
Abstract
Despite numerous studies in the field of congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency, some clinical variability of the presentation and discrepancies in the genotype/phenotype correlation are still unexplained. Some, but not all, discordant phenotypes caused by mutations with known enzyme activity have been explained by in silico structural changes in the 21-hydroxylase protein. The incidence of P30L mutation varies in different populations and is most frequently found in several Central and Southeast European countries as well as Mexico. Patients carrying P30L mutation present predominantly as non-classical CAH; however, simple virilizing forms are found in up to 50% of patients. Taking into consideration the residual 21-hydroxulase activity present with P30L mutation this is unexpected. Different mechanisms for increased androgenization in patients carrying P30L mutation have been proposed including influence of different residues, accompanying promotor allele variability or mutations, and individual androgene sensitivity. Early diagnosis of patients who would present with SV is important in order to improve outcome. Outcome studies of CAH have confirmed the uniqueness of this mutation such as difficulties in phenotype classification, different fertility, growth, and psychologic issues in comparison with other genotypes. Additional studies of P30L mutation are warranted.
Similar content being viewed by others
Introduction
Congenital adrenal hyperplasia (CAH) is a family of autosomal recessive disorders caused by mutations of genes involved in the steroidogenesis pathway [1,2,3,4]. The most common cause of CAH, occurring in 95–99% of cases, is 21-hydroxylase deficiency (21OHD), followed by 11β-hydroxylase deficiency, 17α-hydroxylase/17,20-lyase deficiency, 3β-hydroxy-steroid dehydrogenase type 2 deficiency, P450 oxidoreductase deficiency, lipoid CAH, and cholesterol side chain cleavage enzyme deficiency [4,5,6,7,8,9,10]. Mutations in the cytochrome P450 (CYP) 21A2 (CYP21A2) gene result in 21OHD. The cytochromes P450 comprise a superfamily of heme-containing mono-oxygenases that play central roles in the metabolism of a wide variety of endogenous compounds including steroids, drugs, and carcinogens.
CAH can clinically be manifested in a variety of forms, depending on the amount of the functioning enzyme. The common classification consists of two major forms, classical and nonclassical [1, 11]. Classical form of 21OHD appears with an incidence of 1:10,000–1:20,000 live births in different populations [4, 12,13,14], and is rising with the early detection by neonatal screening [15]. The phenotype of patients with a classical 21OHD is different depending on the degree of the remaining 21-hydroxylase enzyme activity. In a majority of patients with classical 21OHD (75%), 21-hydroxylase activity is completely absent causing life-threatening cortisol and aldosterone deficiency (salt-wasting form [SW]) accompanied with hyperandrogenemia causing sexual ambiguity in affected females [2, 16, 17]. If the remaining enzyme activity is low but present (<2%), simple virilizing (SV) form of the disease occurs which appears in 25% of cases with the classical form. SV is characterized by a cortisol deficiency accompanied with hyperandrogenemia inducing virilization of the external genitalia in females and hyperandrogenemia in boys, often noticed by precocious puberty. The nonclassical form of 21OHD (NC) is common, one of the most common recessive disease in humans (1/200–1/1000 in Caucasians), especially in certain ethnicities such as Eastern European Jews, Finland being an exception with the lowest incidence [18,19,20,21,22]. The enzyme activity in NC CAH is preserved (~20–60%) with symptoms appearing later in life, mostly in preadolescent, adolescent or young adult period [20, 23, 24]. Oligoamenorrhea, hirsutism, and impaired fertility are common symptoms.
The CYP21A2 gene is located on chromosome 6.21p in the region of class III of the human leukocyte antigen [25]. Several other genes are located in this region forming a module. CYP21A2 is located 30 kb upstream of its nonfunctional pseudogene CYP21A1 [26]. Both genes consist of ten exons, and have high sequence homology (98% exonic nucleotide homology and 96% intronic homology) [7, 26, 27]; however, the pseudogene is nonfunctional. Gene changes comprise large deletions, 8 bp deletions, gene conversions due to crossing-over with the adjacent pseudogene, and point mutations in the gene itself [20, 28]. The pseudogene is prone to mutations (e.g., splicing, frameshift, and insertions) which can be transferred to the functional gene by microconversion events. Most frequently CYP21A2 mutations occur as a result of recombination with the pseudogene (75%). The remaining 20–25% of mutations consist of deletions or chimeric genes, both appearing as a result of unequal crossing-over. Only 1% of mutations appear de novo [1]. The number of reported CYP21A2 mutations increases continuously from around 130 [28,29,30,31,32] to over 230 in the last large reports [33, 34]. Most patients with 21OHD are compound heterozygotes carrying different mutated alleles. In consanguineous populations, homozygocity is more common compared with admixed populations. Specific expression variations might also be a problem such as the leaky intron 2 mutation and alternative splicing or duplication of Q318X allele [35, 36].
Mutations are classified as severe, moderate, and mild based upon the enzyme activity and the phenotype they most frequently induce. The phenotype depends on the activity of the milder mutation in compound heterozygotes since it is connected with some enzyme activity [24, 33, 37].
P30L has been classified to the group of the milder mutations with 20–60% of enzyme activity [1, 38]. Most frequently, P30L mutation causes mild, i.e., NC pheontype. However, it can also cause SV form with intensive hyperandrogenism and virilization [20, 22, 39,40,41]. The P30L mutation frequency in different populations and the clinical outcomes in patients carrying P30L mutation have not been extensively reviewed.
The aim of this review is to present clinical, including long-term, outcomes, and molecular findings associated with the P30L mutation.
Molecular structure of P30L mutation and functional analysis
Molecular characterization of the CYP21A2 mutations and their impact upon the structure of the 21-hydroxylase enzyme influence the phenotype and the severity of the disease [7, 31, 39, 42,43,44,45]. The human crystal structure model of the 21-hydroxylase enzyme has been unraveled recently and the impact of numerous different mutations has been explored, explaining the SW, SV, and NC phenotypes [31, 46, 47]. The CYP21A2 molecule resides in the membranes of endoplasmatic reticulum. Different chaperons and cellular proteins assist the proper folding of the protein. The enzyme has a triangular prism shape and contains 16 helices and 9 β-sheets with a heme located centrally [31]. There are two binding sites for 17-hydroxyprogesterone (17OHP), proximal and distal, both in the proximity of the heme moiety. Hydrogen bonds of residues connected to the heme, as well as the electron transfer are crucial for the proper function of the enzyme [31, 33, 47]. Mutations which cause irregular clashes with heme of the enzyme, disruption of hydrogen bonds, substrate binding, mutations causing impaired secondary structure or structural stability, all abrogate the enzyme function and cause the severe SW form of the disease [31, 46, 48,49,50]. On the other hand, mutations causing only reduction in the enzyme anchoring, but allow some residual function, cause SV or NC 21OHD. Moreover, interruption of inter- and intra-protein interactions or hydrophobic environment disruption may cause unexpected phenotypes. Thus I172N can occasionally cause NC, and V281L can be found rarely in SW form [31, 51].
P30L mutation in exon 1 of CYP21A2 gene is a mild missense single base pair mutation (g.89 C>T), which belongs to the pseudogene-derived mutations. It decreases 21-hydroxylase activity to 20–60% according different authors [1, 38, 52], frequently causing NC 21OHD. It is not associated with particular HLA antigens in contrast to V281L which is associated with the haplotype B14;DR1 [1].
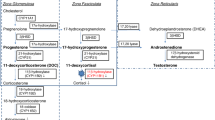
P30 residue is located at the N-terminal site of the enzyme near to the transmembrane region. It is lodged in a hydrophobic cavity of the enzyme and is crucial for attachment of P450 to the membrane. When prolin is replaced with glutamin (P30Q g89 C>A) its hydrophilic properties disrupt the hydrophobic network and affects the attachment of the enzyme to the membrane with SW form as a result. Replacement of prolin with leucine (P30L g89 C>T), which is hydrophobic residue, interferes with the proper orientation of the enzyme with respect to the microsomal membrane segment, orienting the protein away from membrane, but not improper folding of the protein. Therefore P30L mutation is better tolerated [46, 53]. Analyzing the protein stability of P30L mutation has shown that the half-life is significantly reduced compared with other mild mutations [31, 54]. Enzymes carrying P30L mutations were initially structurally studied in detail and confirmed as a cause of NC form in 1991 [53]. The authors used recombinant vaccine virus to express two mutant enzymes carrying P30L as a pathologic mutation and Ser268Thr conferring normal polymorphism as a first control. The wild type of the enzyme was used as a second control. The normal polymorphism showed 100% enzyme activity, the same as the wild type, whereas, the enzyme activity of P30L mutation was 60% for 17OHP and only 25% for binding progesterone as a substrate. The speed of metabolism affected by P30L mutation for 17OHP and progesterone compared with the wild type was 12- and 21-fold lower, respectively [53]. Furthermore, enzymatic activity with P30L mutation was rapidly lost when the cells were lyzed, suggesting relative enzyme instability [1]. Using computational methods Neves Cruz et al. evaluated the structural impact and the effect on the steroid binding as well as protein structural conformation of different CYP21A2 mutations [47]. P30L mutation is located peripherally at the N end of the enzyme and is involved in moderate change of enzyme stability (Fig. 1). However, it showed conserved metallic coordination between the heme group and the Cys428 residue of the polypeptide chain with an average distance of 2.5 Å which is similar with the wild type. This is pivotal for the preservation of the P450 enzymes activity, therefore P30L mutation is frequently associated with the NC phenotype [47]. In a study by Tussie-Luna et al. the mutation was present in 28% (5/18) of patients with hormonal evidence of NC CAH. However, all patients carrying the P30L mutation were symptomatic, in contrast to 69% (9/13) displaying other NC mutations. It is worth mentioning that four out of five patients with P30L had clitoromegaly compared with none of those carrying other NC mutations (e.g., V281L) [53]. Clitoromegaly was also reported in NC patients carrying P30L in China [55]. This shows that the P30L mutation, although the enzyme activity is preserved sufficiently, causes a more severe form of NC or even a SV form. E.g., in one family, the proband had P30L/I172N, and presented as SV form with clitoromegaly, hirsutism, delayed menarche, and severely impaired fertility [40].

Spatial localization of the mutant residues P30L and V281L in the CYP21A2 protein showing their peripheral localization. P30L is localized at the N terminus of the protein responsible for the orientation toward the membrane of the endoplasmatic reticulum
It is still not known whether in certain patients with P30L mutation other residues are unfavorable, and the enzyme, although present, cannot function appropriately causing increased virilization corresponding to SV CAH [31]. Some authors believe that decreased activity in P30L is due to amino acid residues affecting other functions such as heme coordination, posttranslational modifications, or interface with other interacting proteins or ligands [32, 53]. The P30L mutation obviously has additional regulators, or posttranslational inhibitors of enzyme activity. Some additional residues might also have influence [33]. Simultaneous transfer of mutation in the promotor region together with the P30L mutation from the pseudogene has been described causing a fivefold decrease of the gene activity and causing SV form [56]. Moreover, different allele variations in the promotor region of CYP21A2 gene causing 50% lower transcriptional activity of 21-hydroxylase have been identified in SV patients carrying P30L mutation [57]. The presence of unidentified rare mutations modulating the phenotype cannot be excluded; however, it is unlikely due to the frequent occurrence of SV form with P30L mutation in studies with meticulous genotyping. Some other factors modifying phenotype have also been suggested such as the CAG repeats of the androgen receptor and other genes encoding proteins other than cytochrome P450 type II enzyme with a 21-hydroxylase activity, as well as alternative pathway of androgen biosynthesis causing fetal virilization in females [58,59,60].
Prevalence of P30L mutation
The frequency of different CYP21A2 mutations are variable in different populations and ethnicities [7, 16, 45, 61,62,63]. The most common mutations are deletion or large gene conversion, I2 splice, V281L, and I172N [7, 14, 16, 37, 45]. In populations with consanguinity the variability is lower compared with admixed populations [64, 65]. Different reports show that the P30L is not among the most common mutations. Its frequency is between 0 and 46% in different populations (Table 1) [14, 16, 18, 19, 23, 37, 45, 55, 62, 63, 66,67,68,69,70,71,72,73,74,75,76,77,78]. Interestingly, P30L mutation occur more frequently in Central and Southeast Europe, including Balkans, and Mexico [39, 62, 63, 68, 74, 79, 80].
Clinical presentation and genotype/phenotype correlation
Although a good genotype–phenotype correlation has been established in up to 95% of patients with CAH [31, 45, 52], many outliers have been described, of which a significant number is due to the P30L mutation.
The enzyme activity is quite high in individuals with P30L mutations and mild form of CAH would be expected in all patients carrying it independently of the mutation on the other allele. Mild form is indeed the most frequent or the only presentation of P30L mutation in many; however, more severe forms are also present in a significant number of patients in other populations (Table 1) [33, 39, 45].
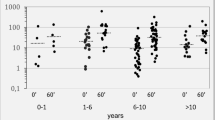
The NC form of CAH was initially called late-onset as clinical presentation was observed in adolescents and adults [38, 81, 82]. Presentation of NC CAH is subtle and diagnosis requires different diagnostic tests to exclude other metabolic problems. Moreover, clinical expression of NC CAH is variable in patients carrying the same mutation [1, 20, 45, 65]. In patients with NC CAH predominant signs are those of mild androgen excess. Therefore, in females the diagnosis is made mostly in late childhood, adolescence, or young adulthood due to symptoms as premature pubarche, acne, hirsutism, male-pattern alopecia, polycystic ovary syndrome (PCOS), and subfertility [11, 65, 79]. However, there are females with minimal or no symptoms, even after a long life [83]. Despite of similar 17OHP levels in patients with other mild mutations, NC patients with P30L mutation can show stronger virilization with clitoromegaly and advanced bone maturation [84]. Although I172N mutation is considered typically associated with SV, it seems that unlike other mild mutations, P30L mutation generates a continuum of phenotypes between NC and SV as well as a typical SV [33, 45, 85]. In a multinational study of 1507 families with CAH, SV form of CAH was found in 17/74 patients having P30L mutation (23%) [45]. Even 66% of patients with P30L mutation express unexpected virilization requiring extensive reconstructive surgery [86]. Moreover, in a study of a large cohort of patients with 21OHD of Greek origin, the phenotype of P30L mutation was equally distributed between SV and NC (19.1% vs 21.4%) [71]. The genotype/phenotype concordance in this study decreased as the severity of the disease diminished. Risk for short stature should be taken into consideration since NC and even SV sometimes are diagnosed late [87, 88], although in the majority final height was within the normal range [89,90,91].
In a Central European study the major genotype–phenotype discrepancies were detected for P30L and I172N mutation [80]. Similar findings have been confirmed in other studies [52, 69, 92]. In a study of 400 families in Argentina, P30L mutation, although rare (0.6%), the SV to NC phenotype ratio was 1:1 [16]. Moreover, in countries with high prevalence of the P30L mutation, in either a homozygous or compound heterozygote state, patients are prone to have increased virilization and fertility issues subsequently placing them in-between the NC and SV forms or even pure SV form [37, 40]. In the Republic of North Macedonia, where the prevalence of P30L mutation is among the highest in the world, the number of patients with SV form is very high [63]. Even when it is not a SV form, the clinical manifestation is with stronger signs of virilization, earlier adrenarche, clitoromegaly, and some patients require higher doses of glucocorticoids compared with other patients with NC form [55, 66, 93]. Moreover, genotypes P30L/I2 splice, P30L/Q318X, and P30L/8Δbp are especially associated with SV form of CAH (Table 2) [45]. Classical presentation of SV in two sisters with clitoromegaly, no breast development, and severely impaired fertility having P30L/I172N genotype has recently been described [40].
SV phenotype in girls is easier to recognize due to clitoromegaly, or severe atypical genital in newborn girls, but in boys the genital pigmentation might be missed, and early pubarche and advanced penile growth may be the first signs [41, 71, 86]. If not treated with glucocorticoids SV progresses steadily during childhood causing early puberty, short adult stature, and fertility issues in both genders including testicular adrenal rest tumors (TARTs) in men.
Thus, it might be advisable to be very cautious with the interpretation of the results of neonatal screening when P30L mutation is detected, since it might be the first sign of the SV phenotype which may manifest clinically later in childhood. Moreover, virilized girls without SW and fast growing boys with advanced bone age may not always have I172N but P30L mutation as shown in different studies [11, 24, 39, 73, 86].
Diagnosis
The diagnosis of classical forms of 21OHD is based upon the clinical picture, blood electrolyte analysis, 17OHP levels, and androgens including testosterone, dehydroepiandrosterone sulfate (DHEAS), and androstenedione. Most patients have a basal morning 17OHP values above 30 nmol/L. However, some patients with suspected NC CAH may have lower basal 17OHP levels and a level between 6 and 30 nmol/L could warrant an ACTH-stimulation test [20, 41, 82, 94]. Levels above 30 nmol/L on ACTH-stimulation test, which is the golden standard for diagnosis of 21OHD, are considered diagnostic [22, 28, 41, 95]. Severe CYP21A2 mutations have higher 17OHP levels both basal and post ACTH-stimulation [23]. Measurement of progesterone, 17-hydroxypregnenolone, 11-deoxycortisol, DHEAS, deoxycorticosterone, and androstenedione may be warranted in order to distinguish other forms of disturbed steroidogenesis. There are limited data on the biochemical parameters in patients with P30L mutation but these show similar 17OHP and testosterone levels as those with other mild mutations and cannot predict the severity of the clinical presentation [53, 96, 97]. Due to late diagnosis of NC and even SV forms, special attention should be given to children who grow faster than expected during early years of life, and follow them thoroughly for signs of early puberty. Bone maturation in these patients is of utmost importance for the diagnosis and follow-up [98]. Differences in the time of presentation and speed of progression in patients with P30L mutation remain to be elucidated. It should be noted that the different national neonatal screening programs are developed to diagnose classic CAH, and many NC newborns will not be detected if additional molecular testing is not applied as a second tier [28, 99]. It might be advisable to preform strict follow-up even in newborns with no symptoms where P30L mutation is detected.
Therapy and follow-up
The goals of therapy in SV and NC forms, including patients with P30L mutation, are to substitute cortisol (especially in SV), reverse hyperandrogenism to ensure normal growth and timely puberty, preserve fertility as well as to avoid the long-term complications [100,101,102]. Hydrocortisone is the drug of choice in newborn and children with confirmed SV form [103]. However, appropriate glucocorticoid regimen (hydrocortisone, prednisolone, dexamethasone, or combinations) with or without mineralocorticoid therapy in adults is still uncertain [104,105,106]. Some SV patients may benefit from adding mineralocorticoids based on the studies showing higher plasma renin activity in SV patients including patients carrying P30L mutation. It is due to accumulation of some steroid precursors which can cause, especially in poorly controlled patients, aldosterone mediated transactivation of the human mineralocorticoid receptor. [107,108,109]. Adding mineralocorticoids in SV patients may provide decreased renin activity and may enable decrease of glucocorticoid dose. Mineralocorticoids have occasionally been used in NC CAH as well [110,111,112,113]. Therapy should be carefully tapered according to the growth pattern and hormonal results. There are trials to simplify frequent blood sampling with adrenal specific androgens measurements in saliva, hair, or urine samples [114,115,116,117]. Therapy changes with aging of the patients, fertility treatment is necessary in some females with CAH, both classic and nonclassic and men who develop TARTs, but also treatment of late complications of the disease and supraphysiological glucocorticoid supplementation [3, 81, 82, 111].
Therapy in patients with P30L mutations depends on the clinical picture, and consist mostly of improving symptoms, not biochemical findings. Achieving normal final height should be among the main goals. Transition from pediatric to adult care is of utmost importance since many patients could be lost from follow-up during this period [100, 118,119,120].
Somatic outcomes
Growth and puberty
Growth is affected in both females and males with CAH. Early prepubertal growth spurt due to early puberty is typical. Final height was affected (−2.5 SD) in earlier works, and significantly less (−1.0 SD for classic and −0.4 SD for NC form) in later works [91, 121,122,123], probably due to improved therapy and compliance. Hyperandrogenism can result in early closure of epiphyses. However, supraphysiological glucocorticoid supplementation may also lead to short adult height [122]. Close follow-up and fine tapering of therapy can improve the adult height, especially if the bone age advancement is detected before 8 years of age [3, 91, 124]. In SV form growth in early childhood in females is normal; however, in boys it is significantly faster and ends within 0.5 SD of target height [121]. The shorter stature in SV form is due to the early puberty, advanced bone age, and lack of important pubertal growth spurt [124, 125]. A high hydrocortisone dose during puberty may affect growth due to deterioration of the metabolic control [126,127,128]. Growth in NC form is often below the target height but within the normal range [96, 129, 130]. Careful tapering of therapy might provide height within the normal range and within the genetic potential.
There are no specific data on the influence of different mutations, yet the growth in patients with P30L mutation is expected to be affected mostly in males with delayed diagnosis of the SV form, and females with early puberty [131].
Puberty in patients with 21OHD occurs earlier compared with age-related peers [129, 132]. It occurs earlier in SW form (9.3 years on average in males and 9.2 years in females) compared with the age-related healthy population. In SV and NC form it occurred on average at ~8.5 years and at ~10 years in males and females, respectively [84, 128, 130]. Central precocious puberty in CAH is rare [41, 133]. There is no specific study but P30L should influence puberty onset causing earlier puberty and may induce secondary central precocious puberty in individuals with the SV form [84, 133]. In females, early puberty would be accompanied with clitoromegaly and impaired breast development [40, 134].
Fertility
Fertility in females with all forms of CAH can be impaired, despite the advances in different therapeutic methods. The role of hyperandrogenism as a cause of impaired fertility has been extensively studied [135,136,137]. In females with SW form spontaneous fertility in older studies has been reported in only 2.5% and in SV 38% [138]. Additional 2% conceive by assisted reproductive technology. Other studies show higher but still low-fertility rate since it has been established that elevated circulating adrenal androgens and elevated serum progesterone concentrations may hinder ovulation and embrio implantation [139,140,141]. However, all agree that some additional factors contribute to the low reproductive rate such as the decreased sexual activity, higher sexual distress, higher prevalence of homosexuality or bisexuality, and unwillingness to pursue motherhood [136, 142,143,144,145]. Due to individualized fertility therapy, newer studies show much higher pregnancy rates in women with CAH approaching the fertility rate in the general population [81, 140]. Women with SV usually seek motherhood six times more frequently compared with those with SW [146]. Women with NC CAH conceive spontaneously in 57.2% [147]. Pregnancy in these women is normal with an outcome similar to the general population [136, 148]. There are no detailed data on fertility issues in patients with P30L mutation except the report of two female patients with P30L mutation and SV phenotype requiring genital surgery and several artificial assisted reproduction cycles [40]. In populations with a high prevalence of NC CAH, many females are diagnosed as PCOS [149,150,151]. NC CAH needs to be excluded before diagnosing PCOS [22, 82, 152, 153]. In one study of hirsute women 10% had NC CAH, some of them with P30L mutation [151]. Whether infertility issues are more frequent, or if they are more difficult to treat in the P30L mutation group remains to be elucidated in future studies.
Fertility issues in males with CAH are mostly due to TARTs. TARTs are common and appear in 40–94% in males, most commonly in severe forms of CAH [154,155,156,157]. They appear earlier and more frequently in patients with a poor metabolic control. TARTs have been described in patients with P30L even in childhood [158, 159]. Spermatogenesis in males with CAH is impaired and deteriorates with the age; however, associated obesity, common in older patients with CAH, might contribute as well [154, 156]. Still fertility was not decreased in 17 males with NC phenotype (none with P30L) or in 12 males with P30L compared with 1700 and 1200 matched controls, respectively [160]. Interestingly, in 221 males with 21OHD studied, only those born before the introduction of neonatal screening had impaired fertility [160]. Thus, early diagnosis may improve fertility in males with 21OHD [3, 160, 161].
Prenatal diagnosis in women with CAH is compromised by the possible genotype/phenotype discordance, especially when mutations causing adverse phenotypes such as P30L are detected [150]. Carriers of mild mutations might end up with unexpectedly high incidence of offspring with SW or SV form of the disease [162].
Metabolic and cardiovascular outcomes
Therapy with glucocorticoids and androgen control influence metabolic status and outcome in all patients with CAH [163,164,165,166]. Long-term glucocorticoid replacement may cause abdominal obesity and hypertension with an onset even in youngsters [131, 167,168,169]. Obesity is frequently associated with high CRP levels, hypercholesterolemia, hyperlipidemia, insulin resistance, diabetes, high leptin, and low adiponectin levels causing a common metabolic syndrome in ~20% of patients with a cardiovascular risk independent of mutations [79, 123, 167, 170]. Very few studies analyzed cardiovascular outcomes according to the genotype [111, 123]. Interestingly, in the population study by Falhammar et al. different mutations had different risk of cardiovascular events [164]. Males with P30L mutation had one of the highest risk for any cardiometabolic condition and obesity as well as a tendency to increased risk for obstructive sleep apnea [164]. However, females with the P30L mutation had no cardiometabolic risk in this study but it should be noted that the number of studied individuals with this mutation was relatively low (n = 24). The metabolic issues were still present in those patients with CAH who were born after the introduction of neonatal screening [164]. Thus, cardiometabolic risk should be carefully monitored in patients with CAH. In fact, mortality has been shown to be increased in CAH (2.3 higher in males and 3.5 times in females compared with matched controls), of which a significant part was cardiovascular mortality [171].
Bone health
Decreased bone mineral density and more fractures have been shown in some studies of CAH, but was absent in others [172,173,174,175,176,177,178]. Bone mineral density and fractures in patients with P30L mutations have not been studied in detail.
Autoimmune diseases
Autoimmune diseases have recently been found to be more frequent in patients with CAH, and their prevalence increases with age. Compared with controls (n = 2900), those with P30L mutations (n = 29) had a tendency to develop more autoimmune disorders in general and especially rheumatoid arthritis [179]. Whether the onset of glucocortiocoid treatment and the duration of therapy have an immunomodulating effect remains to be elucidated [180].
Mental outcomes
Engberg et al. analyzed psychiatric diagnoses in 335 women with CAH compared with 33,500 matched controls [181]. They found that psychiatric diagnoses in general and substance abuse were more common in women with CAH. Interestingly, patients with P30L mutation had much more psychotic disorders and personality disorders in the age group >18 years compared with the carriers of other mutations [181]. Psychotic disorders were increased in both female and male patients with P30L mutation, especially in those born before the neonatal screening [182], with personality disorders being more frequent in women with the P30L mutation [181]. In both genders with 21OHD, alcohol misuse was increased and so were also suicidal attempts in males. However, none in the P30L group has been diagnosed with alcohol misuse or attempted suicide [181, 182]. Similar findings were found in 226 individuals with CAH (almost all females) were psychiatric disorders and suicide attempts were more common than in the general population [166]. Although the genotypes were not described in the latter study it can be suspected that very few had P30L mutations. Whether late diagnosis, glucocorticoid therapy, and/or the androgen exposure contributes to the increased prevalence of psychiatric disorders in CAH, especially in women, remains to be elucidated [182, 183]. Since the level of hyperandrogenism has generally been associated with the alcohol and other substances abuse [184], and patients with P30L are more hyperandrogenic compared with the carriers of other mild mutations, it would be useful with larger studies to investigate addictions and psychiatric issues in this group [183].
Quality of life
Having in mind the complexity of CAH and its complications, it has to be expected that patients with CAH will have affected quality of life (QoL) [11, 185]. Life-long therapy, frequent controls, additional issues as the patient grows, necessary interventions as well as under- or overtreatment and poor metabolic control leading to frequent sick-leave in CAH may result in lower social integration, education, self-confidence, employment, and lower QoL [186, 187]. Late diagnosis is associated with depression and decreased self-control. Many women with CAH are not satisfied with their sexual life and have later sexual debut or complete lack of sexual activity [141, 145]. Males with CAH had impaired sexual well-being in one study [188], but not in another study [186]. The overall psychosexual aspects of life were affected in these patients with later sexual debut, fewer pregnancies and children, and an increased incidence of homosexuality in women [189, 190]. In a QoL study from Norway including 72 adult participants with CAH impaired general health perception, vitality and working ability were found [191]. QoL was correlated to the severity of the mutations [5]. A recent systematic review reported increased psychological and psychiatric issues, impaired QoL, together with reduced satisfaction with reproductive health and sexual function in male with CAH [192]. QoL in patients with SV and NC forms can be similar to the controls as shown in patients diagnosed after the introduction of neonatal screening, probably due to the timely and more sophisticated treatment [154, 186].
Conclusion
Patients with P30L mutations have not been studied extensively. The ethnic variability is wide and P30L mutation affects mostly people from Central Europe, Balkan countries, and Mexico. P30L confers 20–60% 21-hydroxylase activity. However, P30L mutation induces a more severe clinical virilization than the typical NC CAH and clinical presentation is a continuum between NC and SV phenotype. Studies of the structure of the mutated enzyme do not completely explain the discrepancy between the preserved enzyme activity and the phenotype. Therefore, the reclassification of this mutation as mild should be reconsidered. Long-term outcome data in patients with P30L are limited, but some issues such as psychiatric disorders may be more frequent in this group compared with the other mild mutations. Further studies of the genotype/phenotype variations in P30L careers, long-term outcomes, and treatment options are warranted.
References
P.C. White, P.W. Speiser, Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr. Rev. 21(3), 245–291 (2000). https://doi.org/10.1210/edrv.21.3.0398.
P.W. Speiser, W. Arlt, R.J. Auchus, L.S. Baskin, G.S. Conway, D.P. Merke, H.F.L. Meyer-Bahlburg, W.L. Miller, M.H. Murad, S.E. Oberfield, P.C. White, Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 103(11), 4043–4088 (2018). https://doi.org/10.1210/jc.2018-01865.
H. Falhammar, M. Thoren, Clinical outcomes in the management of congenital adrenal hyperplasia. Endocrine 41(3), 355–373 (2012). https://doi.org/10.1007/s12020-011-9591-x
D. El-Maouche, W. Arlt, D.P. Merke, Congenital adrenal hyperplasia. Lancet 390(10108), 2194–2210 (2017). https://doi.org/10.1016/S0140-6736(17)31431-9
W. Arlt, D.S. Willis, S.H. Wild, N. Krone, E.J. Doherty, S. Hahner, T.S. Han, P.V. Carroll, G.S. Conway, D.A. Rees, R.H. Stimson, B.R. Walker, J.M. Connell, R.J. Ross, U.K.C.A.H.A.S.E, Health status of adults with congenital adrenal hyperplasia: a cohort study of 203 patients. J. Clin. Endocrinol. Metab. 95(11), 5110–5121 (2010). https://doi.org/10.1210/jc.2010-0917
B. Dean, G.L.Chrisp, M. Quartararo, A.M. Maguire, S. Hameed, B.R. King, C.F. Munns, D.J. Torpy, H. Falhammar, R.L. Rushworth, P450 oxidoreductase deficiency: a systematic review and meta-analysis of genotypes, phenotypes and their relationships. J. Clin. Endocrinol. Metab. (2019). https://doi.org/10.1210/clinem/dgz255
S. Gidlöf, H. Falhammar, A. Thilén, U. von Döbeln, M. Ritzén, A. Wedell, A. Nordenström,, One hundred years of congenital adrenal hyperplasia in Sweden: a retrospective, population-based cohort study. Lancet Diabetes Endocrinol. 1(1), 35–42 (2013). https://doi.org/10.1016/S2213-8587(13)70007-X
K. Bulsari, H. Falhammar, Clinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. Endocrine 55(1), 19–36 (2017). https://doi.org/10.1007/s12020-016-1189-x
A.M. Al Alawi, A. Nordenström, H. Falhammar, Clinical perspectives in congenital adrenal hyperplasia due to 3β-hydroxysteroid dehydrogenase type 2 deficiency. Endocrine 63(3), 407–421 (2019). https://doi.org/10.1007/s12020-018-01835-3
W.L. Miller, Mechanisms in endocrinology: rare defects in adrenal steroidogenesis. Eur. J. Endocrinol. 179(3), R125–R141 (2018). https://doi.org/10.1530/EJE-18-0279
A. Nordenstrom, H. Falhammar, Management of endocrine disease: diagnosis and management of the patient with non-classic CAH due to 21-hydroxylase deficiency. Eur. J. Endocrinol. 180, R127–R145 (2019). https://doi.org/10.1530/EJE-18-0712
S.Y. Pang, M.A. Wallace, L. Hofman, H.C. Thuline, C. Dorche, I.C. Lyon, R.H. Dobbins, S. Kling, K. Fujieda, S. Suwa, Worldwide experience in newborn screening for classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics 81(6), 866–874 (1988)
A. Tsuji, K. Konishi, S. Hasegawa, A. Anazawa, T. Onishi, M. Ono, T. Morio, T. Kitagawa, K. Kashimada, Newborn screening for congenital adrenal hyperplasia in Tokyo, Japan from 1989 to 2013: a retrospective population-based study. BMC Pediatr. 15, 209 (2015). https://doi.org/10.1186/s12887-015-0529-y
N.M. Stikkelbroeck, L.H. Hoefsloot, I.J. de Wijs, B.J. Otten, A.R. Hermus, E.A. Sistermans, CYP21 gene mutation analysis in 198 patients with 21-hydroxylase deficiency in The Netherlands: six novel mutations and a specific cluster of four mutations. J. Clin. Endocrinol. Metab. 88(8), 3852–3859 (2003)
S. Gidlöf, A. Wedell, C. Guthenberg, U. von Döbeln, A. Nordenström, Nationwide neonatal screening for congenital adrenal hyperplasia in sweden: a 26-year longitudinal prospective population-based study. JAMA Pediatr. 168(6), 567–574 (2014). https://doi.org/10.1001/jamapediatrics.2013.5321
R. Marino, P. Ramirez, J. Galeano, N. Perez Garrido, C. Rocco, M. Ciaccio, D.M. Warman, G. Guercio, E. Chaler, M. Maceiras, I. Bergada, M. Gryngarten, V. Balbi, E. Pardes, M.A. Rivarola, A. Belgorosky, Steroid 21-hydroxylase gene mutational spectrum in 454 Argentinean patients: genotype-phenotype correlation in a large cohort of patients with congenital adrenal hyperplasia. Clin. Endocrinol. 75(4), 427–435 (2011). https://doi.org/10.1111/j.1365-2265.2011.04123.x
Z. Antal, P. Zhou, Congenital adrenal hyperplasia: diagnosis, evaluation, and management. Pediatr. Rev. 30(7), e49–e57 (2009). https://doi.org/10.1542/pir.30-7-e49
P. Carrera, L. Bordone, T. Azzani, V. Brunelli, M.P. Garancini, G. Chiumello, M. Ferrari, Point mutations in Italian patients with classic, non-classic, and cryptic forms of steroid 21-hydroxylase deficiency. Hum. Genet 98(6), 662–665 (1996). https://doi.org/10.1007/s004390050280
J. Jääskeläinen, A. Levo, R. Voutilainen, J. Partanen, Population-wide evaluation of disease manifestation in relation to molecular genotype in steroid 21-hydroxylase (CYP21) deficiency: good correlation in a well defined population. J. Clin. Endocrinol. Metab. 82(10), 3293–3297 (1997). https://doi.org/10.1210/jcem.82.10.4271
M.I. New, Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 91(11), 4205–4214 (2006). https://doi.org/10.1210/jc.2006-1645
S.F. Witchel, The medical home concept and congenital adrenal hyperplasia: a comfortable habitat!. Int J. Pediatr. Endocrinol. 2010, 561526 (2010). https://doi.org/10.1155/2010/561526
H. Falhammar, A. Nordenstrom, Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine 50(1), 32–50 (2015). https://doi.org/10.1007/s12020-015-0656-0
M. Bidet, C. Bellanne-Chantelot, M.B. Galand-Portier, V. Tardy, L. Billaud, K. Laborde, C. Coussieu, Y. Morel, C. Vaury, J.L. Golmard, A. Claustre, E. Mornet, Z. Chakhtoura, I. Mowszowicz, A. Bachelot, P. Touraine, F. Kuttenn, Clinical and molecular characterization of a cohort of 161 unrelated women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency and 330 family members. J. Clin. Endocrinol. Metab. 94(5), 1570–1578 (2009). https://doi.org/10.1210/jc.2008-1582
S. Livadas, M. Dracopoulou, A. Dastamani, A. Sertedaki, M. Maniati-Christidi, A.M. Magiakou, C. Kanaka-Gantenbein, G.P. Chrousos, C. Dacou-Voutetakis, The spectrum of clinical, hormonal and molecular findings in 280 individuals with nonclassical congenital adrenal hyperplasia caused by mutations of the CYP21A2 gene. Clin. Endocrinol. 82(4), 543–549 (2015). https://doi.org/10.1111/cen.12543
P.C. White, M.I. New, B. Dupont, HLA-linked congenital adrenal hyperplasia results from a defective gene encoding a cytochrome P-450 specific for steroid 21-hydroxylation. Proc. Natl. Acad. Sci. USA 81(23), 7505–7509 (1984). https://doi.org/10.1073/pnas.81.23.7505
P.C. White, M.I. New, B. Dupont, Structure of human steroid 21-hydroxylase genes. Proc. Natl. Acad. Sci. USA 83(14), 5111–5115 (1986)
Y. Higashi, H. Yoshioka, M. Yamane, O. Gotoh, Y. Fujii-Kuriyama, Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: a pseudogene and a genuine gene. Proc. Natl. Acad. Sci. USA 83(9), 2841–2845 (1986)
H. Falhammar, A. Wedell, A. Nordenstrom, Biochemical and genetic diagnosis of 21-hydroxylase deficiency. Endocrine 50(2), 306–314 (2015). https://doi.org/10.1007/s12020-015-0731-6
E. Mornet, J.F. Gibrat, A 3D model of human P450c21: study of the putative effects of steroid 21-hydroxylase gene mutations. Hum. Genet 106(3), 330–339 (2000). https://doi.org/10.1007/s004390051046
S. Lajić, S. Clauin, T. Robins, P. Vexiau, H. Blanché, C. Bellanne-Chantelot, A. Wedell, Novel mutations in CYP21 detected in individuals with hyperandrogenism. J. Clin. Endocrinol. Metab. 87(6), 2824–2829 (2002). https://doi.org/10.1210/jcem.87.6.8525
S. Haider, B. Islam, V. D’Atri, M. Sgobba, C. Poojari, L. Sun, T. Yuen, M. Zaidi, M.I. New, Structure-phenotype correlations of human CYP21A2 mutations in congenital adrenal hyperplasia. Proc. Natl. Acad. Sci. USA 110(7), 2605–2610 (2013). https://doi.org/10.1073/pnas.1221133110
C. Minutolo, A.D. Nadra, C. Fernández, M. Taboas, N. Buzzalino, B. Casali, S. Belli, E.H. Charreau, L. Alba, L. Dain, Structure-based analysis of five novel disease-causing mutations in 21-hydroxylase-deficient patients. PLoS One 6(1), e15899 (2011). https://doi.org/10.1371/journal.pone.0015899
P. Concolino, A. Costella, Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency: a comprehensive focus on 233 pathogenic variants of CYP21A2 gene. Mol. Diagn. Ther. 22(3), 261–280 (2018). https://doi.org/10.1007/s40291-018-0319-y
L. Simonetti, C.D. Bruque, C.S. Fernández, B. Benavides-Mori, M. Delea, J.E. Kolomenski, L.D. Espeche, N.D. Buzzalino, A.D. Nadra, L. Dain, CYP21A2 mutation update: comprehensive analysis of databases and published genetic variants. Hum. Mutat. 39(1), 5–22 (2018). https://doi.org/10.1002/humu.23351
M. Kharrat, A. Riahi, F. Maazoul, R. M’rad, H. Chaabouni, Detection of a frequent duplicated CYP21A2 gene carrying a Q318X mutation in a general population with quantitative PCR methods. Diagn. Mol. Pathol. 20(2), 123–127 (2011). https://doi.org/10.1097/PDM.0b013e3181f24807
E. Schulze, G. Scharer, A. Rogatzki, L. Priebe, S. Lewicka, M. Bettendorf, W. Hoepffner, U.E. Heinrich, U. Schwabe, Divergence between genotype and phenotype in relatives of patients with the intron 2 mutation of steroid-21-hydroxylase. Endocr. Res 21(1-2), 359–364 (1995). https://doi.org/10.3109/07435809509030452
N. Krone, A. Braun, A.A. Roscher, D. Knorr, H.P. Schwarz, Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J. Clin. Endocrinol. Metab. 85(3), 1059–1065 (2000). https://doi.org/10.1210/jcem.85.3.6441
F. Hannah-Shmouni, R. Morissette, N. Sinaii, M. Elman, T.R. Prezant, W. Chen, A. Pulver, D.P. Merke, Revisiting the prevalence of nonclassic congenital adrenal hyperplasia in US Ashkenazi Jews and Caucasians. Genet Med. 19(11), 1276–1279 (2017). https://doi.org/10.1038/gim.2017.46
V. Anastasovska, M. Kocova, Genotype-phenotype correlation in CAH patients with severe CYP21A2 point mutations in the Republic of Macedonia. J. Pediatr. Endocrinol. Metab. 23(9), 921–926 (2010). https://doi.org/10.1515/jpem.2010.147
M. Kocova, V. Anastasovska, I. Bitovska, The impact of CYP21A2 (P30L/I172N) genotype on female fertility in one family. Eur. J. Med. Res. 24(1), 21 (2019). https://doi.org/10.1186/s40001-019-0379-4
S.F. Witchel, Congenital adrenal hyperplasia. J. Pediatr. Adolesc. Gynecol. 30(5), 520–534 (2017). https://doi.org/10.1016/j.jpag.2017.04.001
T.A. Bachega, A.E. Billerbeck, G. Madureira, J.A. Marcondes, C.A. Longui, M.V. Leite, I.J. Arnhold, B.B. Mendonca, Molecular genotyping in Brazilian patients with the classical and nonclassical forms of 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 83(12), 4416–4419 (1998). https://doi.org/10.1210/jcem.83.12.5350
F. Baş, H. Kayserili, F. Darendeliler, O. Uyguner, H. Günöz, M. Yüksel Apak, F. Atalar, R. Bundak, R.C. Wilson, M.I. New, B. Wollnik, N. Saka, CYP21A2 gene mutations in congenital adrenal hyperplasia: genotype-phenotype correlation in Turkish children. J. Clin. Res. Pediatr. Endocrinol. 1(3), 116–128 (2009). https://doi.org/10.4008/jcrpe.v1i3.49
T. Robins, J. Carlsson, M. Sunnerhagen, A. Wedell, B. Persson, Molecular model of human CYP21 based on mammalian CYP2C5: structural features correlate with clinical severity of mutations causing congenital adrenal hyperplasia. Mol. Endocrinol. 20(11), 2946–2964 (2006). https://doi.org/10.1210/me.2006-0172
M.I. New, M. Abraham, B. Gonzalez, M. Dumic, M. Razzaghy-Azar, D. Chitayat, L. Sun, M. Zaidi, R.C. Wilson, T. Yuen, Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc. Natl. Acad. Sci. USA 110(7), 2611–2616 (2013). https://doi.org/10.1073/pnas.1300057110
P.S. Pallan, L. Lei, C. Wang, M.R. Waterman, F.P. Guengerich, M. Egli, Research resource: correlating human cytochrome P450 21A2 crystal structure and phenotypes of mutations in congenital adrenal hyperplasia. Mol. Endocrinol. 29(9), 1375–1384 (2015). https://doi.org/10.1210/ME.2015-1127
J. Neves Cruz, K.S. da Costa, T.A.A. de Carvalho, N.A.N. de Alencar, Measuring the structural impact of mutations on cytochrome P450 21A2, the major steroid 21-hydroxylase related to congenital adrenal hyperplasia. J. Biomol. Struct. Dyn. 1–10 (2019). https://doi.org/10.1080/07391102.2019.1607560
A. Baradaran-Heravi, R. Vakili, T. Robins, J. Carlsson, N. Ghaemi, A. A’rabi, M.R. Abbaszadegan, Three novel CYP21A2 mutations and their protein modelling in patients with classical 21-hydroxylase deficiency from northeastern Iran. Clin. Endocrinol. 67(3), 335–341 (2007). https://doi.org/10.1111/j.1365-2265.2007.02886.x
C. Kenaan, H. Zhang, E.V. Shea, P.F. Hollenberg, Uncovering the role of hydrophobic residues in cytochrome P450-cytochrome P450 reductase interactions. Biochemistry 50(19), 3957–3967 (2011). https://doi.org/10.1021/bi1020748
M. Barbaro, S. Lajic, L. Baldazzi, A. Balsamo, P. Pirazzoli, A. Cicognani, A. Wedell, E. Cacciari, Functional analysis of two recurrent amino acid substitutions in the CYP21 gene from Italian patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 89(5), 2402–2407 (2004). https://doi.org/10.1210/jc.2003-031630
B. Zhao, L. Lei, N. Kagawa, M. Sundaramoorthy, S. Banerjee, L.D. Nagy, F.P. Guengerich, M.R. Waterman, Three-dimensional structure of steroid 21-hydroxylase (cytochrome P450 21A2) with two substrates reveals locations of disease-associated variants. J. Biol. Chem. 287(13), 10613–10622 (2012). https://doi.org/10.1074/jbc.M111.323501
M.L. Narasimhan, A. Khattab, Genetics of congenital adrenal hyperplasia and genotype-phenotype correlation. Fertil. Steril. 111(1), 24–29 (2019). https://doi.org/10.1016/j.fertnstert.2018.11.007
M.T. Tusie-Luna, P.W. Speiser, M. Dumic, M.I. New, P.C. White, A mutation (Pro-30 to Leu) in CYP21 represents a potential nonclassic steroid 21-hydroxylase deficiency allele. Mol. Endocrinol. 5(5), 685–692 (1991). https://doi.org/10.1210/mend-5-5-685
Y. Yabusaki, H. Murakami, T. Sakaki, M. Shibata, H. Ohkawa, Genetically engineered modification of P450 monooxygenases: functional analysis of the amino-terminal hydrophobic region and hinge region of the P450/reductase fused enzyme. DNA 7(10), 701–711 (1988). https://doi.org/10.1089/dna.1988.7.701
B. Zhang, L. Lu, Z. Lu, Molecular diagnosis of Chinese patients with 21-hydroxylase deficiency and analysis of genotype-phenotype correlations. J. Int. Med. Res. 45(2), 481–492 (2017). https://doi.org/10.1177/0300060516685204
R.S. Araujo, A.E. Billerbeck, G. Madureira, B.B. Mendonca, T.A. Bachega, Substitutions in the CYP21A2 promoter explain the simple-virilizing form of 21-hydroxylase deficiency in patients harbouring a P30L mutation. Clin. Endocrinol. 62(2), 132–136 (2005). https://doi.org/10.1111/j.1365-2265.2005.02184.x
H.J. Zhang, J. Yang, M.N. Zhang, W. Zhang, J.M. Liu, W.Q. Wang, G. Ning, X.Y. Li, Variations in the promoter of CYP21A2 gene identified in a Chinese patient with simple virilizing form of 21-hydroxylase deficiency. Clin. Endocrinol. 70(2), 201–207 (2009). https://doi.org/10.1111/j.1365-2265.2008.03356.x
L.C. Kaupert, S.H. Lemos-Marini, M.P. De Mello, R.P. Moreira, V.N. Brito, A.A. Jorge, C.A. Longui, G. Guerra, B.B. Mendonca, T.A. Bachega, The effect of fetal androgen metabolism-related gene variants on external genitalia virilization in congenital adrenal hyperplasia. Clin. Genet 84(5), 482–488 (2013). https://doi.org/10.1111/cge.12016
V.O. Moura-Massari, F.S. Cunha, L.G. Gomes, D. Bugano Diniz Gomes, J.A. Marcondes, G. Madureira, B.B. de Mendonca, T.A. Bachega, The presence of clitoromegaly in the nonclassical form of 21-hydroxylase deficiency could be partially modulated by the CAG polymorphic tract of the androgen receptor gene. PLoS ONE 11(2), e0148548 (2016). https://doi.org/10.1371/journal.pone.0148548
N. Reisch, A.E. Taylor, E.F. Nogueira, D.J. Asby, V. Dhir, A. Berry, N. Krone, R.J. Auchus, C.H.L. Shackleton, N.A. Hanley, W. Arlt, Alternative pathway androgen biosynthesis and human fetal female virilization. Proc. Natl. Acad. Sci. USA 116(44), 22294–22299 (2019). https://doi.org/10.1073/pnas.1906623116
R.C. Wilson, S. Nimkarn, M. Dumic, J. Obeid, M.R. Azar, M. Azar, H. Najmabadi, F. Saffari, M.I. New, Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol. Genet Metab. 90(4), 414–421 (2007). https://doi.org/10.1016/j.ymgme.2006.12.005
I. Milacic, M. Barac, T. Milenkovic, M. Ugrin, K. Klaassen, A. Skakic, M. Jesic, I. Joksic, K. Mitrovic, S. Todorovic, S. Vujovic, S. Pavlovic, M. Stojiljkovic, Molecular genetic study of congenital adrenal hyperplasia in Serbia: novel p.Leu129Pro and p.Ser165Pro CYP21A2 gene mutations. J. Endocrinol. Investig 38(11), 1199–1210 (2015). https://doi.org/10.1007/s40618-015-0366-8
V. Anastasovska, T. Milenković, M. Kocova, Direct molecular diagnosis of CYP21A2 point mutations in Macedonian and Serbian patients with 21-hydroxylase deficiency. J. Med. Biochem. 34(1), 52–57 (2015). https://doi.org/10.2478/jomb-2014-0048
M.M. Alswailem, O.S. Alzahrani, D.S. Alhomaidah, R. Alasmari, E. Qasem, A.K. Murugan, A. Alsagheir, I. Brema, B.B. Abbas, M. Almehthel, A. Almeqbali, A.S. Alzahrani, Mutational analysis of rare subtypes of congenital adrenal hyperplasia in a highly inbred population. Mol. Cell. Endocrinol. 461, 105–111 (2018). https://doi.org/10.1016/j.mce.2017.08.022
C. Binay, E. Simsek, O. Cilingir, Z. Yuksel, O. Kutlay, S. Artan, Prevalence of nonclassic congenital adrenal hyperplasia in Turkish children presenting with premature pubarche, hirsutism, or oligomenorrhoea. Int J. Endocrinol. 2014, 768506 (2014). https://doi.org/10.1155/2014/768506
D.F. de Carvalho, M.C. Miranda, L.G. Gomes, G. Madureira, J.A. Marcondes, A.E. Billerbeck, A.S. Rodrigues, P.F. Presti, H. Kuperman, D. Damiani, B.B. Mendonca, T.A. Bachega, Molecular CYP21A2 diagnosis in 480 Brazilian patients with congenital adrenal hyperplasia before newborn screening introduction. Eur. J. Endocrinol. 175(2), 107–116 (2016). https://doi.org/10.1530/EJE-16-0171
C.E. Fardella, H. Poggi, J. Soto, I. Torrealba, A. Cattani, F. Ugarte, A. Cortinez, A. Foradori, Mutations in the CYP21 B gene in a Chilean population with simple virilizing congenital adrenal hyperplasia. J. Endocrinol. Investig 23(6), 412–416 (2000). https://doi.org/10.1007/BF03343746
K. Kotaska, L. Lisá, R. Průsa, Common CYP21 gene mutations in Czech patients and statistical analysis of worldwide mutation distribution. Cent. Eur. J. Public Health 11(3), 124–128 (2003)
K.K. Dumic, Z. Grubic, T. Yuen, R.C. Wilson, V. Kusec, I. Barisic, K. Stingl, I. Sansovic, V. Skrabic, M. Dumic, M.I. New, Molecular genetic analysis in 93 patients and 193 family members with classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency in Croatia. J. Steroid Biochem. Mol. Biol. 165(Pt A), 51–56 (2017). https://doi.org/10.1016/j.jsbmb.2016.03.035
G. Ohlsson, J. Müller, N.E. Skakkebaek, M. Schwartz, Steroid 21-hydroxylase deficiency: mutational spectrum in Denmark, three novel mutations, and in vitro expression analysis. Hum. Mutat. 13(6), 482–486 (1999). https://doi.org/10.1002/(SICI)1098-1004(1999)13:6<482::AID-HUMU8>3.0.CO;2-0
M. Dracopoulou-Vabouli, M. Maniati-Christidi, C. Dacou-Voutetakis, The spectrum of molecular defects of the CYP21 gene in the Hellenic population: variable concordance between genotype and phenotype in the different forms of congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 86(6), 2845–2848 (2001). https://doi.org/10.1210/jcem.86.6.7574
E. Marumudi, A. Sharma, B. Kulshreshtha, R. Khadgawat, M.L. Khurana, A.C. Ammini, Molecular genetic analysis of CYP21A2 gene in patients with congenital adrenal hyperplasia. Indian J. Endocrinol. Metab. 16(3), 384–388 (2012). https://doi.org/10.4103/2230-8210.95679
K. Kashimada, T. Ishii, K. Nagasaki, M. Ono, T. Tajima, I. Yokota, Y. Hasegawa, Clinical, biochemical, and genetic features of non-classical 21-hydroxylase deficiency in Japanese children. Endocr. J. 62(3), 277–282 (2015). https://doi.org/10.1507/endocrj.EJ14-0377
M.L. Ordoñez-Sánchez, S. Ramírez-Jiménez, A.U. López-Gutierrez, L. Riba, S. Gamboa-Cardiel, M. Cerrillo-Hinojosa, N. Altamirano-Bustamante, R. Calzada-León, C. Robles-Valdés, F. Mendoza-Morfin, M.T. Tusié-Luna, Molecular genetic analysis of patients carrying steroid 21-hydroxylase deficiency in the Mexican population: identification of possible new mutations and high prevalence of apparent germ-line mutations. Hum. Genet. 102(2), 170–177 (1998). https://doi.org/10.1007/s004390050672
A. Grigorescu Sido, M.M. Weber, P. Grigorescu Sido, S. Clausmeyer, U. Heinrich, E. Schulze, 21-Hydroxylase and 11beta-hydroxylase mutations in Romanian patients with classic congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 90(10), 5769–5773 (2005). https://doi.org/10.1210/jc.2005-0379
B. Ezquieta, A. Oliver, R. Gracia, P.G. Gancedo, Analysis of steroid 21-hydroxylase gene mutations in the Spanish population. Hum. Genet 96(2), 198–204 (1995). https://doi.org/10.1007/bf00207379
A. Wedell, A. Thilen, E.M. Ritzen, B. Stengler, H. Luthman, Mutational spectrum of the steroid 21-hydroxylase gene in Sweden: implications for genetic diagnosis and association with disease manifestation. J. Clin. Endocrinol. Metab. 78(5), 1145–1152 (1994)
R. Wang, Y. Yu, J. Ye, L. Han, W. Qiu, H. Zhang, L. Liang, Z. Gong, L. Wang, X. Gu, 21-hydroxylase deficiency-induced congenital adrenal hyperplasia in 230 Chinese patients: genotype-phenotype correlation and identification of nine novel mutations. Steroids 108, 47–55 (2016). https://doi.org/10.1016/j.steroids.2016.01.007
D. Macut, V. Zdravkovic, J. Bjekic-Macut, G. Mastorakos, D. Pignatelli, Metabolic perspectives for non-classical congenital adrenal hyperplasia with relation to the classical form of the disease. Front Endocrinol. 10, 681 (2019). https://doi.org/10.3389/fendo.2019.00681
V. Dolzan, J. Sólyom, G. Fekete, J. Kovács, V. Rakosnikova, F. Votava, J. Lebl, Z. Pribilincova, S.M. Baumgartner-Parzer, S. Riedl, F. Waldhauser, H. Frisch, M. Stopar-Obreza, C. Krzisnik, T. Battelino, Mutational spectrum of steroid 21-hydroxylase and the genotype-phenotype association in middle European patients with congenital adrenal hyperplasia. Eur. J. Endocrinol. 153(1), 99–106 (2005). https://doi.org/10.1530/eje.1.01944
S.F. Witchel, Management of CAH during pregnancy: optimizing outcomes. Curr. Opin. Endocrinol. Diabetes Obes. 19(6), 489–496 (2012). https://doi.org/10.1097/MED.0b013e32835a1a2e
E. Carmina, D. Dewailly, H.F. Escobar-Morreale, F. Kelestimur, C. Moran, S. Oberfield, S.F. Witchel, R. Azziz, Non-classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency revisited: an update with a special focus on adolescent and adult women. Hum. Reprod. Update 23(5), 580–599 (2017). https://doi.org/10.1093/humupd/dmx014
H. Falhammar, M. Thoren, An 88-year-old woman diagnosed with adrenal tumor and congenital adrenal hyperplasia: connection or coincidence?. J. Endocrinol. Investig. 28(5), 449–453 (2005).
N. Weintrob, Z. Dickerman, E. Sprecher, A. Galatzer, A. Pertzelan, Non-classical 21-hydroxylase deficiency in infancy and childhood: the effect of time of initiation of therapy on puberty and final height. Eur. J. Endocrinol. 136(2), 188–195 (1997)
V. Tardy, R. Menassa, V. Sulmont, A. Lienhardt-Roussie, C. Lecointre, R. Brauner, M. David, Y. Morel, Phenotype-genotype correlations of 13 rare CYP21A2 mutations detected in 46 patients affected with 21-hydroxylase deficiency and in one carrier. J. Clin. Endocrinol. Metab. 95(3), 1288–1300 (2010). https://doi.org/10.1210/jc.2009-1202
S. Gurgov, K.J. Bernabé, J. Stites, C.M. Cunniff, K. Lin-Su, D. Felsen, M.I. New, D.P. Poppas, Linking the degree of virilization in females with congenital adrenal hyperplasia to genotype. Ann. N. Y Acad. Sci. 1402(1), 56–63 (2017). https://doi.org/10.1111/nyas.13370
L.T. Zeng, B. Han, B.L. Liu, X. Chen, H. Zhu, Y. Chen, M. Chen, J.H. Liu, Y. Liu, J. Qiao, [Clinical features and genetic characteristics of 33 patients with simple virilizing form of 21-hydroxylase deficiency]. Zhonghua Nei Ke Za Zhi 58(6), 428–434 (2019). https://doi.org/10.3760/cma.j.issn.0578-1426.2019.06.006
H. Falhammar, Non-functioning adrenal incidentalomas caused by 21-hydroxylase deficiency or carrier status? Endocrine 47(1), 308–314 (2014). https://doi.org/10.1007/s12020-013-0162-1
W. Hoepffner, A. Kaufhold, H. Willgerodt, E. Keller, Patients with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency can achieve their target height: the Leipzig experience. Horm. Res. 70(1), 42–50 (2008). https://doi.org/10.1159/000129677
P.A. Lee, A. Nordenström, C.P. Houk, S.F. Ahmed, R. Auchus, A. Baratz, K. Baratz Dalke, L.M. Liao, K. Lin-Su, L.H. Looijenga, T. Mazur, H.F. Meyer-Bahlburg, P. Mouriquand, C.A. Quigley, D.E. Sandberg, E. Vilain, S. Witchel, G.D.U. Consortium, Global disorders of sex development update since 2006: perceptions, approach and care. Horm. Res. Paediatr. 85(3), 158–180 (2016). https://doi.org/10.1159/000442975
P. Bretones, B. Riche, E. Pichot, M. David, P. Roy, V. Tardy, B. Kassai, S. Gaillard, D. Bernoux, Y. Morel, P. Chatelain, M. Nicolino, C. Cornu, F.C.C.G.S. Group, Growth curves for congenital adrenal hyperplasia from a national retrospective cohort. J. Pediatr. Endocrinol. Metab. 29(12), 1379–1388 (2016). https://doi.org/10.1515/jpem-2016-0156
V. Anastasovska, M. Kocova, Detected heterozygotes during the molecular analysis of the common CYP21A2 point mutations in Macedonian patients with congenital adrenal hyperplasia and their relatives. Prilozi 31(2), 71–82 (2010)
M. Tankoska, V. Anastasovska, M. Krstevska-Konstantinova, M. Naydenov, M. Kocova, Therapeutic challenges in a patient with the simple virilizing (SV) form of congenital adrenal hyperplasia (CAH) due to the P30L/I172N genotype. J. Pediatr. Endocrinol. Metab. 32(5), 543–547 (2019). https://doi.org/10.1515/jpem-2018-0285
J.B. Armengaud, M.L. Charkaluk, C. Trivin, V. Tardy, G. Breart, R. Brauner, M. Chalumeau, Precocious pubarche: distinguishing late-onset congenital adrenal hyperplasia from premature adrenarche. J. Clin. Endocrinol. Metab. 94(8), 2835–2840 (2009). https://doi.org/10.1210/jc.2009-0314
J.W. Honour, 17-Hydroxyprogesterone in children, adolescents and adults. Ann. Clin. Biochem. 51(Pt 4), 424–440 (2014). https://doi.org/10.1177/0004563214529748
G. Pinto, V. Tardy, C. Trivin, C. Thalassinos, S. Lortat-Jacob, C. Nihoul-Fékété, Y. Morel, R. Brauner, Follow-up of 68 children with congenital adrenal hyperplasia due to 21-hydroxylase deficiency: relevance of genotype for management. J. Clin. Endocrinol. Metab. 88(6), 2624–2633 (2003). https://doi.org/10.1210/jc.2002-021433
B. Ezquieta, E. Cueva, J. Varela, A. Oliver, J. Fernández, C. Jariego, Non-classical 21-hydroxylase deficiency in children: association of adrenocorticotropic hormone-stimulated 17-hydroxyprogesterone with the risk of compound heterozygosity with severe mutations. Acta Paediatr. 91(8), 892–898 (2002). https://doi.org/10.1080/080352502760148595
D.D. Martin, J.M. Wit, Z. Hochberg, R.R. van Rijn, O. Fricke, G. Werther, N. Cameron, T. Hertel, S.A. Wudy, G. Butler, H.H. Thodberg, G. Binder, M.B. Ranke, The use of bone age in clinical practice—part 2. Horm. Res. Paediatr. 76(1), 10–16 (2011). https://doi.org/10.1159/000329374
A. Nordenström, A. Thilén, L. Hagenfeldt, A. Larsson, A. Wedell, Genotyping is a valuable diagnostic complement to neonatal screening for congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 84(5), 1505–1509 (1999). https://doi.org/10.1210/jcem.84.5.5651
A. Bachelot, Transition of care from childhood to adulthood: congenital adrenal hyperplasia. Endocr. Dev. 33, 17–33 (2018). https://doi.org/10.1159/000487523
P.C. White, Update on diagnosis and management of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Curr. Opin. Endocrinol. Diabetes Obes. 25(3), 178–184 (2018). https://doi.org/10.1097/MED.0000000000000402
A. Bachelot, V. Grouthier, C. Courtillot, J. Dulon, P. Touraine, MANAGEMENT OF ENDOCRINE DISEASE: Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: update on the management of adult patients and prenatal treatment. Eur. J. Endocrinol. 176(4), R167–R181 (2017). https://doi.org/10.1530/EJE-16-0888
T. Ishii, M. Anzo, M. Adachi, K. Onigata, S. Kusuda, K. Nagasaki, S. Harada, R. Horikawa, M. Minagawa, K. Minamitani, H. Mizuno, Y. Yamakami, M. Fukushi, T. Tajima, M.S. Committee,Endocrinology, J.S.f.P., Screening, J.S.f.M., Guidelines for diagnosis and treatment of 21-hydroxylase deficiency (2014 revision). Clin. Pediatr. Endocrinol. 24(3), 77–105 (2015). https://doi.org/10.1297/cpe.24.77.
E. Whittle, H. Falhammar, Glucocorticoid regimens in the treatment of congenital adrenal hyperplasia: a systematic review and meta-analysis. J. Endocr. Soc. 3(6), 1227–1245 (2019). https://doi.org/10.1210/js.2019-00136
C.E. Seraphim, J.S. Frassei, B.S. Pessoa, R.C. Scalco, M.C. Miranda, G. Madureira, B.B. Mendonca, T.A.S.S. Bachega, Impact of long-term dexamethasone therapy on the metabolic profile of patients with 21-hydroxylase deficiency. J. Endocr. Soc. 3(8), 1574–1582 (2019). https://doi.org/10.1210/js.2019-00123
N. Reisch, Substitution therapy in adult patients with congenital adrenal hyperplasia. Best. Pr. Res Clin. Endocrinol. Metab. 29(1), 33–45 (2015). https://doi.org/10.1016/j.beem.2014.11.002
C.F. Mooij, S. Parajes, K.J. Pijnenburg-Kleizen, W. Arlt, N. Krone, H.L. Claahsen-van der Grinten, Influence of 17-hydroxyprogesterone, progesterone and sex steroids on mineralocorticoid receptor transactivation in congenital adrenal hyperplasia. Horm. Res. Paediatr. (2015). https://doi.org/10.1159/000374112
R.J. Auchus, Management considerations for the adult with congenital adrenal hyperplasia. Mol. Cell Endocrinol. 408, 190–197 (2015). https://doi.org/10.1016/j.mce.2015.01.039
H. Frisch, T. Battelino, E. Schober, S. Baumgartner-Parzer, P. Nowotny, H. Vierhapper, Salt wasting in simple virilizing congenital adrenal hyperplasia. J. Pediatr. Endocrinol. Metab. 14(9), 1649–1655 (2001). https://doi.org/10.1515/jpem.2001.14.9.1649
H. Falhammar, M. Thoren, K. Hagenfeldt, A 31-year-old woman with infertility and polycystic ovaries diagnosed with non-classic congenital adrenal hyperplasia due to a novel CYP21 mutation. J. Endocrinol. Investig. 31(2), 176–180 (2008).
H. Falhammar, H. Filipsson Nystrom, A. Wedell, M. Thoren, Cardiovascular risk, metabolic profile, and body composition in adult males with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Eur. J. Endocrinol. 164(2), 285–293 (2011). https://doi.org/10.1530/EJE-10-0877
H. Falhammar, H. Filipsson, G. Holmdahl, P.O. Janson, A. Nordenskjold, K. Hagenfeldt, M. Thoren, Metabolic profile and body composition in adult women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 92(1), 110–116 (2007). https://doi.org/10.1210/jc.2006-1350
R.M. Williams, A. Deeb, K.K. Ong, W. Bich, P.R. Murgatroyd, I.A. Hughes, C.L. Acerini, Insulin sensitivity and body composition in children with classical and nonclassical congenital adrenal hyperplasia. Clin. Endocrinol. 72(2), 155–1602010). https://doi.org/10.1111/j.1365-2265.2009.03587.x
C. Kamrath, L. Wettstaedt, C. Boettcher, M.F. Hartmann, S.A. Wudy, The urinary steroidome of treated children with classic 21-hydroxylase deficiency. J. Steroid Biochem. Mol. Biol. 165(Pt B), 396–406 (2017). https://doi.org/10.1016/j.jsbmb.2016.08.006
E. Pussard, S. Travers, C. Bouvattier, Q.Y. Xue, C. Cosson, S. Viengchareun, L. Martinerie, M. Lombès, Urinary steroidomic profiles by LC-MS/MS to monitor classic 21-hydroxylase deficiency. J. Steroid Biochem. Mol. Biol. 198, 105553 (2019). https://doi.org/10.1016/j.jsbmb.2019.105553
M. Auer, A. Krumbholz, M. Bidlingmaier, D. Thieme, N. Reisch, Steroid 17-hydroxyprogesterone in hair is a potential long-term biomarker of androgen control in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Neuroendocrinology (2019). https://doi.org/10.1159/000504672.
I. Bacila, J. Adaway, J. Hawley, S. Mahdi, R. Krone, L. Patel, S. Alvi, T. Randell, E. Gevers, M. Dattani, T. Cheetham, A. Kyriakou, L. Schiffer, F. Ryan, E. Crowne, J.H. Davies, S.F. Ahmed, B. Keevil, N. Krone, Measurement of salivary adrenal-specific androgens as biomarkers of therapy control in 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 104(12), 6417–6429 (2019). https://doi.org/10.1210/jc.2019-00031
A. Bachelot, M. Vialon, A. Baptiste, I. Tejedor, C. Elie, M. Polak, P. Touraine, Impact of transition on quality of life in patients with congenital adrenal hyperplasia diagnosed during childhood. Endocr. Connect 6(7), 422–429 (2017). https://doi.org/10.1530/EC-17-0094
J.H. Choi, H.W. Yoo, Management issues of congenital adrenal hyperplasia during the transition from pediatric to adult care. Korean J. Pediatr. 60(2), 31–37 (2017). https://doi.org/10.3345/kjp.2017.60.2.31
B. McCann-Crosby, M.J. Chen, S.K. Lyons, Y. Lin, M. Axelrad, J.E. Dietrich, V.R. Sutton, C.G. Macias, S. Gunn, L. Karaviti, Nonclassical congenital adrenal hyperplasia: targets of treatment and transition. Pediatr. Endocrinol. Rev. 12(2), 224–238 (2014)
A. Balsamo, A. Cicognani, L. Baldazzi, M. Barbaro, F. Baronio, M. Gennari, M. Bal, A. Cassio, K. Kontaxaki, E. Cacciari, CYP21 genotype, adult height, and pubertal development in 55 patients treated for 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 88(12), 5680–5688 (2003). https://doi.org/10.1210/jc.2003-030123
Z. Aycan, S. Akbuğa, E. Cetinkaya, G. Ocal, M. Berberoğlu, O. Evliyaoğlu, P. Adiyaman, Final height of patients with classical congenital adrenal hyperplasia. Turk. J. Pediatr. 51(6), 539–544 (2009)
G.P. Finkielstain, M.S. Kim, N. Sinaii, M. Nishitani, C. Van Ryzin, S.C. Hill, J.C. Reynolds, R.M. Hanna, D.P. Merke, Clinical characteristics of a cohort of 244 patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 97(12), 4429–4438 (2012). https://doi.org/10.1210/jc.2012-2102
L. Juan, M. Huamei, S. Zhe, L. Yanhong, C. Hongshan, C. Qiuli, Z. Jun, G. Song, D. Minlian, Near-final height in 82 Chinese patients with congenital adrenal hyperplasia due to classic 21-hydroxylase deficiency: a single-center study from China. J. Pediatr. Endocrinol. Metab. 29(7), 841–848 (2016). https://doi.org/10.1515/jpem-2015-0406
G. Hargitai, J. Solyom, T. Battelino, J. Lebl, Z. Pribilincova, R. Hauspie, J. Kovacs, F. Waldhauser, H. Frisch, M.-C.S. Group, Growth patterns and final height in congenital adrenal hyperplasia due to classical 21-hydroxylase deficiency. Results of a multicenter study. Horm. Res. 55(4), 161–171 (2001). https://doi.org/10.1159/000049990
I. Manoli, C. Kanaka-Gantenbein, A. Voutetakis, M. Maniati-Christidi, C. Dacou-Voutetakis, Early growth, pubertal development, body mass index and final height of patients with congenital adrenal hyperplasia: factors influencing the outcome. Clin. Endocrinol. 57(5), 669–676 (2002). https://doi.org/10.1046/j.1365-2265.2002.01645.x
H.J. Van der Kamp, B.J. Otten, N. Buitenweg, S.M. De Muinck Keizer-Schrama, W. Oostdijk, M. Jansen, H.A. Delemarre-de Waal, T. Vulsma, J.M. Wit, Longitud inal analysis of growth and puberty in 21-hydroxylase deficiency patients. Arch. Dis. Child. 87(2), 139–144 (2002).
W. Bonfig, S. Bechtold, H. Schmidt, D. Knorr, H.P. Schwarz, Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J. Clin. Endocrinol. Metab. 92(5), 1635–1639 (2007). https://doi.org/10.1210/jc.2006-2109
P.C. White, P.W. Speiser, Long-term consequences of childhood-onset congenital adrenal hyperplasia. Best. Pr. Res. Clin. Endocrinol. Metab. 16(2), 273–288 (2002). https://doi.org/10.1053/beem.2002.0198
L. Trinh, S. Nimkarn, M.I. New, K. Lin-Su, Growth and pubertal characteristics in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Pediatr. Endocrinol. Metab. 20(8), 883–891 (2007). https://doi.org/10.1515/jpem.2007.20.8.883
T.D. Nebesio, E.A. Eugster, Growth and reproductive outcomes in congenital adrenal hyperplasia. Int J. Pediatr. Endocrinol. 2010, 298937 (2010). https://doi.org/10.1155/2010/298937
T.M. Völkl, L. Öhl, M. Rauh, C. Schöfl, H.G. Dörr, Adrenarche and puberty in children with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Horm. Res. Paediatr. 76(6), 400–410 (2011). https://doi.org/10.1159/000333696
C. Dacou-Voutetakis, N. Karidis, Congenital adrenal hyperplasia complicated by central precocious puberty: treatment with LHRH-agonist analogue. Ann. N. Y Acad. Sci. 687, 250–254 (1993). https://doi.org/10.1111/j.1749-6632.1993.tb43873.x
T.C. van de Grift, B.P.C. Kreukels, dsd-LIFE: Breast development and satisfaction in women with disorders/differences of sex development. Hum. Reprod. 34(12), 2410–2417 (2019). https://doi.org/10.1093/humrep/dez230
G. Robin, C. Decanter, H. Baffet, S. Catteau-Jonard, D. Dewailly, [Steroid 21-hydroxylase deficiencies and female infertility: pathophysiology and management]. Gynecol. Obstet. Fertil. 42(6), 422–428 (2014). https://doi.org/10.1016/j.gyobfe.2014.04.005
L.G. Gomes, T.A.S.S. Bachega, B.B. Mendonca, Classic congenital adrenal hyperplasia and its impact on reproduction. Fertil. Steril. 111(1), 7–12 (2019). https://doi.org/10.1016/j.fertnstert.2018.11.037
K.A. Walters, V. Rodriguez Paris, A. Aflatounian, D.J. Handelsman, Androgens and ovarian function: translation from basic discovery research to clinical impact. J. Endocrinol. 242(2), R23–R50 (2019). https://doi.org/10.1530/JOE-19-0096
R.M. Mulaikal, C.J. Migeon, J.A. Rock, Fertility rates in female patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N. Engl. J. Med. 316(4), 178–182 (1987). https://doi.org/10.1056/NEJM198701223160402
J. Jaaskelainen, M. Hippelainen, O. Kiekara, R. Voutilainen, Child rate, pregnancy outcome and ovarian function in females with classical 21-hydroxylase deficiency. Acta Obstet. Gynecol. Scand. 79(8), 687–692 (2000)
O. Lekarev, K. Lin-Su, M.G. Vogiatzi, Infertility and reproductive function in patients with congenital adrenal hyperplasia: pathophysiology, advances in management, and recent outcomes. Endocrinol. Metab. Clin. North Am. 44(4), 705–722 (2015). https://doi.org/10.1016/j.ecl.2015.07.009
F. Gastaud, C. Bouvattier, L. Duranteau, R. Brauner, E. Thibaud, F. Kutten, P. Bougnères, Impaired sexual and reproductive outcomes in women with classical forms of congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 92(4), 1391–1396 (2007). https://doi.org/10.1210/jc.2006-1757
J. Słowikowska-Hilczer, A.L. Hirschberg, H. Claahsen-van der Grinten, N. Reisch, C. Bouvattier, U. Thyen, P. Cohen Kettenis, R. Roehle, B. Köhler, A. Nordenström, d-L. Group, Fertility outcome and information on fertility issues in individuals with different forms of disorders of sex development: findings from the dsd-LIFE study. Fertil. Steril. 108(5), 822–831 (2017). https://doi.org/10.1016/j.fertnstert.2017.08.013
H.F. Meyer-Bahlburg, C. Dolezal, S.W. Baker, M.I. New, Sexual orientation in women with classical or non-classical congenital adrenal hyperplasia as a function of degree of prenatal androgen excess. Arch. Sex. Behav. 37(1), 85–99 (2008). https://doi.org/10.1007/s10508-007-9265-1
M.H. Schernthaner-Reiter, S. Baumgartner-Parzer, H.C. Egarter, M. Krebs, A. Kautzky-Willer, K. Kirchheiner, A. Luger, M. Bayerle-Eder, Influence of genotype and hyperandrogenism on sexual function in women with congenital adrenal hyperplasia. J. Sex. Med. 16(10), 1529–1540 (2019). https://doi.org/10.1016/j.jsxm.2019.07.009
K. Hagenfeldt, P.O. Janson, G. Holmdahl, H. Falhammar, H. Filipsson, L. Frisen, M. Thoren, A. Nordenskjold, Fertility and pregnancy outcome in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum. Reprod. 23(7), 1607–1613 (2008). https://doi.org/10.1093/humrep/den118
A. Casteràs, P. De Silva, G. Rumsby, G.S. Conway, Reassessing fecundity in women with classical congenital adrenal hyperplasia (CAH): normal pregnancy rate but reduced fertility rate. Clin. Endocrinol. 70(6), 833–837 (2009). https://doi.org/10.1111/j.1365-2265.2009.03563.x
M. Bidet, C. Bellanné-Chantelot, M.B. Galand-Portier, J.L. Golmard, V. Tardy, Y. Morel, S. Clauin, C. Coussieu, P. Boudou, I. Mowzowicz, A. Bachelot, P. Touraine, F. Kuttenn, Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 95(3), 1182–1190 (2010). https://doi.org/10.1210/jc.2009-1383
J.C. Lo, M.M. Grumbach, Pregnancy outcomes in women with congenital virilizing adrenal hyperplasia. Endocrinol. Metab. Clin. North Am. 30(1), 207–229 (2001)
G. Papadakis, E.A. Kandaraki, E. Tseniklidi, O. Papalou, E. Diamanti-Kandarakis, Polycystic ovary syndrome and NC-CAH: distinct characteristics and common findings. a systematic review. Front Endocrinol. 10, 388 (2019). https://doi.org/10.3389/fendo.2019.00388
M.I. New, L. Ghizzoni, H. Meyer-Bahlburg, A. Khattab, D. Reichman, Z. Rosenwaks, Fertility in patients with nonclassical congenital adrenal hyperplasia. Fertil. Steril. 111(1), 13–20 (2019). https://doi.org/10.1016/j.fertnstert.2018.11.023
E. Trakakis, C. Loghis, D. Kassanos, Congenital adrenal hyperplasia because of 21-hydroxylase deficiency. A genetic disorder of interest to obstetricians and gynecologists. Obstet. Gynecol. Surv. 64(3), 177–189 (2009). https://doi.org/10.1097/OGX.0b013e318193301b
M. Pall, R. Azziz, J. Beires, D. Pignatelli, The phenotype of hirsute women: a comparison of polycystic ovary syndrome and 21-hydroxylase-deficient nonclassic adrenal hyperplasia. Fertil. Steril. 94(2), 684–689 (2010). https://doi.org/10.1016/j.fertnstert.2009.06.025
D. Pignatelli, Non-classic adrenal hyperplasia due to the deficiency of 21-hydroxylase and its relation to polycystic ovarian syndrome. Front. Horm. Res. 40, 158–170 (2013). https://doi.org/10.1159/000342179
H. Falhammar, H.F. Nyström, U. Ekström, S. Granberg, A. Wedell, M. Thorén, Fertility, sexuality and testicular adrenal rest tumors in adult males with congenital adrenal hyperplasia. Eur. J. Endocrinol. 166(3), 441–449 (2012). https://doi.org/10.1530/EJE-11-0828
N.M. Stikkelbroeck, B.J. Otten, A. Pasic, G.J. Jager, C.G. Sweep, K. Noordam, A.R. Hermus, High prevalence of testicular adrenal rest tumors, impaired spermatogenesis, and Leydig cell failure in adolescent and adult males with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 86(12), 5721–5728 (2001)
C. Bouvattier, L. Esterle, P. Renoult-Pierre, A.B. de la Perriere, F. Illouz, V. Kerlan, V. Pascal-Vigneron, D. Drui, S. Christin-Maitre, F. Galland, T. Brue, Y. Reznik, F. Schillo, D. Pinsard, X. Piguel, G. Chabrier, B. Decoudier, P. Emy, I. Tauveron, M.L. Raffin-Sanson, J. Bertherat, J.M. Kuhn, P. Caron, M. Cartigny, O. Chabre, D. Dewailly, Y. Morel, P. Touraine, V. Tardy-Guidollet, J. Young, Clinical outcome, hormonal status, gonadotrope axis, and testicular function in 219 adult men born with classic 21-hydroxylase deficiency. A French National Survey. J. Clin. Endocrinol. Metab. 100(6), 2303–2313 (2015). https://doi.org/10.1210/jc.2014-4124
M. Engels, K. Gehrmann, H. Falhammar, E.A. Webb, A. Nordenström, F.C. Sweep, P.N. Span, A.E. van Herwaarden, J. Rohayem, A. Richter-Unruh, C. Bouvattier, B. Köhler, B.B. Kortmann, W. Arlt, N. Roeleveld, N. Reisch, N.M.M.L. Stikkelbroeck, H.L. Claahsen-van der Grinten, d-L. group, Gonadal function in adult male patients with congenital adrenal hyperplasia. Eur. J. Endocrinol. 178(3), 285–294 (2018). https://doi.org/10.1530/EJE-17-0862
H.L. Claahsen-van der Grinten, B.J. Otten, M.M. Stikkelbroeck, F.C. Sweep, A.R. Hermus, Testicular adrenal rest tumours in congenital adrenal hyperplasia. Best practice & research. Clin. Endocrinol. Metab. 23(2), 209–220 (2009). https://doi.org/10.1016/j.beem.2008.09.007
M. Kocova, V. Janevska, V. Anastasovska, Testicular adrenal rest tumors in boys with 21-hydroxylase deficiency, timely diagnosis and follow-up. Endocr. Connect 7(4), 544–552 (2018). https://doi.org/10.1530/EC-18-0097
H. Falhammar, L. Frisen, C. Norrby, C. Almqvist, A.L. Hirschberg, A. Nordenskjold, A. Nordenstrom, Reduced frequency of biological and increased frequency of adopted children in males with 21-hydroxylase deficiency: a Swedish population-based national cohort study. J. Clin. Endocrinol. Metab. 102(11), 4191–4199 (2017). https://doi.org/10.1210/jc.2017-01139
T.F. King, M.C. Lee, E.E. Williamson, G.S. Conway, Experience in optimizing fertility outcomes in men with congenital adrenal hyperplasia due to 21 hydroxylase deficiency. Clin. Endocrinol. 84(6), 830–836 (2016). https://doi.org/10.1111/cen.13001
D. Pignatelli, B.L. Carvalho, A. Palmeiro, A. Barros, S.G. Guerreiro, D. Maçut, The complexities in genotyping of congenital adrenal hyperplasia: 21-hydroxylase deficiency. Front Endocrinol. 10, 432 (2019). https://doi.org/10.3389/fendo.2019.00432
L. Paizoni, M.K. Auer, H. Schmidt, A. Hübner, M. Bidlingmaier, N. Reisch, Effect of androgen excess and glucocorticoid exposure on metabolic risk profiles in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Steroid Biochem. Mol. Biol. 197, 105540 (2019). https://doi.org/10.1016/j.jsbmb.2019.105540
H. Falhammar, L. Frisen, A.L. Hirschberg, C. Norrby, C. Almqvist, A. Nordenskjold, A. Nordenstrom, Increased cardiovascular and metabolic morbidity in patients with 21-hydroxylase deficiency: a Swedish Population-Based National Cohort Study. J. Clin. Endocrinol. Metab. 100(9), 3520–3528 (2015). https://doi.org/10.1210/JC.2015-2093
L. de Vries, Y. Lebenthal, M. Phillip, S. Shalitin, A. Tenenbaum, R. Bello, Obesity and cardiometabolic risk factors in children and young adults with non-classical 21-hydroxylase deficiency. Front Endocrinol. 10, 698 (2019). https://doi.org/10.3389/fendo.2019.00698
H. Falhammar, H. Claahsen-van der Grinten, N. Reisch, J. Slowikowska-Hilczer, A. Nordenström, R. Roehle, C. Bouvattier, B.P.C. Kreukels, B. Köhler, d-L. group, Health status in 1040 adults with disorders of sex development (DSD): a European multicenter study. Endocr. Connect 7(3), 466–478 (2018). https://doi.org/10.1530/EC-18-0031
K. Ariyawatkul, S. Tepmongkol, S. Aroonparkmongkol, T. Sahakitrungruang, Cardio-metabolic risk factors in youth with classical 21-hydroxylase deficiency. Eur. J. Pediatr. 176(4), 537–545 (2017). https://doi.org/10.1007/s00431-017-2875-2
M.A. Alwashih, D.G. Watson, R. Andrew, R.H. Stimson, M. Alossaimi, G. Blackburn, B.R. Walker, Plasma metabolomic profile varies with glucocorticoid dose in patients with congenital adrenal hyperplasia. Sci. Rep. 7(1), 17092 (2017). https://doi.org/10.1038/s41598-017-17220-5
M.S. Kim, A. Ryabets-Lienhard, A. Dao-Tran, S.D. Mittelman, V. Gilsanz, S.M. Schrager, M.E. Geffner, Increased abdominal adiposity in adolescents and young adults with classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 100(8), E1153–E1159 (2015). https://doi.org/10.1210/jc.2014-4033
P. Sartorato, E. Zulian, S. Benedini, B. Mariniello, F. Schiavi, F. Bilora, G. Pozzan, N. Greggio, A. Pagnan, F. Mantero, C. Scaroni, Cardiovascular risk factors and ultrasound evaluation of intima-media thickness at common carotids, carotid bulbs, and femoral and abdominal aorta arteries in patients with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 92(3), 1015–1018 (2007). https://doi.org/10.1210/jc.2006-1711
H. Falhammar, L. Frisen, C. Norrby, A.L. Hirschberg, C. Almqvist, A. Nordenskjold, A. Nordenstrom, Increased mortality in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 99(12), E2715–E2721 (2014). https://doi.org/10.1210/jc.2014-2957
M. Delvecchio, L. Soldano, A. Lonero, A. Ventura, P. Giordano, L. Cavallo, M. Grano, G. Brunetti, M.F. Faienza, Evaluation of impact of steroid replacement treatment on bone health in children with 21-hydroxylase deficiency. Endocrine 48(3), 995–1000 (2015). https://doi.org/10.1007/s12020-014-0332-9
N.M. Stikkelbroeck, W.J. Oyen, G.J. van der Wilt, A.R. Hermus, B.J. Otten, Normal bone mineral density and lean body mass, but increased fat mass, in young adult patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 88(3), 1036–1042 (2003)
P. Christiansen, C. Mølgaard, J. Müller, Normal bone mineral content in young adults with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Horm. Res. 61(3), 133–136 (2004). https://doi.org/10.1159/000075588
H. Falhammar, H. Filipsson, G. Holmdahl, P.O. Janson, A. Nordenskjold, K. Hagenfeldt, M. Thoren, Fractures and bone mineral density in adult women with 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 92(12), 4643–4649 (2007). https://doi.org/10.1210/jc.2007-0744
D. El-Maouche, S. Collier, M. Prasad, J.C. Reynolds, D.P. Merke, Cortical bone mineral density in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin. Endocrinol. 82(3), 330–337 (2015). https://doi.org/10.1111/cen.12507
H. Falhammar, H. Filipsson Nystrom, A. Wedell, K. Brismar, M. Thoren, Bone mineral density, bone markers, and fractures in adult males with congenital adrenal hyperplasia. Eur. J. Endocrinol. 168(3), 331–341 (2013). https://doi.org/10.1530/EJE-12-0865
F. Ceccato, M. Barbot, N. Albiger, M. Zilio, P. De Toni, G. Luisetto, M. Zaninotto, N.A. Greggio, M. Boscaro, C. Scaroni, V. Camozzi, Long-term glucocorticoid effect on bone mineral density in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Eur. J. Endocrinol. 175(2), 101–106 (2016). https://doi.org/10.1530/EJE-16-0104
H. Falhammar, L. Frisen, A.L. Hirschberg, A. Nordenskjold, C. Almqvist, A. Nordenstrom, Increased risk of autoimmune disorders in 21-hydroxylase deficiency: A Swedish Population-Based National Cohort Study. J. Endocr. Soc. 3(5), 1039–1052 (2019). https://doi.org/10.1210/js.2019-00122
L. Karlsson, M. Barbaro, E. Ewing, D. Gomez-Cabrero, S. Lajic, Epigenetic alterations associated with early prenatal dexamethasone treatment. J. Endocr. Soc. 3(1), 250–263 (2019). https://doi.org/10.1210/js.2018-00377
H. Engberg, A. Butwicka, A. Nordenstrom, A.L. Hirschberg, H. Falhammar, P. Lichtenstein, A. Nordenskjold, L. Frisen, M. Landen, Congenital adrenal hyperplasia and risk for psychiatric disorders in girls and women born between 1915 and 2010: a total population study. Psychoneuroendocrinology 60, 195–205 (2015). https://doi.org/10.1016/j.psyneuen.2015.06.017
H. Falhammar, A. Butwicka, M. Landen, P. Lichtenstein, A. Nordenskjold, A. Nordenstrom, L. Frisen, Increased psychiatric morbidity in men with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 99(3), E554–E560 (2014). https://doi.org/10.1210/jc.2013-3707
B. Lenz, C.P. Müller, C. Stoessel, W. Sperling, T. Biermann, T. Hillemacher, S. Bleich, J. Kornhuber, Sex hormone activity in alcohol addiction: integrating organizational and activational effects. Prog. Neurobiol. 96(1), 136–163 (2012). https://doi.org/10.1016/j.pneurobio.2011.11.001
S.C. Mueller, E.M. Grissom, G.P. Dohanich, Assessing gonadal hormone contributions to affective psychopathologies across humans and animal models. Psychoneuroendocrinology 46, 114–128 (2014). https://doi.org/10.1016/j.psyneuen.2014.04.015
A. Bachelot, P. Touraine, [Health status of adults with congenital adrenal hyperplasia due to 21-hydroxylase deficiency]. Presse Med 43(4 Pt 1), 428–437 (2014). https://doi.org/10.1016/j.lpm.2013.10.006
H. Falhammar, H.F. Nystrom, M. Thoren, Quality of life, social situation, and sexual satisfaction, in adult males with congenital adrenal hyperplasia. Endocrine 47(1), 299–307 (2014). https://doi.org/10.1007/s12020-013-0161-2
A. Strandqvist, H. Falhammar, P. Lichtenstein, A.L. Hirschberg, A. Wedell, C. Norrby, A. Nordenskjold, L. Frisen, A. Nordenstrom, Suboptimal psychosocial outcomes in patients with congenital adrenal hyperplasia: epidemiological studies in a nonbiased national cohort in Sweden. J. Clin. Endocrinol. Metab. 99(4), 1425–1432 (2014). https://doi.org/10.1210/jc.2013-3326
B. Dudzińska, J. Leubner, M. Ventz, M. Quinkler, Sexual well-being in adult male patients with congenital adrenal hyperplasia. Int J. Endocrinol. 2014, 469289 (2014). https://doi.org/10.1155/2014/469289
A. Nordenskjold, G. Holmdahl, L. Frisen, H. Falhammar, H. Filipsson, M. Thoren, P.O. Janson, K. Hagenfeldt, Type of mutation and surgical procedure affect long-term quality of life for women with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 93(2), 380–386 (2008). https://doi.org/10.1210/jc.2007-0556
C.F. Mooij, J.M. Kroese, H.L. Claahsen-van der Grinten, C.J. Tack, A.R:. Hermus, Unfavourable trends in cardiovascular and metabolic risk in paediatric and adult patients with congenital adrenal hyperplasia? Clin. Endocrinol. (Oxf.) 73(2), 137–146 (2010). https://doi.org/10.1111/j.1365-2265.2009.03690.x
I. Nermoen, E.S. Husebye, J. Svartberg, K. Løvås, Subjective health status in men and women with congenital adrenal hyperplasia: a population-based survey in Norway. Eur. J. Endocrinol. 163(3), 453–459 (2010). https://doi.org/10.1530/EJE-10-0284
E. Daae, K.B. Feragen, I. Nermoen, H. Falhammar, Psychological adjustment, quality of life, and self-perceptions of reproductive health in males with congenital adrenal hyperplasia: a systematic review. Endocrine 62(1), 3–13 (2018). https://doi.org/10.1007/s12020-018-1723-0
Acknowledgements
We thank Jorddy N Cruz for the help in designing the structural model of the CYP21A2 enzyme. Open access funding provided by Karolinska Institute.
Funding
This study was funded by Magnus Bergvall Foundation (Grant Number 2017–02138, 2018–02566, and 2019-03149).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kocova, M., Anastasovska, V. & Falhammar, H. Clinical outcomes and characteristics of P30L mutations in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrine 69, 262–277 (2020). https://doi.org/10.1007/s12020-020-02323-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-020-02323-3