
Abstract
Inference of the evolutionary histories of species, commonly represented by a species tree, is complicated by the divergent evolutionary history of different parts of the genome. Different loci on the genome can have different histories from the underlying species tree (and each other) due to processes such as incomplete lineage sorting (ILS), gene duplication and loss, and horizontal gene transfer. The multispecies coalescent is a commonly used model for performing inference on species and gene trees in the presence of ILS. This paper introduces Lily-T and Lily-Q, two new methods for species tree inference under the multispecies coalescent. We then compare them to two frequently used methods, SVDQuartets and ASTRAL, using simulated and empirical data. Both methods generally showed improvement over SVDQuartets, and Lily-Q was superior to Lily-T for most simulation settings. The comparison to ASTRAL was more mixed—Lily-Q tended to be better than ASTRAL when the length of recombination-free loci was short, when the coalescent population parameter \(\theta \) was small, or when the internal branch lengths were longer.





















Similar content being viewed by others


References
Avni E, Cohen R, Snir S (2015) Weighted Quartets Phylogenetics. Systematic Biology 64(2):233–242
Chifman J, Kubatko L (2014) Quartet Inference from SNP Data Under the Coalescent Model. Bioinformatics 30(23):3317–3324
Chifman J, Kubatko L (2015) Identifiability of the unrooted species tree topology under the coalescent model with time-reversible substitution processes, site-specific rate variation, and invariable sites. Journal of Theoretical Biology 374(1):35–47
Chou J, Gupta A, Yaduvanshi S, Davidson R, Nute M, Mirarab S, Warnow T (2015) A comparative study of SVD quartets and other coalescent-based species tree estimation methods. BMC Genomics 16(S2). https://doi.org/10.1186/1471-2164-16-S10-S2
DeGiorgio M, Degnan JH (2010) Fast and consistent estimation of species trees using supermatrix rooted triples. Mol Biol Evol 27(3):552–569
Degnan JH, Salter LA (2005) Gene tree distributions under the coalescent process. Evolution 59(1):24–37
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7(214):1. https://doi.org/10.1186/1471-2148-7-214
Efron B (1979) Bootstrap methods: another look at the jackknife. Ann Stat 7(1):1–26
Gatesy J, Meredith RW, Janecka JE, Simmons MP, Murphy WJ, Springer MS (2017) Resolution of a concatenation/coalescence kerfuffle: partitioned coalescence support and a robust family-level tree for Mammalia. Cladistics 33:295–332
Gatesy J, Springer MS (2014) Phylogenetic analysis at deep timescales: Unreliable gene trees, bypassed hidden support, and the coalescence/concatalescence conundrum. Mol Phylogenetics Evol 80:231–266
Harding EF (1971) The probabilities of rooted tree-shapes generated by random bifurcation. Adv Appl Probab 3(1):44–77
Hobolth A, Dutheil JY, Hawks J, Schierup MH, Mailund T (2011) Incomplete lineage sorting patterns among human, chimpanzee, and orangutan suggest recent orangutan speciation and widespread selection. Genome Res 21:349–356
Hudson RR (2003) Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics 18(2):337–338
Jennings WB, Edwards SV (2005) Speciational history of Australian grass finches (Pephila) inferred from thirty gene trees. Evolution 59(9):2033–2047
Jukes TH, Cantor CR (1969) Evolution of protein molecules. Academic Press, New York
Kingman JFC (1982) The coalescent. Stoch Process Appl 13(3):235–248
Kopp A, Barmina O (2005) Evolutionary history of the drosophila bipectinata species complex. Genetical Res 85(1):23–46
Kubatko LS, Carstens BC, Knowles LL (2009) STEM: species tree estimation using maximum likelihood for gene trees under coalescence. Bioinformatics 25(7):971–973
Kubatko LS, Degnan JH (2007) Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst Biol 56(1):14–24
Liu L, Pearl DK (2007) Species trees from gene trees: reconstructing Bayesian posterior distributions of a species phylogeny using estimated gene tree distributions. Syst Biol 56(3):504–514
Liu L, Yu L, Edwards SV (2010) A maximum qseudo-likelihood approach for estimating species trees under the coalescent model. BMC Evol Biol. https://doi.org/10.1186/1471-2148-10-302
Mahim M, Zahin W, Rezwana R, Bayzid MS (2020) wQFM: statistically consistent genome-scale species tree estimation from weighted quartets. bioRxiv. https://www.biorxiv.org/content/early/2020/12/01/2020.11.30.403352
Mirarab S, Reaz R, Bayzid MS, Zimmermann T, Swenson MS, Warnow T (2014) ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics 30(17):i541–i548
Mirarab S, Warnow T (2015) ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 31(12):i44–i52
Oglivie HA, Bouckaert RR, Drummond AJ (2017) StarBEAST2 brings faster species tree inference and accurate estimates of substitution rates. Mol Biol Evol 34(8):2101–2114
Paradis E, Schliep K (2019) ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35:526–528
Peng J, Swofford D, Kubatko L (2021) Estimation of speciation times under the multispecies coalescent (in review)
Price MN, Dehal PS, Arkin AP (2009) FastTree: computing large minimum-evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26:1641–1650
Rambaut A, Grassly NC (1997) Seq-Gen: an application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comput Appl Biosci 13(3):235–238
Rannala B, Yang Z (2003) Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 164(4):1645–1656
Robinson DF, Foulds LR (1981) Comparison of phylogenetic trees. Math Biosci 53(1):131–147
Roch S, Steel M (2015) Likelihood-based tree reconstruction on a concatenation of aligned sequence data sets can be statistically inconsistent. Theor Popul Biol 100c:56–62
Rokas A, Williams BL, Carroll S (2003) Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 425:798–804
Rosenberg NA (2007) Counting coalescent histories. J Comput Biol 14(3):360–377
Salter L (2001) Complexity of the likelihood surface for a large DNA data set. Syst Biol 50(6):970–978
Schliep KP (2011) Phangorn: phylogenetic analysis in R. Bioinformatics 27(4):592–593
Sevillya G, Frenkel Z, Snir S (2016) TripletMaxCut: a new toolkit for rooted supertree. Methods Ecol Evol 7(11):1359–1365
Springer MS, Gatesy J (2016) Phylogenetic analysis at deep timescales: Unreliable gene trees, bypassed hidden support, and the coalescence/concatalescence conundrum. Mol Phylogenetics Evol 94:1–33
Stadler T, Steel M (2011) Distribution of branch lengths and pylogenetic diversity under homogeneous speciation models. J Theor Biol 297:33–40
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313, 01
Steel M, Penny D (1993) Distribution of tree comparison metrics—some new results. Syst Biol 42(2):136–141
Swofford DL (2003) Paup*. phylogenetic analysis using parsimony (*and other methods), version 4. Sinauer Associates. Sunderland, Massachusetts
Thawornwattana Y, Dalquen D, Yang Z (2018) Coalescent analysis of phylogenomic data confidently resolves the species relationships in the anopheles gambiae species complex. Mol Biol Evol 35(10):2512–2527
Wakeley J (2009) Coalescent Theory: An Introduction. Roberts & Company Publishers, Greenwood Village
Wascher M, Kubatko L (2021) Consistency of SVDQuartets and maximum likelihood for coalescent-based species tree estimation. Syst Biol 70(1):33–48
Wen D, Nakhleh L (2018) Coestimating reticulate phylogenies and gene trees from multilocus sequence data. Syst Biol 67(1):439–457
Whidden C, Matsen IV FA (2015) Quantifying MCMC exploration of phylogenetic tree space. Syst Biol 64(3):472–491
Yang Z (2014) Molecular evolution: a statistical approach. Oxford University Press, New York
Yang Z (2015) The BPP program for species tree estimation and species delimitation. Curr Zool 61(5):854–865
Yang Z, Rannala B (1997) Bayesian phylogenetic inference using DNA sequences: a Markov Chain Monte Carlo Method. Mol Biol Evol 14:717–724
Zhang C, Rabiee M, Sayyari E, Mirarab S (2018) ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19(Supp 6):15–30
Acknowledgements
The authors thank the Associate Editor and three anonymous reviewers for helpful comments on an earlier draft of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Appendix
Appendix
1.1 Calculation of \(\varvec{\delta }|S\) for 4-Taxon Trees
From Chifman and Kubatko (2015), as in the 3-taxon case \(P(\varvec{\delta }|(S,\varvec{\tau }))\) is the inner product \(\mathbf{c }^{T}\mathbf{b }\) where \(\mathbf{c }\) is a vector of constants times a function of \(\varvec{\tau }\) that is of the form \(e^{-a\tau _1-b\tau _2-g\tau _3}\).
1.1.1 Asymmetric Case
From (Chifman and Kubatko 2015) we have the following, converting to coalescent units from mutation units and using 4 characters \(\{A,C,G,T\}\):
where the coefficients are given by Table 4.
We have from the Yule model that \(\tau _1 \sim Exp(4\beta )\), \((\tau _2-\tau _1) \sim Exp(3\beta )\), and \((\tau _3-\tau _2) \sim Exp(2\beta )\). Then:
Then, integrating over \(f(\varvec{\tau })\) gives:
Applying this formula to each term above gives:
1.1.2 Symmetric Case
From (Chifman and Kubatko 2015) we have the following, converting to coalescent units from mutation units and using 4 characters \(\{A,C,G,T\}\):
where the coefficients are given by Table 5.
The prior differs from the asymmetric case in that \(\tau _1<\tau _2\) and \(\tau _2<\tau _1\) occurs with equal probability. The form of the integrals are the same in either case, just substituting the coefficients in front of \(\tau _1\) and \(\tau _2\). As a result, integrating over the prior gives (where the only difference between the terms is the a or b in the middle factor of the denominator):
Applying this formula to each term above gives:
1.2 Calculation of \(\varvec{\delta }|(S,\varvec{\tau }))\) for 3-Taxon Trees
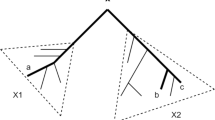
The fifteen site pattern probabilities shown above for the 4-taxon case can be mapped down to the five site patterns for three taxa by summing over the fourth taxon we are not interested in. Any of the four taxa can be used, however, we found it simplest to marginalize over the outgroup of the asymmetric tree (see Fig. 3) to get the following relationships (dropping the conditioning on \((S, \varvec{\tau })\) for clarity):
These probabilities take the form:
where (measuring \(\tau _1\) and \(\tau _2\) in coalescent units):
It is easily verified that:
The coefficients in Eq. 8 arise as follows. X can represent any of A, C, G, or T. Y can represent any of the remaining three characters, and Z any of the remaining two. Finally, the coefficient of \(p_2\) is doubled to account for both \(\delta _{XYX}\) and \(\delta _{XXY}\).
1.3 Calculation of \(\varvec{\delta }|S\) for 3-Taxon Trees
One can see from Eq. 6 that \(\delta _k|(S,\varvec{\tau })\) is a linear combination of terms, so \(\delta _k|S\) is also linear combination of terms. Given \(\tau _1 \sim Exp(3\beta )\) and \((\tau _2-\tau _1) \sim Exp(2\beta )\), the prior is:
Then to integrate over \(f(\varvec{\tau }|\beta )\), each term has the form:
Taken together, \(\delta _k|S\) has the linear form of Eq. 6 with the following terms replacing those of Eq. 7:
where \(\alpha =4/3\) from the JC69 model. Comparison to the results in the supplementary material reveals the computational advantages of the Yule model approach.
1.4 Proof of Theorem 1
Proof
Without loss of generality, let \(S_0=(a,(b,c))\) be the true tree and \(\varvec{\tau }_0\) be the true value of \(\varvec{\tau }\). We want to show that \(\forall \epsilon>0 \, \, \exists J_0 : \forall J>J_0 \, \, P({\hat{S}}=S)>1-\epsilon \). First, note that for all values of \((\varvec{\tau }, \theta )\), we have \(\delta _{XXY}=\delta _{XYX}\).
Second, we note using the results of Eq. 6
which holds w.p.1 by assumption.
Next, note
by properties of expectations from Eq. 10 and since \(f(\varvec{\tau })>0\) a.e. under the prior.
Since each topology (a, (b, c)), (b, (a, c)), and (c, (a, b)) has a prior probability of 1/3, the maximum posterior topology will be the maximum likelihood topology. Recall from Sect. 2.4 that we can permute any site pattern probability given one topology to find a site pattern probability given another topology—e.g., \(\delta _{YXX}|(a,(b,c))=\delta _{XYX}|(b,(a,c))\). So, the likelihood of trees (a, (b, c)) and (b, (a, c)) given the data are:
By cancelling terms in common, one can see that the likelihood ratio comes down to permuting the number of sites that follow the XYX and YXX patterns:
Together \(p_{yxx}>p_{xyx}\) and \(d_{yxx}>d_{xyx}\) imply that \(\frac{L((a,(b,c))|{\varvec{d}})}{L((b,(a,c))|{\varvec{d}})}>1\), which implies that \({\hat{S}}=S\). As we have shown that \(p_{yxx}>p_{xyx}\), it is sufficient to show that \(\frac{d_{yxx}}{J} \rightarrow p_{yxx}\) and \(\frac{d_{xyx}}{J} \rightarrow p_{xyx}\) for sufficiently large number of sites.
Let \(p_{yxx,0}=\delta _{yxx}|((a,(b,c)),\varvec{\tau }_0)\) and \(p_{xyx,0}=\delta _{xyx}|((a,(b,c)),\varvec{\tau }_0)\). From the SLLN, we have
Choose \(\delta _1,\delta _2\) such that \(\delta _1+\delta _2<p_{yxx,0}-p_{xyx,0}\), \(\epsilon _1=\epsilon _2=\epsilon /2\), and \(J_0=max\{J_1,J_2\}\) by applying the Bonferroni inequality we have that \(P(\frac{d_yxx}{J}>p_{yxx,0}-\delta _1, p_{xyx,0}+\delta _2>\frac{d_{xyx}}{J})>1-\epsilon \) and the result holds.
\(\square \)
Rights and permissions
About this article

Cite this article
Richards, A., Kubatko, L. Bayesian-Weighted Triplet and Quartet Methods for Species Tree Inference. Bull Math Biol 83, 93 (2021). https://doi.org/10.1007/s11538-021-00918-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11538-021-00918-z