
Abstract
Corticobasal syndrome (CBS) is characterised by asymmetrical parkinsonism and cognitive impairment. The underlying pathology varies between corticobasal degeneration, progressive supranuclear palsy, Alzheimer’s disease, Creutzfeldt–Jakob disease and frontotemporal lobar degeneration sometimes in association with GRN mutations. A 61-year-old male underwent neurological examination, neuropsychological assessment, MRI, and HMPAO-SPECT at our medical centre. After his death at the age of 63, brain autopsy, genetic screening and mRNA expression analysis were performed. The patient presented with slow progressive walking disabilities, non-fluent language problems, behavioural changes and forgetfulness. His family history was negative. He had primitive reflexes, rigidity of his arms and postural instability. Later in the disease course he developed dystonia of his left leg, pathological crying, mutism and dysphagia. Neuropsychological assessment revealed prominent ideomotor and ideational apraxia, executive dysfunction, non-fluent aphasia and memory deficits. Neuroimaging showed symmetrical predominant frontoparietal atrophy and hypoperfusion. Frontotemporal lobar degeneration (FTLD)-TDP type 3 pathology was found at autopsy. GRN sequencing revealed a novel frameshift mutation c.314dup, p.Cys105fs and GRN mRNA levels showed a 50% decrease. We found a novel GRN mutation in a patient with an atypical (CBS) presentation with symmetric neuroimaging findings. GRN mutations are an important cause of CBS associated with FTLD-TDP type 3 pathology, sometimes in sporadic cases. Screening for GRN mutations should also be considered in CBS patients without a positive family history.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Corticobasal syndrome (CBS) is characterised by progressive asymmetric rigidity and apraxia, accompanied by additional signs of cortical and extrapyramidal dysfunction such as alien limb phenomenon, cortical sensory loss, myoclonus, bradykinesia and focal dystonia unresponsive to dopaminergic treatment (Boeve et al. 2003). Although corticobasal degeneration (CBD) is the most frequent underlying pathology of CBS, other neuropathological causes, including frontotemporal lobar degeneration (FTLD), Alzheimer’s disease (AD), progressive supranuclear palsy (PSP) and Creutzfeldt–Jakob disease have been reported (Boeve et al. 1999; Wadia and Lang 2007; Tartaglia et al. 2010).
Although family history is usually negative in CBS (Spina et al. 2007), a familial form has been described which can be caused by progranulin (GRN) mutations (Wadia and Lang 2007; van Swieten and Heutink 2008). GRN mutations show a high phenotypic variability reflected by diagnoses of frontotemporal dementia, progressive non-fluent aphasia or AD within the same families (Gass et al. 2006; Beck et al. 2008; Le Ber et al. 2008; van Swieten and Heutink 2008; Kelley et al. 2009). The underlying pathology of GRN mutations is FTLD with TAR DNA-binding protein of 43 kDA (TDP-43) positive neuronal cytoplasmic and intranuclear inclusions (Gass et al. 2006; Whitwell et al. 2007; Beck et al. 2008; Kelley et al. 2009) which is known as FTLD-TDP type 3 (Cairns et al. 2007).
In this report, we describe the clinical, radiological, pathological and genetic features of a 61-year-old patient with an initial presentation of symmetric CBS caused by a novel GRN c.314dup mutation in the absence of a positive family history.
Materials and Methods
Subjects
A 61-year-old male was referred to the outpatient clinic of our neurology department. On the basis of neurological examination, neuropsychological assessment, brain MRI and single photon emission computed tomography with 99mTc-hexamethyl propyleneamine oxine–single-photon emission computed tomography (SPECT), he was diagnosed with atypical parkinsonism with dementia. He died from bronchopneumonia at the age of 63.
Neuropathological Analysis
Brain autopsy was performed by the Netherlands Brain Bank according to their legal and ethical code of conduct. Paraffin-embedded sections from all brain regions were stained with haematoxylin and eosin, Bodian, methenamine silver, and Congo Red. Immunostaining with antibodies for tau (Innogenetics), ubiquitin (DAKO), β-amyloid (DAKO), α-synuclein (Zymed Laboratories), p62 (BD Biosciences Pharmingen), FUS (Sigma-Aldrich anti-FUS) and TDP-43 (Biotech) was performed. Neuropathological diagnosis was made by a neuropathologist (A.J.M.R.).
Genetic Analysis
DNA was extracted from peripheral blood using the DNA Blood Kit Special (Chemagen, Baesweiler), which is based on DNA extraction and purification with magnetic beads (Smit et al. 2000). All coding exons (2–13) and exon/intron boundaries of GRN (NM_002087.2) were amplified from genomic DNA by PCR and directly sequenced in both strands using the ABI 3730XL automated sequencer (Applied Biosystems, Foster City, CA). Data were analysed using SeqScape software (version 2.6; Applied Biosystems).
mRNA Expression Analysis
Total RNA was extracted from cerebellar brain tissue using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA), and quality and quantity was assessed on the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA samples were normalised to 50 ng/μl, and using 200 ng as the template, a reverse transcription reaction was performed using a 1:1 mix of random hexamers and oligo(dT) primers and the SuperScript III system (Invitrogen, Carlsbad, CA). Gene expression assays were ordered from Applied Biosystems for GRN (Hs00963703_g1) and GAPDH (Hs00266705_g1) as an endogenous control. Real-time PCR was performed on an ABI 7900 using the TaqMan method. Reactions contained 1 μl of cDNA amplified with 0.25 μl primer/probe mix and 2.5 μl TaqMan 2× Universal PCR Master Mix (Applied Biosystems, Foster City, CA). The cycling parameters recommended by the manufacturer were followed. All samples were run in triplicate and normalised to GAPDH. The carboxyfluorescein (FAM)-fluorescent signal was analysed using the SDS2.2.2 software, and relative quantities of GRN mRNA were determined using the ΔΔct method.
Results
Clinical, Neuropsychological and Imaging Features
A 61-year-old male barber presented with a 2-year duration of progressive walking disability, frequent falls and difficulty with rising from a chair. This motor decline was later followed by loss of initiative, emotional lability, more compliant behaviour and increasing forgetfulness. He made inappropriate sexual remarks, and his speech became non-fluent with perseverations, word-finding difficulties and comprehension deficits. Financial matters were taken over by his wife. Furthermore, he needed assistance for dressing and personal hygiene and was occasionally incontinent of urine and faeces. His symptoms were unresponsive to galantamine and levodopa. The patient’s family history was negative for neurodegenerative disorders. His father died at the age of 72 from a stroke and his mother at the age of 53 due to liver cancer.
Initial neurological examination showed normal eye movements, a mask-like facial expression, hypokinetic dysarthria, positive primitive reflexes, subtle axial rigidity, hypokinetic-rigid syndrome with cogwheeling of the upper extremities, mildly increased tone of the lower extremities, postural instability and impaired tandem gait.
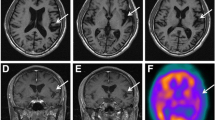
Neuropsychological testing revealed a Mini Mental State Examination score of 24/30 with intact orientation in time and place. The patient was highly distractible and exhibited sparse spontaneous speech with stuttering and perseverations. Ideomotor and ideational apraxia were prominent, the patient did not succeed in imitation, constructional tasks, and rapid alternating hand movements. Furthermore, he had severely impaired executive and memory functions, poor semantic word fluency and naming deficits but intact visuospatial functions. Neuroimaging revealed mild symmetrical cerebral atrophy, most prominent frontoparietal and perisylvian on MRI and symmetrical frontoparietal hypoperfusion on SPECT, which is shown in Fig. 1.

Mild symmetric frontoparietal atrophy on MRI (a) and symmetric parietal hypoperfusion on SPECT scan (b)
The patient’s condition further declined over the subsequent years. Neurological examination at a follow-up visit 1 year later revealed slowed saccadic eye movements, severe symmetrical rigidity of the upper extremities, an increased tone with dystonic flexion posturing of the left leg and pathological crying. Finally, he developed mutism and dysphagia, which did not respond to amitriptyline treatment, and he died from bronchopneumonia at the age of 63 years.
Neuropathological Examination
Gross brain (1,327 g) examination showed mild generalised atrophy of the cortex with atrophy of caudate nucleus and putamen and normal pigmentation of the substantia nigra. Microscopic examination revealed moderate to severe neuronal loss, gliosis and spongiosis in the frontal and parietal cortices but mild changes in the temporal cortex, hippocampus and basal ganglia. Immunohistochemistry showed TDP-43-positive dystrophic neurites, neurocytoplasmatic and intranuclear inclusions most prominent in the second superficial layer of the frontal and parietal cortices and to a lesser extent in the temporal cortex, hippocampus and basal ganglia, consistent with a diagnosis of FTLD-TDP type 3 (Fig. 2) (Cairns et al. 2007).

TDP-43-positive dystrophic neurites in the frontal cortex (a) a TDP-43-positive neuronal intranuclear inclusion in the parietal cortex (b), and TDP-43-positive neuronal cytoplasmatic inclusions in hippocampus (c), and caudate nucleus and putamen (d). Scale bar = 20 μm
Genetic Analysis
After receiving the autopsy results, we screened GRN and identified a novel c.314dup, p.Cys105fs mutation. This frameshift mutation causes premature termination of the coding sequence, which is likely to result in loss of function of Granulin.
mRNA Expression Analysis
GRN mRNA expression analysis in cerebellar tissue from this patient showed a 50% decrease in GRN mRNA levels compared to a pathologically-confirmed normal control, confirming the pathogenicity of the mutation.
Discussion
We described a patient with an atypical CBS presentation with symmetrical neuroimaging findings with FTLD-TDP type 3 pathology due to a novel c.314dup, p.Cys105fs mutation in the absence of a positive family history.
The clinical presentation of our case showed strong similarities with the symmetric CBD described by Hassan et al., except for the presence of ideomotor and ideational apraxia (Hassan et al. 2010). The symmetric rigidity and the absence of alien hand phenomenon and myoclonus did not fit with the clinical diagnosis CBS in the initial phase of the disease (Boeve et al. 2003). Moreover, the symmetric pattern of frontoparietal atrophy on MRI and parietal hypoperfusion on SPECT scan was inconsistent with the typical asymmetric distribution in CBS but was in line with the observations in symmetric CBD (Boeve et al. 2003; Whitwell et al. 2010a; Hassan et al. 2010). In a later stage of the disease, our patient developed dystonia in the left leg, which made the clinical diagnosis CBS more likely (Boeve et al. 2003). However, we did not have the opportunity to visualize an asymmetric pattern of atrophy, as an attempt to obtain additional neuroimaging was unsuccessful.
The neuropathological findings of the present case were similar to those in the case by Spina et al., which showed TDP-43 pathology most prominent in parietal and frontal cortex and to a lesser extent in the temporal cortex, with involvement of the basal ganglia (Spina et al. 2007). Their patient had a more typical CBS presentation with an alien hand phenomenon and marked asymmetry of symptoms as well as neuroimaging findings, which is more often seen in CBS patients with GRN mutations (Masellis et al. 2006; Spina et al. 2007; Benussi et al. 2008). The combination of parkinsonism, apraxia, behavioural changes, non-fluent speech and memory impairment in our patient could retrospectively have indicated a GRN mutation because these symptoms have been more frequently observed in FTLD patients with GRN mutations (Beck et al. 2008; Le Ber et al. 2008; Kelley et al. 2009).
The absence of a positive family history in the current case emphasises the possibility of reduced penetrance of GRN mutations or concealed family history due to the wide range in onset age of GRN mutations (Gass et al. 2006; Le Ber et al. 2007; Whitwell et al. 2007; Seelaar et al. 2008). We only screened for GRN mutations after the observation of FTLD-TDP type 3 pathology at autopsy. This case therefore underlines that genetic screening should be considered even when family history appears to be negative. The novel GRN frameshift mutation in this patient led to a premature stop codon and thereby to loss of function of Granulin, which was confirmed by reduced mRNA expression levels in the cerebellar tissue. Recent studies have shown reduced Granulin levels in the serum of patients with GRN mutations (Coppola et al. 2008; Ghidoni et al. 2008; Finch et al. 2009; Sleegers et al. 2009; Schofield et al. 2010). We could not confirm this in our patient, however, since his serum was not available.
Based on the clinical presentation of this patient, we did not predict FTLD-TDP pathology. Although non-fluent aphasia and parkinsonism can be seen in FTLD-TDP, an asymmetric presentation and pattern of atrophy is associated with all underlying pathologies of CBS, including CBD, PSP, FTLD and AD (Whitwell et al. 2010a). In addition, the extension of atrophy into the frontal cortex is typically asymmetric in patients with CBS with FTLD-TDP pathology, including those with GRN mutations (Whitwell et al. 2010a; Beck et al. 2008; Le Ber et al. 2008; van Swieten and Heutink 2008; Rohrer et al. 2010; Whitwell et al. 2010b). Therefore the present case is unique by its atypical CBS presentation and symmetric atrophy pattern associated with FTLD-TDP type 3 pathology with a novel GRN mutation.
To conclude, we found a novel GRN mutation in a patient with an atypical CBS presentation with symmetric neuroimaging findings without a positive family history. GRN mutations are an important cause of CBS associated with FTLD-TDP pathology. Genetic screening should also be considered in CBS patients with a negative or concealed family history.
References
Beck J, Rohrer JD, Campbell T et al (2008) A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain 131(Pt 3):706–720
Benussi L, Binetti G, Sina E et al (2008) A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol Aging 29(3):427–435
Boeve BF, Maraganore DM, Parisi JE et al (1999) Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology 53(4):795–800
Boeve BF, Lang AE, Litvan I (2003) Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 54(Suppl 5):S15–S19
Cairns NJ, Bigio EH, Mackenzie IR et al (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol (Berl) 114(1):5–22
Coppola G, Karydas A, Rademakers R et al (2008) Gene expression study on peripheral blood identifies progranulin mutations. Ann Neurol 64(1):92–96
Finch N, Baker M, Crook R et al (2009) Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 132(Pt 3):583–591
Gass J, Cannon A, Mackenzie IR et al (2006) Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 15(20):2988–3001
Ghidoni R, Benussi L, Glionna M, Franzoni M, Binetti G (2008) Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 71(16):1235–1239
Hassan A, Whitwell JL, Boeve BF et al (2010) Symmetric corticobasal degeneration (S-CBD). Parkinsonism Relat Disord 16(3):208–214
Kelley BJ, Haidar W, Boeve BF et al (2009) Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol Aging 30(5):739–751
Le Ber I, van der Zee J, Hannequin D et al (2007) Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum Mutat 28(9):846–855
Le Ber I, Camuzat A, Hannequin D et al (2008) Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 131(Pt 3):732–746
Masellis M, Momeni P, Meschino W et al (2006) Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain 129(Pt 11):3115–3123
Rohrer JD, Geser F, Zhou J et al (2010) TDP-43 subtypes are associated with distinct atrophy patterns in frontotemporal dementia. Neurology 75(24):2204–2211
Schofield EC, Halliday GM, Kwok J, Loy C, Double KL, Hodges JR (2010) Low serum progranulin predicts the presence of mutations: a prospective study. J Alzheimers Dis 22(3):981–984
Seelaar H, Kamphorst W, Rosso SM et al (2008) Distinct genetic forms of frontotemporal dementia. Neurology 71(16):1220–1226
Sleegers K, Brouwers N, Van Damme P et al (2009) Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol 65(5):603–609
Smit ML, Giesendorf BA, Heil SG, Vet JA, Trijbels FJ, Blom HJ (2000) Automated extraction and amplification of DNA from whole blood using a robotic workstation and an integrated thermocycler. Biotechnol Appl Biochem 32(Pt 2):121–125
Spina S, Murrell JR, Huey ED et al (2007) Corticobasal syndrome associated with the A9D Progranulin mutation. J Neuropathol Exp Neurol 66(10):892–900
Tartaglia MC, Sidhu M, Laluz V et al (2010) Sporadic corticobasal syndrome due to FTLD-TDP. Acta Neuropathol 119(3):365–374
van Swieten JC, Heutink P (2008) Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol 7(10):965–974
Wadia PM, Lang AE (2007) The many faces of corticobasal degeneration. Parkinsonism Relat Disord 13(Suppl 3):S336–S340
Whitwell JL, Jack CR Jr, Baker M et al (2007) Voxel-based morphometry in frontotemporal lobar degeneration with ubiquitin-positive inclusions with and without progranulin mutations. Arch Neurol 64(3):371–376
Whitwell JL, Jack CR Jr, Boeve BF et al (2010a) Imaging correlates of pathology in corticobasal syndrome. Neurology 75(21):1879–1887
Whitwell JL, Jack CR Jr, Parisi JE et al (2010b) Does TDP-43 type confer a distinct pattern of atrophy in frontotemporal lobar degeneration? Neurology 75(24):2212–2220
Competing interests
None.
Funding
This work was supported by Stichting Dioraphte grant 09-02-03-00 (to J.C.v.S.) and NIH grant R01 NS065782-01 (to R.R.).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Dopper, E.G.P., Seelaar, H., Chiu, W.Z. et al. Symmetrical Corticobasal Syndrome Caused by a Novel c.314dup Progranulin Mutation. J Mol Neurosci 45, 354–358 (2011). https://doi.org/10.1007/s12031-011-9626-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-011-9626-z