
Abstract
Wolbachia pipientis (Rickettsiales), an obligate intracellular alphaproteobacterium in insects, manipulates host reproduction to maximize invasion of uninfected insect populations. Modification of host population structure has potential applications for control of pest species, particularly if Wolbachia can be maintained, manipulated, and genetically engineered in vitro. Although Wolbachia maintains an obligate mutualism with genome stability in nematodes, arthropods can be co-infected with distinct Wolbachia strains, and horizontal gene transfer between strains is potentially mediated by WO phages encoded within Wolbachia genomes. Proteomic analysis of a robust, persistent infection of a mosquito cell line with wStr from the planthopper, Laodelphax striatellus, revealed expression of a full array of WO phage genes, as well as nine of ten non-phage genes that occur between two distinct clusters of WOMelB genes in the genome of wMel, which infects Drosophila melanogaster. These non-phage genes encode potential host-adaptive proteins and are expressed in wStr at higher levels than phage structural proteins. A subset of seven of the non-phage genes is flanked by highly conserved non-coding sequences, including a putative promoter element, that are not present in a syntenically arranged array of homologs in plasmids from three tick-associated Rickettsia spp. These studies expand our understanding of wStr in a host cell line derived from the mosquito, Aedes albopictus, and provide a basis for investigating conditions that favor the lytic phase of the WO phage life cycle and recovery of infectious phage particles.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Wolbachia (Anaplasmataceae; Rickettsiales) is an obligate intracellular alpha proteobacterium that engages in distinctive interactions with invertebrate hosts, depending on whether they are nematodes or arthropods. In nematodes, Wolbachia functions as a mutualist (Taylor et al. 2005; Comandatore et al. 2013) and is required for host survival. The genomes of nematode-associated Wolbachia supergroup C- and D-strains coevolve with their host genomes and lack mobile genetic elements that are abundant in strains from the arthropod-associated supergroups A and B, hereafter designated as WOL-A and WOL-B (Wu et al. 2004; Cordaux et al. 2008; Ishmael et al. 2009; Newton and Bordenstein 2011). Arthropod-associated Wolbachia are typically vertically transmitted parasites that manipulate reproduction to invade uninfected populations of their hosts (Zug and Hammerstein 2014), but can also be transmitted horizontally to new host species by predators, parasites, and parasitoids (reviewed in Zug et al. 2012). Arthropod hosts that are co-infected with multiple Wolbachia strains have provided an arena for genetic recombination, which is reflected in present-day strains by mosaic gene sequences, a lack of phylogenetic congruence between Wolbachia strains and arthropod hosts, and the presence of mobile genetic elements in Wolbachia genomes.
Arthropod-associated Wolbachia genomes typically contain one or more WO prophages consisting of a complete set of genes with a modular organization and encoding head, baseplate, tail, and all other proteins required for the lytic cycle and packaging of a potentially infectious phage (Kent et al. 2011). WO prophages have been likened to lysogenic forms of bacteriophage lambda in Escherichia coli (Kent and Bordenstein 2010). Recombination or transposition near prophage termini is thought to be a major mechanism by which Wolbachia acquires DNA from other prokaryotic taxa (Ishmael et al. 2009; Tanaka et al. 2009; Kent et al. 2011; Duplouy et al. 2013). Foreign DNA associated with WO prophages encodes ankyrin repeat proteins, host cell adhesion and invasion factors, and type IV secretion system effectors, which have potential host-adaptive functions (Tanaka et al. 2009; Kent et al. 2011; Siozios et al. 2013). In addition to the complete prophages typically present in WOL-A and WOL-B genomes, degenerate prophages are common and may occur in isolation as in A wSol (hereafter, supergroup designations are indicated by superscripts preceding the strain name) from the fig wasp, Ceratosolen solmsi, whose degenerate prophage has the highest known proportion (27.6%) of pseudogenes and lacks the tail module (Wang et al. 2013). Genome reduction in Wolbachia may in fact target prophage sequences, as has been suggested for A wRec from Drosophila recens (Metcalf et al. 2014).
Here, we define the prophage proteome from B wStr, which maintains a robust infection in C/wStr1 mosquito cells. Proteomic analyses suggest that the B wStr genome contains a prophage resembling WOMelB from the A wMel genome that infects Drosophila melanogaster. DNA sequence analyses verified detection of peptides corresponding to proteins encoded by a syntenic array of genes present in the B wStr genome and in the WOMelB prophage as well as in plasmids from three Rickettsia spp. associated with ixodid ticks. Expression of proteins representing one or more complete prophages suggests that a lytic cycle occurs in C/wStr1 cells, which provide advantages of scale and ease of manipulation for future identification of conditions that favor isolation of infectious phage particles.
Materials and Methods
Cell culture.
Uninfected Aedes albopictus C7-10 and B wStr-infected C/wStr1 cells were maintained in Eagle’s minimal medium supplemented with 5% fetal bovine serum at 28–30°C in a 5% CO2 atmosphere as described previously (Shih et al. 1998; Fallon et al. 2013).
Polymerase chain reaction, DNA cloning and sequencing, and sequence identity comparisons.
The polymerase chain reaction (PCR) was used to amplify the B wStr homologs of the A wMel loci WD0611–WD620 from template DNA prepared from Wolbachia enriched by fractionation of C/wStr1 cells by density gradient centrifugation (GF-50/60) as detailed elsewhere (Baldridge et al. 2014). We obtained 21 PCR products using a panel of 69 primers (Table S1), GoTaq DNA polymerase (Promega, Madison, WI), and a Techne TC-312 cycler (Staffordshire, UK). Cycle parameters were 1 cycle at 94°C for 2 min, 35 cycles at 94°C for 35 s, 53°C for 35 s, 72°C for 1 min, followed by 1 cycle at 72°C for 5 min. Extension time was increased to 2 min for products ≥1000 bp. PCR products were cloned in the pCR4-TOPO vector with the TOPO-TA Cloning Kit for Sequencing (Life Technologies, Grand Island, NY), and two or more clones each were sequenced at the University of Minnesota BioMedical Genomics Center. DNA and protein alignments were executed with the Clustal Omega program (Sievers et al. 2011). Alignments were edited and modified using Microsoft Word. All nucleotide and protein sequence identity comparisons were executed with the BLASTn and BLASTp algorithms available at http://blast.ncbi.nlm.nih.gov.
Mass spectrometry, peptide detection, and protein identification.
LC–MS/MS on LTQ and Orbitrap Velos mass spectrometers was executed as described previously (Baldridge et al. 2014). Tandem mass spectra were extracted by Sequest (Thermo Fisher Scientific, San Jose, CA; version SRF v.3 or version 27, rev. 12); Sequest parameters and protein sequence database information have been reported previously (see Table S4 in Baldridge et al. 2014). Data were searched against an rs_wolbachia_aedes_v200808_cRAP_flavivirusREV database that contained 74,570 protein entries from sequenced Wolbachia genomes, the Aedes aegypti genome, and flavivirus genomes available as of July 2011. Assembled Wolbachia genomes included those of the B wPip WOL-B strain associated with Culex pipiens quinquefasciatus Pel mosquitoes from Sri Lanka (Klasson et al. 2008), the Drosophila-associated WOL-A strains, A wMel (Wu et al. 2004) and A wRi (Klasson et al. 2009), and the nematode-associated WOL-D strain, D wBm (Foster et al. 2005). Incomplete genomes included the Drosophila-associated A wAna and A wWill WOL-A strains. Scaffold (version 4.2.1, Proteome Software Inc., Portland, OR) was used to validate MS/MS-based peptide and protein identifications. As detailed in the “Results,” the original MS search database was modified to support a refined search by inclusion of proteins deduced from sequenced B wStr genes.
Relative abundance estimation and statistical analysis.
RAL, or relative abundance level, is based on counts of unique peptides in four MS data sets, as shown for data sets MS-D, MS-E, MS-F, and MS-G in Table S2. In each column, values indicate number of peptides\% protein coverage (the percentage of amino acids in the full-length protein represented by MS peptides). As detailed in Baldridge et al. (2014), RAL scores indicate the relative abundance of a particular protein relative to the total of 790 Wolbachia proteins detected by mass spectrometry in C/wStr1 cells. For proteins grouped according to functional class, RAL scores ranged from a maximum of 7.7 to a minimum of 1.0 (see Table 4 in Baldridge et al. 2014). SR, or studentized residuals, are derived from a statistical analysis of RAL scores normalized to protein mass and indicate deviance from expected values adjusted for estimated standard deviation from the mean. A protein of average abundance relative to all other Wolbachia proteins identified in C/wStr1 cells has an SR of 0, while above-average abundance is associated with a positive SR value and below-average abundance is associated with a negative value, with overall scores ranging from −2.36 to +3.69. The details of the statistical analysis are given in Baldridge et al. (2014). All tests of association were performed with SAS v9.3 (Cary, NC; http://www.sas.com/en_us/home.html/).
Results
The WO phage proteome from the C/wStr1 cell line.
We have previously shown that exponentially growing A. albopictus C/wStr1 cells express a B wStr proteome of nearly 800 proteins, using a stringent threshold requiring detection of multiple peptides from the same protein within at least one of four MS data sets (Baldridge et al. 2014). Based on those criteria, we identified 39 WO phage proteins, some of which were mosaic in the sense that they were represented by peptides corresponding to homologs from distinct Wolbachia genomes. As a group, these 39 phage/virus related proteins were expressed at relatively low abundance, with a relative abundance level (RAL) score of 1.5, compared to 7.7 for the most highly abundant functional group with protein modification/chaperone activities, and the lowest RAL of 1.0 for proteins of unknown function (Baldridge et al. 2014). We attribute the aggregate low RAL score for phage-related proteins to our efforts to harvest B wStr from cells in exponential growth phase. To a first approximation, these observations indicate that under optimal conditions for host cells, WO phage genes are expressed at detectable levels and possibly contribute to variation in Wolbachia levels among individual cells within a population.
Here, we examined WO phage expression more closely to determine whether the data supported expression of packaged phage. WO phage genes evolve rapidly (Kent et al. 2011), and our stringent criteria for inclusion in the original B wStr proteome therefore underestimated proteins encoded by prophage genomes. Re-examination of the original data including proteins represented by multiple homologs uncovered a final B wStr prophage proteome of 119 proteins that included previously unreported proteins represented by single peptides (Tables 1 and S2). In aggregate, head (20), baseplate (9), and tail (10) proteins necessary for formation of viral particles accounted for 33% of the phage proteome, while 19 proteins (16%) have functions in DNA recombination (7 proteins), replication (5 proteins), or modification (7 proteins). An additional 19 proteins (16%) have known or likely roles in phage or Wolbachia interactions with host cells and virulence, while the remaining 41 proteins (34%) are homologs of WO phage proteins whose functions remain unknown. The prophage proteome included 16 proteins encoded by orphan genes in sequenced Wolbachia genomes and nine proteins encoded by foreign genes believed to have been acquired by a WO-B phage (Ishmael et al. 2009; Kent et al. 2011), seven of which occur as syntenic arrays on rickettsial plasmids. Thus C/wStr1 cells express proteins representing all modules and functional classes encoded by WO prophages in annotated genomes of representative Wolbachia strains, most of which correspond to intact prophages with potential lytic activity. For example, of 61 proteins that matched WO prophage genes in B wPip (Table S2), only four correspond to orphan genes.
Similarities to WOMelB and rickettsial plasmids.
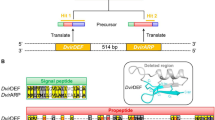
The most abundantly represented B wStr WO phage proteins corresponded to homologs encoded by WOMelB (Fig. 1), which are depicted schematically by crosses below arrows representing open reading frames. WOMelB is organized as two blocks of genes: B1: WD0565 to WD0610 and B2: WD0633 to WD0644, relative to a single block of genes in the degenerate WOMelA (WD0261 to WD0288; see Fig. 1 in Kent et al. 2011). The 35-kb gap separating the B1 and B2 regions includes ten non-phage genes (WD0611–WD0620), of which nine were represented by B wStr peptides. WD0612–WD0618 also occur in syntenic arrangement on rickettsial plasmids (see below). In addition, we recovered B wStr peptides corresponding to ten proteins encoded by WOMelA and 12 proteins encoded by phage orphan genes in the A wMel, A wAna, and A wSim genomes (Tables 1 and S2). Overall, the WO phage peptides recovered in this analysis suggest that intact prophages similar to WOMelB (Fig. 1) and WOPip4/Pip5 (Tables 1 and S2) occur in the B wStr genome. We note that the mean relative abundance levels (RAL; for details see Baldridge et al. 2014) of proteins from potentially intact prophages range from 0.97 to 1.26 (Table S2), and are two to fourfold higher than those of degenerate prophage and orphan proteins (range 0.30–0.62). These expression levels, combined with detection of critical proteins such as the phage recombinase/integrase, RepA, and others involved in DNA replication, modification, and packaging, suggest that B wStr encodes at least one prophage capable of excision and completion of the lytic life cycle.

Schematic depiction of proteins in the BwStr WO phage proteome matched to peptides from homologs encoded by the A wMel WO-B prophage. As detailed by Kent et al. (2011), A wMel WO-B occurs as two blocks of phage genes (B1 and B2) separated by ∼35 kb of non-phage DNA. The central schematic represents the complete WO-B phage extending from WD0565 to WD0644. The region labeled “gap” corresponds to 35 kb separating B1 and B2 that contains foreign genes (WD0611 to WD0620) shown at top as light green arrows representing open reading frames. The bracket indicates genes that occur as syntenic arrays of homologs on three rickettsial plasmids. At the bottom, open reading frames of genes in the B1 and B2 blocks are indicated by color-coded arrows oriented in the direction of transcription (phage head, purple; baseplate, dark blue; tail, green; recombinase, pink; replication, light blue; transposase, yellow; virulence factors, orange as depicted in Fig. 1 in Kent et al. 2011). Stars designate proteins matched to B wStr peptides.
Homologs of rickettsial plasmid genes.
The phage-associated WD0611–WD0620 gene array in WOMelB (Fig. 1) is of special interest because it encodes proteins that may mediate host–microbe interactions and is conserved in other WO phages, such as AwRi and AwVitA2 (Kent et al. 2011), and because homologs of WD0612–WD0618 occur as syntenic arrays on plasmids from Rickettsia spp. associated with ixodid ticks (see Table S3 for all accession numbers). We used PCR amplification and DNA sequencing to validate proteomic evidence for expression of WD0611–WD0620 homologs and to determine whether they occur as a block of contiguous genes in the B wStr genome (Fig. 2 and Table S2, entries 51–59). In the B wStr genome, a 13,127-bp sequence based on 21 overlapping PCR products (Fig. 2B and Table S3) begins near the 5′ end of the WD0611 homolog and ends 115 bp upstream of the WD0620 start codon (Fig. 2A ). In addition to 53 unique peptides detected originally (Baldridge et al. 2014) and represented by star symbols below the genes in Fig. 2A , the B wStr sequence data resulted in detection of 42 new peptides (stars above genes) including 35 from WD0611–WD0616, three from the highly conserved WD0617 and WD0618, and four from the slightly less conserved WD0620. BLASTn comparisons (Table 2) confirmed that the WD0611–WD0620 genes from B wStr have homologs in WOL-A and WOL-B genomes and that the WD0612–WD0618 genes have homologs on three rickettsial plasmids. These genes do not occur in nematode-associated WOL-C/D genomes, which lack prophages.

Schematic map of PCR-amplified B wStrB homologs of prophage-associated genes and genes on rickettsial plasmids. (A) The B wStr genes are depicted as arrowheads oriented in the direction of transcription. Genes are identified by WOMelB locus tags (Genbank Acc. # NC_002978.6). Light shading indicates genes present in both WO prophages and in plasmids from Rickettsia spp. Downward arrows indicate sites of conserved non-coding regions (see Fig. 3). The diamond symbols below and above the arrows indicate unique peptides identified in the original and refined searches of the MS data sets, respectively. See Table 3 for protein identities. (B) Horizontal lines represent cloned PCR amplification products. See Table S1 for primers. (C) Scale marker in 1-kb increments.
Sequence comparisons between Wolbachia strains.
Nucleotide identities of the B wStr WD0611–WD0616 homologs to WOL-B genes ranged from 96 to 99% (Table 2; italicized values), with the exception of WD0614–WD0616 in B wNo, which fell into the range (85–92%) of WOL-A strains. Pairwise comparisons between A wHa and B wNo homologs were 99% identical, suggesting that B wNo has acquired the WOL-A genes through genetic exchange during co-infection, which occurs in some populations of Drosophila simulans (James et al. 2002). In contrast to the differential pattern of strain-related identities of WD0611–WD0616, WD0617–WD0620 were 96–99% identical among all strains with the exception of the degenerate prophage from A wSol, in which WD0616 and 0617 are pseudogenes. Finally, deduced amino acid identities were similar to nucleotide identities for all genes except WD0611, where values were 5–6% lower in WOL-A strain comparisons. We noted that WOL-A homologs were slightly longer and contained conserved amino acid substitutions relative to WOL-B homologs. These differences may reflect differential selection pressures on WD0611 versus other genes in WOL-A versus B strains.
Sequence comparisons between B wStr and rickettsial plasmids.
B wStr WD0612–0618 nucleotide identities to homologs from three rickettsial endosymbionts of Ixodes ticks ranged from 70 to 80% (Table 2), with the internal genes WD0614, 0615, and 0616 having the lowest identities. Concatenated sequences of WD0612–0618 from the three plasmids shared 95–96% identity, and pairwise comparisons of individual pREIS2 genes to those from the representative WOL-A (A wHa) and WOL-B (B wStr) strains showed differences of ≤1% among the rickettsial plasmid genes. In comparisons to the B wStr sequences, the rickettsial WD0612, 0614, 0615, and 0616 homologs had deduced amino acid identities that were 7–9% lower than nucleotide identities versus only 0–3% lower in WD0613, 0617, and 0618 comparisons.
Conserved sequences flanking the Wolbachia WD0612–0618 homologs contain potential transcriptional regulatory elements.
In B wStr, the WD0612–WD0618 homologs were arranged as directly adjacent or overlapping ORFs, suggesting possible organization as an operon. At the 5′ end, WD0612 was separated from the upstream WD0611 by 125 bp, and at the 3′ end, WD0618 was separated from WD0619 by 95 bp (Fig. 2A ). These flanking sequences had no significant BLAST similarities to one another and were not self-complementary. The 125-bp non-coding sequence (Fig. 3) is highly conserved among WOL-B strains (97–98% nucleotide identities) but less so (range 82–83%) with WOL-A strains (Table 2). The B wStr 125-bp sequence contains two short direct repeats, a –35 box and –10 box in a potential prokaryotic promoter, and interaction sites for several E. coli DNA-binding proteins and transcription factors (Fig. 3A ), including ihf, involved in bacteriophage lambda integration and recombination; hns, bacterial chromosome organization and global modulation of gene expression; fnr, an oxygen-responsive regulator; rpoD17, a “housekeeping” sigma factor that interacts with RNA polymerase; cytR, involved in nucleoside uptake and metabolism; cpxR, involved in expression of outer membrane porin proteins; and lrp, a metabolic regulator involved in amino acid metabolism. Although the presence of these potential transcriptional regulatory elements is intriguing, their functional context is unclear because the WD0612–WD0618 proteins are expressed at variable levels and represent a diverse array of potential functions. Moreover, the highly reduced genomes of Wolbachia strains encode a small suite of known transcription factors relative to E. coli, and their functions are largely unexplored. The B wStr 95-bp sequence (Fig. 3B ) following the 3′ end of WD0618 is also highly conserved (98–100% identity) among all ten Wolbachia strains except the degenerate A wSol (89%). Although the sequence has a potential promoter, it lacks transcription factor binding sites. Uniquely, the consecutive gene, WD0619, was not represented in the B wStr proteome, while WD0620, encoded on the opposite strand, was represented by 12 peptides (Fig. 2A ).

Non-coding sequences in the WD0611–WD0620 syntenic array. Sequences of the non-coding 125-bp region upstream of WD0612 (A) and the 95-bp region downstream of WD0618 (B) were annotated using Softberry BPROM prediction (http://linux1.softberry.com/berry.phtml?topic=bprom&group=programs&subgroup=gfindb; Solovyev and Salamov 2011). In A and B, the –35 and –10 boxes associated with predicted promoters are indicated in white font with black shading, and an arrowhead above the sequence marks the promoter site. (A) black and gray horizontal arrows above the sequence indicate two sets of direct repeats; positions of DNA-binding protein and transcription factor interaction sites described in the “Results” are indicated by bars below the sequence. The ATG start site for WD0612 is indicated in white font with black shading. (B) No predicted transcription factor binding sites occur in the 95-bp sequence.
Relative abundance of WD0611–WD0620 homologs in C/wStr1 cells.
With the exception of WD0619, for which no B wStr peptides were recovered, protein coverage by MS/MS detected peptides from WD0611–WD0620 homologs ranged from 5 to 58% (Table 3). We estimated protein relative abundance levels (RAL) using studentized residuals (SR), a measure of deviance from expected values adjusted for estimated standard deviation from the mean as reported in Table S4, in which the WD0611–0620 homologs are in blue font. WD0612 and WD0613 were the most abundantly expressed, while WD0614, WD0615, and WD0616 were among the least abundant (Table 3).
WD0611 encodes a UDP-N-acetylglucosamine pyrophosphorylase, with slightly above-average abundance (RAL = 4.0; SR = 0.20). The enzyme contains N- and C-terminal domains with uridyl- and acetyltransferase activities and catalyzes the final steps in synthesis of UDP-N-acetylglucosamine, which is an essential precursor in lipopolysaccharide metabolic pathways. Although Wolbachia does not synthesize a cell wall, it expresses all proteins necessary for lipid II synthesis, which is required for cell division (Vollmer et al. 2013). WD0612 encodes an abundant NAD-dependent epimerase/dehydratase (RAL = 8.0; SR = 1.11) that is a member of the WcaG group of proteins (COG0451) involved in lipopolysaccharide biosynthesis, bacterial cell envelope biogenesis, and modification of surface carbohydrates. WD0612 from B wStr has 58% BLASTp identity to UDP-glucose 4-epimerase from Chondromyces apiculatus (EYF00501.1; Table 4), and its probable role in cell surface polysaccharide modification may intersect with that of the slightly below-average abundance UDP-glucose 6-dehydrogenase (WD0620; RAL = 3.5; SR = −0.02).
WD0613 (RAL = 10; SR = 0.49) is particularly interesting because it contains a glycosyl transferase type 1 ExpE7-like domain (cd03823) in the N-terminal half of the protein. In Sinorhizobium meliloti, ExpE7 is involved in biosynthesis of galactoglucan exopolysaccharide II (Becker et al., 1997), which facilitates plant host cell adhesion and invasion, provides anti-oxidant defense, and may modulate plant defense responses during bacterial colonization (Lehman and Long 2013). The C-terminal half of WD0613 contains a superfamily domain (cd01335) typical of enzymes that use a molecule of S-adenosylmethionine in close proximity to 4Fe-4Fs iron–sulfur clusters to generate a deoxyadenosyl radical. A SPASM domain (pfam13186) located near the C-terminus provides an additional binding site for iron–sulfur clusters. These domains often occur in enzymes involved in biosynthesis of vitamins, coenzymes, and antibiotics or modification of other proteins. WD0613 is thus a potential multi-functional protein that may influence Wolbachia host interactions through modification of membrane proteins and surface interactions or supplementation of host cells with vitamins or coenzymes.
SR values indicate that WD0614 and 0615 have below-average abundances in C/wStr1 cells. WD0614 contains an unusual fusion of an N-terminal KWG Leptospira repeat domain (pfam14903) to an O-methyltransferase type 2 domain (Gillespie et al. 2012). In many prokaryotes, methylation of DNA protects against degradation by restriction enzymes, which have not been identified in Wolbachia. WD0615 is a phytanoyl-CoA dioxygenase, which catalyzes conversion of alpha-ketoglutarate and phytanoyl-CoA to succinate and 2-hydroxyphytanoyl-CoA. In oceanic cyanobacteria, generation of succinate by a cyanophage-borne phytanoyl-CoA dioxygenase was suggested to play a role in energy generation under nutritional stress conditions (Sullivan et al. 2010).
WD0616 (SR = −2.68), an ABC transporter permease/ATP binding protein with a possible role in multidrug efflux and/or iron–sulfur cluster transport, is one of the least abundant proteins in the B wStr proteome. The WD0619 GlpT/PgpT/UhpT transporter with a predicted function in phosphoglycerate uptake, if expressed, was below the limit of detection by LC–MS/MS. We note that the majority of transporter proteins in the B wStr proteome were of below-average abundance (SR < 0, Table S4, Baldridge et al. 2014).
Two l-allo-threonine aldolases (WD0617 and 0618) with below-average abundances interconvert l-3-hydroxy-α-amino acids to glycine and an aldehyde in reactions similar to those catalyzed in B wStr by an abundant serine hydroxymethyltransferase (Baldridge et al. 2014). WD0617 and 0618 share only 31% amino acid identity and may be specialized for different and as yet unknown functions (Contestabile et al. 2001; diSalvo et al. 2014).
Although BLASTn comparisons indicated insufficient sequence identities to support broad phylogenetic analyses of WD0611–WD0620 from B wStr, BLASTp comparisons suggest that homologs may be present in distantly related bacteria (Table 4). To evaluate these relationships, A wMel values are in bold as representative Wolbachia reference values. Species in which two or more syntenically arranged homologs may occur are underlined. These include marine sulfate-reducing Desulfovibrio hydrothermalis (WD0612–0614), for which BLASTp comparisons indicated sequence coverages ≥91% with identities that ranged from 33 to 50%. Similarly, genomes from the chemoautotroph Haliangium ochraceum, Capnocytophaga granulosa, and Saprospira grandis encode potential syntenically arranged homologs of WD0613, WD0614, and/or WD0615. Among all of the proteins, sequence coverages, E values ≤1e−125, and percent identities ≥44% indicated that WD0613 followed by WD0616 and WD0620 were most similar to proteins from other taxa.
Discussion
In the absence of a B wStr genome, the proteomics approach described here provides strong evidence for expression of WO phage genes in C/wStr cells harboring relatively high levels of Wolbachia (Baldridge et al. 2014). Because Wolbachia abundance is inversely correlated with increasing WO phage densities in the wasp Nasonia vitripennis (Bordenstein et al. 2006; Bordenstein and Bordenstein 2011), it will be of interest to learn whether phage activity can be induced to higher levels in C/wStr1 cells. Detection of peptides corresponding to the phage-related WD0611–WD0620 homologs with potential host-adaptive functions (Kent et al. 2011) further supports the presence of one or more active WO phages in B wStr. Sequenced Wolbachia genomes indicate that phage-associated host-adaptive functions potentially involve proteins with ankyrin repeat domains (Table S2, entries 15, 23, 65, 72, 93, 94, 123, 144) that may mediate protein–protein interactions (Pan et al. 2008; Siozios et al. 2013). Other proteins with likely roles in phage infectivity or host interactions include patatin-like phospholipases (Table S2, entries 22 and 114) known to be involved in rickettsial infection of host cells (Rahman et al. 2013), as well as homologs of the VrlC virulence-related protein from the sheep pathogen, Dichelobacter nodosus (entries 32, 33 and 128), that were first identified in WOCauB2/B3 (Tanaka et al. 2009). Finally, we note that a single unique peptide matched a “phage host specificity protein” (Table S2, entry 152) that contains domains found in a small phage-like particle containing DNA that is released from Rhodobacter capsulatus cells (Leung et al. 2010).
Although annotation of sequenced Wolbachia genomes provides firm evidence for potentially active WO phages, physical detection of viral particles associated with Wolbachia has been achieved in only a few instances. Transmission electron micrographs of ovaries from Culex pipiens mosquitoes provided the first evidence for association of Wolbachia with particles resembling bacteriophages (Wright et al. 1978). More than 20 yr later, Masui et al. (2000) characterized phage-related sequences in a DNA library from B wTai, which infects the cricket Teleogryllus taiwanemma. Hybridization of a probe from orf7, which has homology to a lambda minor capsid protein, to DNA from each of eight Wolbachia genomes, suggested that WO phages are widespread. RT-PCR experiments further showed that orf7 was expressed, and phylogenetic comparisons of orf7 sequences indicated that WO phages have mosaic structures suggestive of horizontal transmission within a common pool of double-stranded DNA phages (Masui et al. 2000). Gavotte et al. (2004) extended these observations, using orf7 to identify WO phages in additional Wolbachia strains, and in some cases observed phage particles captured on 0.22-μm filters. Completion of the A wMel genome, and annotation of WOMelA and WOMelB, provided the basis for characterization of prophage sequences in a wide range of Wolbachia genomes (reviewed in Metcalf and Bordenstein 2012).
The WO phage is believed to be similar to the E. coli bacteriophage lambda (Tanaka et al. 2009; Kent and Bordenstein 2010), and Bordenstein and Bordenstein (2011) have shown that in the wasp, N. vitripennis, temperature influences the balance between lysogenic/lytic development of the WO phage, Wolbachia densities, and cytoplasmic incompatibility. It will be of interest to investigate whether heat or cold treatments that increase phage abundance in N. vitripennis can be adapted to C/wStr1 cells to optimize recovery of WO phage particles, which may be facilitated by advantages of scale provided by cell culture. We are encouraged by the successful isolation of B wCauB from gram quantities of host insects, without enrichment of infected tissues by dissection (Fujii et al. 2004; Tanaka et al. 2009). Successful isolation of physical particles will allow direct sequencing of B wStr WO phage. Finally, we note that efforts to transform Wolbachia with transposon and plasmid-based technologies developed for related Rickettsiales genera (Felsheim et al. 2006, 2010; Baldridge et al. 2007; Burkhardt et al. 2011) have been unsuccessful, further underscoring the need to investigate phage-mediated genetic exchange as a means to facilitate genetic manipulation of Wolbachia (Metcalf and Bordenstein 2012).
References
Baldridge GD, Burkhardt NY, Felsheim RF, Kurtti TJ, Munderloh UG (2007) Transposon insertion reveals prM, a plasmid of Rickettsia monacensis. Appl Environ Microbiol 73:4984–4995
Baldridge GD, Witthuhn BA, Higgins L, Markowski TW, Fallon AM (2014) Proteomic analysis of a robust Wolbachia infection in an Aedes albopictus cell line. Mol Microbiol 94:537–556. doi:10.1111/mmi.12768
Becker A, Rüberg S, Küster H, Roxlau AA, Keller M, Ivashina T, Cheng HP, Walker GC, Pühler A (1997) The 32-kilobase gene cluster of Rhizobium meliloti directing the biosynthesis of galactoglucan: genetic organization and properties of the encoded gene products. J Bacteriol 179:1375–84
Bordenstein SR, Bordenstein SR (2011) Temperature affects the tripartite interactions between bacteriophage WO, Wolbachia, and cytoplasmic incompatibility. PLoS One 6(12):e29106. doi:10.1371/journal.pone.0029106
Bordenstein SR, Marshall ML, Fry AJ, Kim U, Wernegreen JJ (2006) The tripartite associations between bacteriophage, Wolbachia, and arthropods. PLoS Pathog 2(5):e43
Burkhardt NY, Baldridge GD, Williamson PC, Billingsley PM, Heu CC, Felsheim RF et al (2011) Development of shuttle vectors for transformation of diverse Rickettsia species. PLoS One 6(12):e29511. doi:10.1371/journal.pone.0029511
Comandatore F, Sassera D, Montagna M, Kumar S, Darby A, Blaxter M et al (2013) Phylogenomics and analysis of shared genes suggest a single transition to mutualism in Wolbachia of nematodes. Genome Biol Evol 5:1668–1674
Contestabile R, Paiardini A, Pascarella S, di Salvo ML, D’Aguanno S, Bossa F (2001) L-Threonine aldolase, serine hydroxymethyltransferase and fungal alanine racemase. A subgroup of strictly related enzymes specialized for different functions. Eur J Biochem 268:6508–6525
Cordaux R, Pichon S, Ling A, Perez P, Delaunay C, Vavre F et al (2008) Intense transpositional activity of insertion sequences in an ancient endosymbiont. Mol Biol Evol 25:1889–1895. doi:10.1093/molbev/msn134
diSalvo ML, Remesh SG, Vivoli M, Ghatge MS, Paidardini A, D’Aguanno S et al (2014) On the catalytic mechanism and stereospecificity of Escherichia coli L-threonone aldolase. FEBS J 281:129–145. doi:10.1111/febs.12581
Dong X, Karkouri KE, Robert C, Gavory F, Raoult D, Fournier P-E (2012) Genomic comparison of Rickettsia helvetica and other Rickettsia species. J Bacteriol 194:2751–2755. doi:10.1128/JB.00299-12
Duplouy A, Iturbe-Ormaetxe I, Beatson SA, Szubert JM, Brownlie JC, McMeniman CJ et al (2013) Draft genome sequence of the male-killing Wolbachia strain wBol1 reveals recent horizontal gene transfers from diverse sources. BMC Genomics 14:20. doi:10.1186/1471-2164-14-20
Fallon AM, Baldridge GD, Higgins LA, Witthuhn BA (2013) Wolbachia from the planthopper Laodelphax striatellus establishes a robust, persistent, streptomycin-resistant infection in clonal mosquito cells. Vitro Cell Dev Biol Anim 49:66–73
Felsheim RF, Herron MJ, Nelson CM, Burkhardt NY, Barbet AF, Kurtti TJ et al (2006) Transformation of Anaplasma phagocytophilum. BMC Biotechnol 6:42. doi:10.1186/1472-6750-6-42
Felsheim RF, Chávez AS, Palmer GH, Crosby L, Barbet AF, Kurtti TJ, Munderloh UG (2010) Transformation of Anaplasma marginale. Vet Parasitol 10:167–174. doi:10.1016/j.vetpar.2009.09.018
Foster J, Ganatra M, Kamal I, Ware J, Makarova K, Ivanova N et al (2005) The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol 3(4):e121. doi:10.1371/journal.pbio.0030121
Fujii Y, Kubo T, Ishikawa H, Sasaki T (2004) Isolation and characterization of the bacteriophage WO from Wolbachia, an arthropod endosymbiont. Biochem Biophys Res Comm 317:1183–1188
Gavotte L, Vavre F, Henri H, Ravallec M, Stouthamer R, Boulétreau M (2004) Diversity, distribution and specificity of WO phage infection in Wolbachia of four insect species. Insect Mol Biol 13:147–153
Gillespie JJ, Joardar V, Williams KP, Driscoll T, Hostetler B, Nordberg E et al (2012) A Rickettsia genome overrun by mobile genetic elements provides insight into the acquisition of genes characteristic of an obligate intracellular lifestyle. J Bacteriol 194:376–394. doi:10.1128/JB.06244-11
Ishmael N, Dunning-Hotopp JC, Ioannidis P, Biber S, Sakamoto J, Siozios S et al (2009) Extensive genomic diversity of closely related Wolbachia strains. Microbiology 155:2211–2222
James AC, Dean MD, McMahon ME, Ballard JW (2002) Dynamics of double and single Wolbachia infections in Drosophila simulans from New Caledonia. Heredity 88:182–189
Kent BN, Bordenstein SR (2010) Phage WO of Wolbachia: lambda of the endosymbiont world. Trends Microbiol 18:173–181. doi:10.1016/j.tim.2009.12.011
Kent BN, Funkhouser LJ, Setia S, Bordenstein SR (2011) Evolutionary genomics of a temperate bacteriophage in an obligate intracellular bacteria (Wolbachia). PLoS One 6(9):e24984. doi:10.1371/journal.pone.0024984
Klasson L, Walker T, Sebaihia M, Sanders MJ, Quail MA, Lord A et al (2008) Genome evolution of Wolbachia strain wPip from the Culex pipiens group. Mol Biol Evol 25:1877–1887
Klasson L, Westberg J, Sapountzis P, Naslund K, Lutnaes Y, Darby AC et al (2009) The mosaic structure of the Wolbachia wRi strain infecting Drosophila simulans. Proc Natl Acad Sci U S A 106:5725–5730
Lehman AP, Long SR (2013) Exopolysaccharides from Sinorhizobium meliloti can protect against H2O2-dependent damage. J Bacteriol 195:5362–5369
Leung MM, Florizone SM, Taylor TA, Lang AS, Beatty JT (2010) The gene transfer agent of Rhodobacter capsulatus. Adv Exp Med Biol 675:253–264. doi:10.1007/978-1-4419-1528-3_14
Masui S, Kamoda S, Sasaki T, Ishikawa H (2000) Distribution and evolution of bacteriophage WO in Wolbachia, the endosymbiont causing sexual alterations in arthropods. J Mol Evol 51:491–497
Metcalf JA, Bordenstein SR (2012) The complexity of virus systems: the case of endsymbionts. Curr Opin Microbiol 15:546–552. doi:10.1016/j.mib.2012.04.010
Metcalf JA, Jo M, Bordenstein SR, Jaenike J, Bordenstein SR (2014) Recent genome reduction of Wolbachia in Drosophila recens targets phage WO and narrows candidates for reproductive parasitism. Peer J 2:e529. doi:10.7717/peerj.529
Newton LG, Bordenstein SR (2011) Correlations between bacterial ecology and mobile DNA. Curr Microbiol 62:198–208. doi:10.1007/s00284-010-9693-3
Pan X, Satoh A, Laskowski-Arce MA, Roy CR (2008) Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science 320:1651–1654. doi:10.1126/science.1158160
Rahman MS, Gillespie JJ, Kaur SJ, Sears KT, Ceraul SM, Beier-Sexton M et al (2013) Rickettsia typhi possesses phospholipase A2 enzymes that are involved in infection of host cells. PLoS Pathog. doi:10.1371/journal.ppat.1003399
Shih KM, Gerenday A, Fallon AM (1998) Culture of mosquito cells in Eagle’s medium. Vitro Cell Dev BiolAnim 34:629–630
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W et al (2011) Fast, scalable generation of high–quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi:10.1038/msb.2011.75
Siozios S, Ioannidis P, Klasson L, Andersson SGE, Braig HR, Bourtzis K (2013) The diversity and evolution of Wolbachia ankyrin repeat domain genes. PLoS One. doi:10.1371/journal.pon.0055390
Solovyev, V, Salamov A (2011) Automatic annotation of microbial genomes and metagenomic sequences. In Metagenomics and its applications in agriculture, biomedicine and environmental studies. Li RW (Ed.), Nova Science Publishers 61–78
Sullivan MB, Huang K, Ignacio-Espinoza JC, Berlin AM, Kelly L, Weigele PR, DeFrancesco AS et al (2010) Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ Microbiol 12:3035–3056. doi:10.1111/j.1462-2920.2010.02280.x
Tanaka K, Furukawa S, Nikoh N, Sasaki T, Fukatsu T (2009) Complete WO phage sequences reveal their dynamic evolutionary trajectories and putative functional elements required for integration into the Wolbachia genome. Appl Environ Microbiol 75:5676–5686. doi:10.1128/AEM.01172-09
Taylor MJ, Bandi C, Hoerauf A (2005) Wolbachia bacterial endosymbionts of filarial nematodes. Adv Parasitol 60:245–284
Vollmer J, Scheifer A, Schneider T, Julcher K, Johnstone KL, Taylor MJ et al (2013) Requirement for lipid II biosynthesis for cell division in cell wall-less Wolbachia, endobacteria of arthropods and filarial nematodes. Int J Med Microbiol 303:140–149. doi:10.1016/j.ijmm.2013.01.002
Wang G-H, Xiao J-H, Xiong T-L, Li Z, Murphy RW, Haing D-W (2013) High-efficiency thermal asymmetric interlaced PCR (hTail-PCR) for determination of a highly degenerated prophage WO genome in a Wolbachia strain infecting a fig wasp species. Appl Environ Microbiol 79:7476–7481. doi:10.1128/AEM.02261-13
Wright JD, Sjostrand FS, Portaro JK, Barr R (1978) The ultrastructure of the rickettsia-like microorganism Wolbachia pipientis and associated virus-like bodies in the mosquito Culex pipiens. J Ultrastruct Res 63:79–85
Wu M, Sun LV, Vamathevan J, Riegler M, Deboy R, Brownlie JC et al (2004) Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile gentic elements. PLoS Biol 2:0327–0341
Zug R, Hammerstein P (2014) Bad guys turned nice? A critical assessment of Wolbachia mutualisms in arthropod hosts. Biol Rev. doi:10.1111/brv.12098
Zug R, Koehncke A, Hammerstein P (2012) Epidemiology in evolutionary time: the case of Wolbachia horizontal transmission between arthropod species. J Evol Biol 25:2149–2160. doi:10.1111/j.1420-9101.2012.02601.x
Acknowledgments
This work was supported by R01 AI 081322 from the National Institutes of Health and by the University of Minnesota Agricultural Experiment Station, St. Paul, MN.
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: Tetsuji Okamoto
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental Table S1
Polymerase chain reaction (PCR) primers and amplification products from B wStr homologs of WD0611–WD0620. Products are depicted schematically in Fig. 2b. (DOCX 116 kb)
Supplemental Table S2
LC-MS/MS detection of 95% confidence unique peptides matched to proteins encoded by WO prophage and prophage-related genes. Columns A and B show WO prophage-encoded proteins and locus tags (as identified by Kent et al. 2011) as well as total numbers and proportions of encoded proteins matched to unique peptides in MS/MS data sets D, E, F and G in corresponding columns. Total number of matched peptides and percent protein sequence coverage are reported in columns D to G; e.g., 2/5 indicates 2 peptides constituting 5% protein sequence coverage. Column H reports the mean number of detected peptides, a rough estimate of the protein’s relative abundance level (RAL), which can be normalized to expected values for all proteins of that same mass (see SR values in Tables S3 and S4) as described by Baldridge et al. 2014. (XLSX 30 kb)
Supplemental Table S3
Genbank accession numbers for PCR-amplified and cloned wStr genes and homologs from other Wolbachia strains. (XLS 271 kb)
Supplemental Table S4
Results of Univariable and Multivariable Analyses after log transformation of the outcome, Peptide Count, and predictor, Molecular Weight. This table reports results for the original (Baldridge et al. 2014), and refined searches of the LC-MS/MS data sets D, E, F and G. See tabs at bottom: Sheet 1 reports mean SR values for all proteins in the original and refined searches in columns M and R. Runs 1, 2, 3 and 4 correspond to MS data sets D, E, F and G, respectively; Univariable Model and Multivariable Model (adjusted for functional class and MS Dataset) for results of tests of association. Proteins encoded by cloned genes from B wStr reported in this manuscript (WD0611 – WD0620 homologs) are in blue font and the ♦ symbols below and above the corresponding arrowheads in Fig. 2 indicate unique peptides identified in the original and refined searches of the MS data sets, respectively. Proteins encoded by cloned genes from B wStr reported in a previous refined search (Baldridge et al., submitted Archs Microbiol) are highlighted in yellow. (XLSX 24 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Baldridge, G.D., Markowski, T.W., Witthuhn, B.A. et al. The Wolbachia WO bacteriophage proteome in the Aedes albopictus C/wStr1 cell line: evidence for lytic activity?. In Vitro Cell.Dev.Biol.-Animal 52, 77–88 (2016). https://doi.org/10.1007/s11626-015-9949-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-015-9949-0