracy Mass Spectrometry. Food Anal. Methods 13, 1099–1110 (2020). https://doi.org/10.1007/s12161-020-01727-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-020-01727-1