
Abstract
Background
Varlitinib is a highly potent, small-molecule, pan-HER inhibitor targeting HER1, HER2, and HER4. It has demonstrated activity in gastric, biliary tract, and breast cancers.
Objective
We conducted a phase Ib dose confirmation study to determine safety and early efficacy signals of varlitinib in combination with chemotherapy (paclitaxel ± carboplatin) ± subcutaneous trastuzumab.
Methods
Eligible patients had advanced or metastatic solid tumors. A 3+3 dose de-escalation study design was used and pharmacokinetic analyses of varlitinib and paclitaxel were performed.
Results
Thirty-seven patients were enrolled into eight cohorts with median 4 (0–14) prior lines of palliative systemic therapies. Carboplatin area under the curve 1.5 and paclitaxel 80 mg/m2 weekly with varlitinib 500 mg twice daily continuously was de-escalated over four dose levels to 300 mg twice daily intermittently (4 days on, 3 days off) due to dose-limiting toxicities, most commonly neutropenia, febrile neutropenia, and electrolyte disturbances, with the triplet combination deemed intolerable and unable to be developed further. Varlitinib was then combined with paclitaxel alone; the recommended phase II dose of varlitinib was 300 mg twice daily intermittently. The addition of subcutaneous trastuzumab 600 mg was safe with no dose-limiting toxicities. Thirty-one patients were evaluable for response: 35.5% partial response, 41.9% stable disease. Twenty patients had HER2+ metastatic breast cancer with a median of 4 (0–14) treatment lines, 8/20 continued on single-agent varlitinib after completing chemotherapy for a median of 5.1 (range 2.0–13.3) months. A pharmacokinetic analysis showed that plasma exposure of varlitinib was dose dependent. Varlitinib administration did not significantly affect the maximum concentration or area under the curve of paclitaxel.
Conclusions
The recommended phase II dose of varlitinib with paclitaxel is 300 mg twice daily intermittently dosed. This is active in HER2+ metastatic breast cancer. Subcutaneous trastuzumab can be added safely to varlitinib and paclitaxel. This combination is currently being evaluated as neoadjuvant therapy in HER2+ breast cancer (NCT02396108).
Clinical Trial Registration: NCT02396108, date of registration: 25 March, 2015.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The recommended phase II dose of varlitinib in combination with weekly paclitaxel with or without subcutaneous trastuzumab is 300 mg dosed intermittently. |
Activity in heavily pretreated HER2+ metastatic breast cancer is promising with a clinical benefit rate of 81.3% and a partial response rate of 56.3%. |
Varlitinib was well tolerated and did not significantly affect the pharmacokinetics of paclitaxel. |
1 Introduction
Up to 20% of patients with breast cancer have HER2-amplified tumors and strategies to block HER2 signaling pathways have been developed to improve treatment of these patients [1, 2]. HER2 is a member of the human epidermal growth factor receptor (EGFR) family [3,4,5]. Activated members of the EGFR family (HER1, HER2, and HER4) lie upstream of several signal transduction pathways, which orchestrate excessive proliferation and uncontrolled cell growth, and are responsible for the development of many malignancies [6,7,8].
The first drug targeting HER2 approved by the US Food and Drug Administration was trastuzumab, a humanized monoclonal antibody that binds to the extracellular domain of HER2 [9, 10]. Small molecules that specifically inhibit EGFR tyrosine kinases were later developed, including lapatinib that inhibits the intracellular domains of both HER1 and HER2, and neratinib that irreversibly binds to HER1, HER2, and HER4 [11,12,13,14,15].
Varlitinib is a highly potent, orally administered, small-molecule inhibitor targeting HER1, HER2, and HER4 [16]. Varlitinib inhibits the phosphorylation and activation of these receptors, and has demonstrated activity in gastric, biliary tract, and breast cancers [17,18,19,20,21]. It has been granted orphan drug designation in the USA since 2015 for the treatment of cholangiocarcinoma and gastric cancer [22].
The half maximal inhibitory concentration of varlitinib for inhibiting the phosphorylation of HER1, HER2, and HER4 are 7nM, 2nM, and 4nM, respectively, which is lower, and thus more potent, than other small-molecule EGFR inhibitors such as lapatinib (HER1: 10.8 nM, HER2: 9.3 nM) and neratinib (HER1: 12 nM, HER2: 39 nM, HER4: 19 nM) [17, 23,24,25,26]. In prior studies of varlitinib as monotherapy (ARRAY 543-103) and as combination therapy with docetaxel (ARRAY 543-104), the continuous dose of 500 mg twice a day was found to be the maximum tolerated dose (MTD) [26].
The primary objective of this phase Ib study is to determine the MTD of varlitinib when combined with carboplatin and paclitaxel in patients with metastatic solid organ malignancies. The secondary objectives are to evaluate the safety of combining varlitinib with chemotherapy and trastuzumab, and early efficacy signals.
2 Patients and Methods
2.1 Patient Eligibility
Eligible patients were required to have advanced or metastatic solid organ malignancies of any histologic type with evaluable disease defined by Response Evaluation Criteria in Solid Tumors, for which treatment with weekly paclitaxel and carboplatin was deemed indicated by their physician. Patients were 21 years of age and above, had a Karnofsky Performance Status or 70 or higher, an estimated life expectancy of at least 12 weeks, and adequate organ and bone marrow function before study enrollment. This was defined as an absolute neutrophil count ≥1.5 × 109/L, platelets ≥100 × 109/L, total bilirubin ≤1.5 times the upper limit of normal, aspartate transaminase and alanine transaminase ≤2.5 times the upper limit of normal, calculated creatinine clearance of >30 mL/minute, and a left ventricular ejection fraction of ≥50% measured by a transthoracic echocardiogram or multi-gated acquisition scan. All patients had to be able to swallow tablets, and provide written informed consent. Patients with reproductive potential had to use an approved contraceptive method during and for 3 months after the study. Female individuals with childbearing potential were required to have a negative serum pregnancy test within 7 days prior to enrollment into the study.
Exclusion criteria included clinically detectable second primary malignancies, poorly controlled diabetes mellitus, active hepatic or biliary disease (except patients with Gilbert’s syndrome, asymptomatic gallstones, or stable chronic liver disease as assessed by the investigator), concurrent administration of other anti-tumor therapies including cytotoxic chemotherapy, endocrine therapy, and immunotherapy within 21 days prior to study drug administration, serious cardiac disease (left ventricular ejection fraction < 50%, high-risk uncontrolled arrhythmias, significant ischemic heart disease, significant valvular heart disease, poorly controlled hypertension), or serious medical conditions. Patients who had an active infection that would compromise the ability to tolerate therapy, or had major surgery within 28 days of planned study drug administration, or were breastfeeding, or had a history of significant neurological or mental disorders, including seizures or dementia, were ineligible.
2.2 Study Design
All study participants were enrolled from a single institution, the National University Cancer Institute, Singapore. Patients were enrolled between April 2015 and December 2017. Data were collected up till December 2018. This trial was approved by the institutional review board prior to commencement. This single-arm, open-label phase I trial was conducted in three sequential cohorts: varlitinib administered in combination with carboplatin are under the curve (AUC) 1.5 and paclitaxel 80 mg/m2 dosed weekly in cohort A, varlitinib in combination with single-agent paclitaxel 80 mg/m2 dosed weekly in cohort B, and varlitinib in combination with paclitaxel 80 mg/m2 dosed weekly and subcutaneous trastuzumab 600 mg administered every 3 weeks (cohort C; Table 1 of the Electronic Supplementary Material [ESM]). The initial starting dose of varlitinib was 500 mg twice daily administered with a continuous daily dosing schedule. This dose was selected as the starting dose based on previous studies of varlitinib as monotherapy and as combination therapy with docetaxel. A dose de-escalation study design was planned to be employed in the event the 500-mg twice-daily dose was found to be intolerable in combination with weekly carboplatin and paclitaxel. As the eventual intention is to develop the combination as neoadjuvant therapy in HER2+ breast cancer, if the triplet combination was deemed to be intolerable, it was planned to open cohorts studying the combination of varlitinib with weekly paclitaxel, dropping carboplatin from the combination as paclitaxel is the standard chemotherapy backbone drug in HER2+ breast cancer. Depending on the number of patients developing dose-limiting toxicities (DLTs), the dose of varlitinib could be escalated in these cohorts to find the MTD. In some of the subsequent dose levels, varlitinib was administered intermittently, 4 days per week, starting 24–36 h after and ending 24–36 h before each weekly paclitaxel dose administration.
Using a modified 3+3 design, patients were enrolled in cohorts of three to six patients. If two or more of six patients developed DLTs, three subjects were enrolled into the next lower dose cohort. If DLTs were observed in one of three subjects in the cohort, or if two or more subjects experienced drug-related adverse events (AEs) of more than or equal to grade 2, an additional three subjects were enrolled at the same dose level. Cohorts were enrolled at de-escalating dose levels until the MTD was identified, defined as the dose level at which DLTs developed in fewer than one of three, or two of six patients.
Dose-limiting toxicity was defined as any of the following: grade 4 neutropenia lasting ≥ 7 days, grade 3 and above febrile neutropenia or neutropenic infection, grade 4 thrombocytopenia, grade 3 thrombocytopenia lasting for ≥ 7 days, grade 3 and above nausea, vomiting or diarrhea despite optimal use of anti-emetic and anti-diarrheal medications, grade 2 abnormal alanine transaminase or aspartate transaminase in the presence of grade 2 raised bilirubin attributed to study treatment with or without signs and symptoms of drug-induced liver injury, or any other grade 3 or 4 treatment-related non-hematological toxicities except alopecia. If patients received less than 75% of the intended varlitinib dose because of any toxicities during cycle 1, they were also deemed to have a DLT. Patients who had received at least four cycles of varlitinib in combination with chemotherapy and are benefitting from the treatment regimen (sustained stable disease or objective response) and who experience unacceptable toxicity from paclitaxel/carboplatin may discontinue paclitaxel/carboplatin and be maintained on anti-HER2 therapy alone with single-agent varlitinib (with or without trastuzumab for cohort C) until documented disease progression or unacceptable toxicity.
2.3 Study Assessments
Safety evaluations occurred weekly during cycle 1 and on the first day of each subsequent 3-weekly cycle. Evaluations included physical examination and laboratory tests (hematology and blood chemistries: bilirubin, alanine transaminase, aspartate transaminase, creatinine, electrolytes). Adverse events were graded according to the Common Terminology Criteria for Adverse Events version 4.03 following each cycle of treatment.
Radiological assessments were conducted during screening, after every 2 cycles (6 weeks) for the first four cycles, and every three cycles (9 weeks) thereafter with computed tomography of the thorax, abdomen, with or without pelvis. Repeat isotope bone scans were performed every 9–12 weeks if there were known bone metastases or clinical suspicion of new bony metastases. In addition, at the discretion of the investigator, other imaging modalities were used if clinically indicated. Lesions were evaluated using Response Evaluation Criteria in Solid Tumors version 1.1.
Patients were treated until disease progression, intolerable toxicities, or subject withdrawal. In the presence of intolerable toxicities to one or more drugs in the regimen but not all study drugs, the drug in question may be discontinued and the other drug(s) continued until disease progression or intolerable toxicity, if the patient is deemed to be benefiting, at the discretion of the investigator.
2.4 Pharmacokinetic Assessments
Pharmacokinetic assessments were carried out for paclitaxel and varlitinib during the first cycle of administration of treatment. Seven-timepoint pharmacokinetic analyses of paclitaxel was performed on cycle 1 day 1 and cycle 1 day 8 at 0, 0.5, 1, 2, 5, 7, and 24 h after paclitaxel administration.
For patients receiving continuous daily dosing of varlitinib, seven-point pharmacokinetic analyses of varlitinib was performed on cycle 1 day 1 and cycle 1 day 8 at 0, 0.5, 1, 2, 5, 7, and 24 h after varlitinib administration. For patients in whom varlitinib was administered intermittently, the seven-point pharmacokinetic analyses was carried out on cycle 1 day 5 after dosing of varlitinib. The timepoints at which blood was taken for this analysis was at 0, 0.5, 1, 2, 5, 7, and 24 h.
3 Results
3.1 Patient Characteristics (Table 1)
A total of 37 patients were enrolled between April 2015 and December 2017; two had advanced non-metastatic breast cancer and received study treatment as neoadjuvant therapy; all others received the treatment as palliative systemic therapy. The most common tumor type was breast cancer (n = 28; 75.6%) with 20/37 (54%) patients having HER2+ breast cancer. Patients were heavily pretreated, with a median of four prior lines of palliative systemic therapies (range 0–14). Other tumor types include lung, pancreatic, colon, nasopharyngeal, cervical, endometrial, and malignant peripheral nerve sheath cancers. Eighteen patients were treated in cohort A (varlitinib + carboplatin + paclitaxel), 16 in cohort B (varlitinib + paclitaxel), and three in cohort C (varlitinib + paclitaxel + trastuzumab).
3.2 Dose De-Escalation and MTD
In cohort A (varlitinib + carboplatin + paclitaxel), 18 patients were enrolled at four dose levels, starting with varlitinib 500 mg continuously (level A1, n = 3), then de-escalating to 400 mg continuously (level A2, n = 5), 400 mg intermittently (level A3, n = 4), and 300 mg intermittently (level A4, n = 6). All four dose levels within this cohort were deemed intolerable, with 3/3, 3/5, 2/4, and 2/6 patients at levels A1, A2, A3, and A4, developing DLTs respectively (Table 2). The most common DLTs were G3/4 neutropenia (n = 6; 33.3%); G3 hyperbilirubinemia occurred in one patient.
As it was deemed not clinically meaningful to reduce the dose of varlitinib to less than 300 mg intermittently, the triplet combination of varlitinib, carboplatin, and paclitaxel was declared intolerable and could not be developed further. The combination of varlitinib with single-agent paclitaxel was studied next in cohort B.
Sixteen patients were treated at three dose levels in cohort B, starting with varlitinib 300 mg intermittently, 4 days on, 3 days off, with weekly paclitaxel (level B1), then escalating to level B2 (varlitinib 400 mg intermittent dosing) and level B3 (varlitinib 300 mg continuous dosing). Level B1 was tolerable with 0/6 DLTs, while levels B2 and B3 were deemed intolerable, with 2/4 and 2/6 DLTs, respectively. The most common DLTs were G3/4 neutropenia (n = 3; 18.8%), G3 transaminitis (n = 1; 6.25%), and G3 diarrhea (n = 1; 6.25%) (Table 2). The dose level of intermittent dosing varlitinib at 300 mg twice daily (days 3–6 each week) in combination with weekly paclitaxel (80 mg/m2 on days 1, 8, and 15 of a 21-day cycle) was thus determined to be the maximum tolerated and recommended phase II dose.
Having established the recommended phase II dose of varlitinib with weekly paclitaxel, we proceeded to determine the safety of adding 3-weekly subcutaneous trastuzumab 600 mg to the combination in cohort C given on day 1 of each 3-weekly cycle. Three patients were treated in cohort C without any DLTs. Thus varlitinib dosed at 300 mg intermittently with weekly paclitaxel and 3-weekly subcutaneous trastuzumab 600 mg was determined to be the recommended phase II dose for the triplet combination. The DLTs of all three cohorts are summarized in Table 2.
3.3 Safety and Tolerability
Table 3 summarizes the treatment-related AEs observed. Overall, the most common any grade AEs were diarrhea (70.2%), fatigue (67.5%), and neutropenia (45.9%). The most frequent grade 3 and above AEs were neutropenia (40.5%), hyponatremia (16.2%), and hypophosphatemia (13.5%). None of the patients experienced a reduction in left ventricular ejection fraction or cardiomyopathy. There was no treatment-related mortality. One patient died from suicide that was deemed not to be related to treatment.
3.4 Tumor Response
All patients with metastatic cancer had progressed at the time of reporting. Thirty-one patients were evaluable for radiologic response: 30 received at least two cycles of varlitinib while one patient progressed while on the first cycle of study treatment. Six other patients discontinued treatment prior to cycle 2; five because of toxicities, and one patient committed suicide before a radiologic efficacy assessment could be performed.
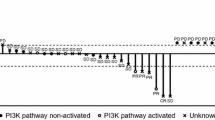
Among the patients evaluable for response, 11/31 (35.5%) patients achieved confirmed partial response and 13/31 (41.9%) achieved stable disease as their best response. Excluding the two subjects who had non-metastatic breast cancer, the partial response rate is 10/30 (33.3%) and 13/30 (43.3%) achieved stable disease. The overall response rate in the entire cohort is 11/37 (29.7%). Figure 1 summarizes the responses to treatment. The median duration on study treatment was 3.0 months (0.1–17.1 months). In patients who achieved a partial response, the median duration on study treatment was 5.0 months (2.2–17.1 months) Fig. 2.

Responses to treatment. Waterfall plot summarizes the best percentage change from the baseline sum of target lesions (Response Evaluation Criteria for Solid Tumors version 1.1) during treatment of the patients evaluable for radiologic response. 35.5% (11/31) achieved confirmed partial response (PR), 41.9% (13/31) had stable disease. The majority of patients with a PR had HER2+ metastatic breast cancer. Each bar is labeled with the dose cohort the patient was enrolled into. Responses to therapy were observed in patients across all cohorts of differing varlitinib doses. HER2+ MBC HER2+ metastatic breast cancer, PD progressive disease, * indicates PD with new lesions

Swimmer’s plot represents the progression-free survival (PFS) of individual patients (median PFS 3.7 months, range 0.3–17.1 months) and the duration of treatment while on maintenance single-agent varlitinib (median duration 5.1 months, range 2.0–13.3 months). Patients with HER2+ metastatic breast cancer (*) [HER2+ MBC] tended to have longer PFS (median PFS 5.4 months, range 1.2–17.1 months). PD progressive disease, PR partial response, SD stable disease
Twenty patients had HER2+ metastatic breast cancer with a median four lines of prior systemic palliative therapies (range 0–14). Sixteen had received two or more cycles of varlitinib and were evaluable for response; one received study treatment as neoadjuvant therapy. All four patients who were not evaluable for a response had discontinued treatment within the first cycle because of intolerable toxicities. 9/16 (56.3%) evaluable patients achieved a partial response while 4/16 (25.0%) achieved stable disease as their best response, with a clinical benefit rate of 81.3%.
Eight patients were maintained on single-agent varlitinib (cohort A: 4, cohort B: 2, cohort C: 2) after receiving a median of 7.5 cycles (range 2–15 cycles) of chemotherapy combined with varlitinib. The median duration of maintenance single-agent varlitinib therapy was 5.1 months (range 2.0–13.3 months) without disease progression in these patients. All eight patients had HER2+ metastatic breast cancer.
3.5 Varlitinib and Paclitaxel Pharmacokinetics Analysis (Table 4)
Blood was drawn to study the pharmacokinetics of varlitinib in patients from all cohorts, and of paclitaxel from cohorts A3, A4, and B1. In patients receiving continuous dosing of varlitinib, a trend of a dose-dependent increment was observed in varlitinib plasma exposure. Mean AUC from time 0 to 24 h (AUC0–24h) was 21574 ± 7750 vs 37292 ± 2044 vs 49493 ± 22186 h*ng/mL for 300-mg vs 400-mg vs 500-mg continuous dosing, respectively (p = 0.434). A linear relationship between varlitinib dose and AUC0–24h was observed. The mean maximum plasma concentration (Cmax) of varlitinib was 1540 ± 636 vs 2155 ± 970 vs 2305 ± 1293 ng/mL for 300-mg vs 400-mg vs 500-mg continuous dosing, respectively, although the difference was not statistically significant (p = 0.614). The mean Cmax of the 500-mg cohort was lower than predicted based on that of the 400-mg cohort (Fig. 1 of the ESM). This lower than proportionate increase in Cmax may be because absorption saturation had been achieved.
A similar trend was seen in patients receiving intermittent dosing of varlitinib (mean varlitinib Cmax 2737 ± 1441 vs 4669 ± 3014 ng/mL for 300-mg vs 400-mg intermittent dosing, p = 0.149; mean AUC0–24h 43,960 ± 22,567 vs 75,636 ± 43,698 h*ng/mL for 300-mg vs 400-mg intermittent dosing, p = 0.110).
The Cmax of paclitaxel on cycle 1 day 1 was not significantly different between the varlitinib 400-mg intermittent dosing cohorts (A3, B2) and 300-mg intermittent dosing cohorts (A4, B1, C1); mean Cmax 3525 ± 917 vs 2961 ± 1120 ng/mL, p = 0.241). Similarly, AUC0–24h on cycle 1 day 1 did not differ significantly (mean AUC0–24h 6126 ± 2054 vs 5007 ± 1863 ng/mL, p = 0.206). This suggests that varlitinib did not significantly affect the pharmacokinetics of paclitaxel.
4 Discussion and Conclusions
In this phase I study, varlitinib was administered orally in combination with chemotherapy in patients with advanced solid cancers. When varlitinib was administered together with carboplatin and paclitaxel, the combination was intolerable across four dose levels of varlitinib, with more than half the patients experiencing DLTs, mainly related to neutropenia and electrolyte disturbances. Weekly carboplatin and paclitaxel is a myelosuppressive regimen with grade 3 and above neutropenia observed in 42% of patients with ovarian cancer; febrile neutropenia was observed in 7% of patients receiving varlitinib in combination with 3-weekly docetaxel in a phase I study [26, 27]. In our study, we similarly observed a high rate of neutropenia, impeding the administration of higher doses of varlitinib. The most common DLT is grade 3 and 4 neutropenia in cohort A (n = 6, 33.3%), and this decreased to 18.8% (n = 3) in cohort B when carboplatin was dropped from the study regimen, confirming that carboplatin contributed to the high rates of neutropenia. A pharmacokinetic analysis did not demonstrate any interaction between varlitinib and paclitaxel, the backbone chemotherapy drug in HER2+ breast cancer. Hence, we inferred that carboplatin was the major contributor to the high rates of neutropenia in cohort A and dropped carboplatin in subsequent dose cohorts. A limitation to this conclusion is that we did not study the pharmacokinetics of carboplatin, and it is uncertain if there could be any interaction between carboplatin and varlitinib, which may explain the increased myelosuppression rates with the combination.
In combination with weekly paclitaxel, varlitinib 300 mg dosed intermittently was tolerable. However, further escalation of the dose or frequency of varlitinib dosing proved to be poorly tolerated with neutropenia and transaminitis being the main DLTs. The occurrence of grade 3 and above diarrhea was low at 5.6%, and was easily managed with anti-diarrheal medications; this is lower in comparison with the grade 3 and 4 diarrhea rate of 12% for the combination of lapatinib and capecitabine, and 8% for the combination of lapatinib and letrozole [11, 28]. Of interest, the all grade pneumonitis rate was low at 5.4%.
This initial clinical experience with varlitinib in combination with weekly paclitaxel ± carboplatin demonstrates promising anti-tumor activity. Objective responses were seen in patients evaluable for a response across all varlitinib doses: 500 mg continuous (1/1; 100%), 400 mg continuous (2/5; 40%), 300 mg continuous (1/5; 20%), 400 mg intermittent (3/6; 50%), 300 mg intermittent (4/14; 28.6%). Comparing objective responses seen in patients receiving varlitinib 300 mg intermittently vs higher doses of varlitinib, there was no significant difference (p = 0.707). Clinical benefit rates were also not significantly different (p = 0.671).
As expected, varlinitib was active in patients with HER2+ metastatic breast cancer, with 81.3% (13/16) achieving clinical benefit (56.3% partial response and 25.0% stable disease). Eight patients were maintained on single-agent varlitinib after completing chemotherapy, and achieved durable disease control for up to 17.1 months (range 5.2–17.1 months), despite not responding to prior HER2-directed therapies.
Pharmacokinetic analysis showed that varlitinib Cmax and AUC increased in a dose-dependent manner. The linear increase in drug concentration accumulation is consistent with the clinical observation of more intolerable toxicities when varlitinib was dosed continuously. The Cmax and AUC in our study are consistent with what has been reported in previous early-phase studies [26]. Our analysis suggests that there is no interaction between the pharmacokinetics of paclitaxel and varlitinib. However, a limitation of this study is that the stipulated sampling timepoints were insufficient to capture the actual time to Cmax of varlitinib. The median half-life of varlitinib is 5–7 h, which has been reported in other studies [26].
Of note, Cmax at the recommended phase II dose of 300 mg twice daily administered intermittently was higher than the half maximal inhibitory concentration of varlitinib, suggesting that future studies can be carried out to evaluate the pharmacokinetics of once-daily dosing of varlitinib [26]. Once-daily administration of varlitinib may allow continuous dosing to be utilized while sustaining an adequate Cmax yet reducing the degree of drug accumulation, hence ameliorating severe toxicities.
The manageable toxicity profile, pharmacokinetic properties, and encouraging antitumor activity in patients with advanced solid tumors, particularly HER2+ breast cancer, warrant further evaluation of varlitinib in larger phase studies. In this study, we observed that subcutaneous trastuzumab could be added safely to intermittent dosing varlitinib and weekly paclitaxel. The use of dual HER2-targeted agents in combination with chemotherapy is currently the standard of care in the treatment of metastatic HER2 over-expressed breast cancer, and is frequently used as neoadjuvant therapy in the earlier stages of HER2+ breast cancer [30,31,32]. Varlitinib has the potential to be an effective oral drug in the armamentarium of agents used to treat HER2 over-expressed breast cancer. For this reason, we are now studying the triplet combination of trastuzumab, varlitinib, and paclitaxel in an ongoing phase II trial as neoadjuvant therapy in HER2+ breast cancers (ClinicalTrials.gov: NCT02396108).
References
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–82. https://doi.org/10.1126/science.3798106.
Howlader N, Altekruse SF, Li CI, Chen VW, Clarke C, et al. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. JNCI J Natl Cancer Inst. 2014;106:5. https://doi.org/10.1093/jnci/dju055.
Hsu JL, Hung M-C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016;35(4):575–88. https://doi.org/10.1007/s10555-016-9649-6.
Casalini P, Iorio MV, Galmozzi E, Ménard S. Role of HER receptors family in development and differentiation. J Cell Physiol. 2004;200(3):343–50. https://doi.org/10.1002/jcp.20007.
Roskoski R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. https://doi.org/10.1016/j.phrs.2013.11.002.
Wieduwilt MJ, Moasser MM. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci. 2008;65(10):1566–84. https://doi.org/10.1007/s00018-008-7440-8.
Normanno N, Maiello MR, De Luca A. Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs): simple drugs with a complex mechanism of action? J Cell Physiol. 2003;194(1):13–9. https://doi.org/10.1002/jcp.10194.
Mishra R, Hanker AB, Garrett JT. Genomic alterations of ERBB receptors in cancer: clinical implications. Oncotarget. 2017;8(69):114371–92. https://doi.org/10.18632/oncotarget.22825.
Shepard HM, Jin P, Slamon DJ, Pirot Z, Maneval DC. Herceptin. Handb Exp Pharmacol. 2008;181:183–219. https://doi.org/10.1007/978-3-540-73259-4_9.
Hudis CA. Trastuzumab: mechanism of action and use in clinical practice. N Engl J Med. 2007;357(1):39–51. https://doi.org/10.1056/NEJMra043186.
Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–43. https://doi.org/10.1056/NEJMoa064320.
Martin M, Holmes FA, Ejlertsen B, Delaloge S, Moy B, Iwata H, et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688–700. https://doi.org/10.1016/S1470-2045(17)30717-9.
Montemurro F, Valabrega G, Aglietta M. Lapatinib: a dual inhibitor of EGFR and HER2 tyrosine kinase activity. Expert Opin Biol Ther. 2007;7(2):257–68. https://doi.org/10.1517/14712598.7.2.257.
Tiwari SR, Mishra P, Abraham J. Neratinib, a novel HER2-targeted tyrosine kinase inhibitor. Clin Breast Cancer. 2016;16(5):344–8. https://doi.org/10.1016/j.clbc.2016.05.016.
Whenham N, D’Hondt V, Piccart MJ. HER2-positive breast cancer: from trastuzumab to innovatory anti-HER2 strategies. Clin Breast Cancer. 2008;8(1):38–49. https://doi.org/10.3816/CBC.2008.n.002.
Javle MM, Oh D-Y, Ikeda M, Qin SK, Yong WP, Chao Y, et al. TREETOPP: a phase 2/3 study of varlitinib plus capecitabine versus placebo plus capecitabine as second-line treatment in patients with advanced or metastatic biliary tract cancers (BTCs). J Clin Oncol. 2018;36(15):TPS4143. https://doi.org/10.1200/JCO.2018.36.15_suppl.TPS4143.
Kim J, Im S, Lee K, Kim JW, Lee K, Han S, et al. Phase IIa study to evaluate the biological activity of ASLAN001 in HER-1/2 co-expressing or HER-2 amplified advanced gastric cancer. Ann Oncol. 2014;25:4. https://doi.org/10.1093/annonc/mdu334.49.
Lee SC, Chen SC, Dai MS, Lee GE, Liu CL, Chan A, et al. Multicenter phase 2 trial of varlitinib versus lapatinib in combination with capecitabine in patients with HER2+ metastatic breast cancer (MBC) who failed prior trastuzumab therapy. Ann Oncol. 2017;28:10. https://doi.org/10.1093/annonc/mdx654.010.
Tan AC, Oh D-Y, Chao Y, Hsieh C-Y, Chang W-L, Isanto F, et al. Efficacy and safety of varlitinib, a reversible pan-HER tyrosine kinase inhibitor, in combination with platinum-based regimens in biliary tract cancers: a pooled analysis from three phase I studies. J Clin Oncol. 2019;37(4):331. https://doi.org/10.1200/JCO.2019.37.4_suppl.331.
Liu C-Y, Chu P-Y, Huang C-T, Chen J-L, Yang H-P, Wang W-L, et al. Varlitinib downregulates HER/ERK signaling and induces apoptosis in triple negative breast cancer cells. Cancers (Basel). 2019;11(1):105. https://doi.org/10.3390/cancers11010105.
Tan AC, Seet AOL, Choo SP, Tai SWM, Lam YCJ, Teng WT, et al. A phase I study of varlitinib (VAR; ASLAN001) an oral pan-HER tyrosine kinase inhibitor (TKI) combined with mFOLFIRI chemotherapy in advanced solid tumours. Ann Oncol. 2019;30:v178. https://doi.org/10.1093/annonc/mdz244.034.
Gras J. Varlitinib tosylate. Pan-HER tyrosine kinase inhibitor, treatment of cancer. Drugs Future. 2018;43(11):815. https://doi.org/10.1358/dof.2018.043.11.2817971.
Hsieh C-Y, Ooi L, Ong RW, Lindmark B, McHale M, Huynh HT. Varlitinib to demonstrate anti-tumour efficacy in patient-derived hepatocellular carcinoma xenograft models. J Clin Oncol. 2016;34(15):e15598. https://doi.org/10.1200/JCO.2016.34.15_suppl.e15598.
Wainberg ZA, Anghel A, Desai AJ, Ayala R, Luo T, Safran B, et al. Lapatinib, a dual EGFR and HER2 kinase inhibitor, selectively inhibits HER2-amplified human gastric cancer cells and is synergistic with trastuzumab in vitro and in vivo. Clin Cancer Res. 2010;16(5):1509–19. https://doi.org/10.1158/1078-0432.CCR-09-1112.
Center for Drug Evaluation and Research. NDA/BLA multi-disciplinary review and evaluation NDA 208051. 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208051Orig1s000MultidisciplineR.pdf. Accessed 6 Feb 2022.
ASLAN Pharmaceuticals Pte Ltd. Varlitinib investigator’s Brochure, Edition 13. 2019.
Pignata S, Scambia G, Katsaros D, Gallo C, Pujade-Lauraine E, De Placido S, et al. Carboplatin plus paclitaxel once a week versus every 3 weeks in patients with advanced ovarian cancer (MITO-7): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2014;15(4):396–405. https://doi.org/10.1016/S1470-2045(14)70049-X.
Schwartzberg LS, Schwarzberg LS, Franco SX, Florance A, O’Rourke L, Maltzman J, et al. Lapatinib plus letrozole as first-line therapy for HER-2+ hormone receptor-positive metastatic breast cancer. Oncologist. 2010;15(2):122–9. https://doi.org/10.1634/theoncologist.2009-0240.
Guo X, Loibl S, Untch M, Möbus V, Schwedler K, Fasching P, et al. Re-challenging taxanes in recurrent breast cancer in patients treated with (neo-)adjuvant taxane-based therapy. Breast Care Basel. 2020;6(4):279–83. https://doi.org/10.1159/000330946.
Swain SM, Miles D, Kim S-B, Im Y-H, Im S-A, Semiglazov V, et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): end-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020;21(4):519–30. https://doi.org/10.1016/S1470-2045(19)30863-0.
Gianni L, Pienkowski T, Im Y-H, Tseng L-M, Liu M-C, Lluch A, et al. 5-year analysis of neoadjuvant pertuzumab and trastuzumab in patients with locally advanced, inflammatory, or early-stage HER2-positive breast cancer (NeoSphere): a multicentre, open-label, phase 2 randomised trial. Lancet Oncol. 2016;17(6):791–800. https://doi.org/10.1016/S1470-2045(16)00163-7.
Schneeweiss A, Chia S, Hickish T, Harvey V, Eniu A, Hegg R, et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA). Ann Oncol. 2013;24(9):2278–84. https://doi.org/10.1093/annonc/mdt182.
Acknowledgments
We thank the patients for their participation in this study, and their families and caregivers.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by research grants from the Singapore Ministry of Health’s National Medical Research Council under its Clinician Scientist Awards [Grant numbers: NMRC/CSA-SI/0004/2015 and NRMC/CSA/015/2009] and the National University Cancer Institute, Singapore’s Centre Grant [Grant number: NMRC/CG/012/2013]. Varlitinib was kindly provided by ASLAN Pharmaceuticals Ltd (Singapore) through a research collaboration.
Conflict of interest
Andrea Wong has received honoraria from Pfizer, Novartis, and Eisai; and has advisory activity with Otsuka. Samuel Ow has received honoraria from Pfizer, AstraZeneca, Eli Lilly, Novartis, and Roche. Raghav Sundar has received honoraria from Bristol-Myers Squibb, Lilly, Roche, Taiho, Astra Zeneca, DKSH, and MSD; has advisory activity with Bristol-Myers Squibb, Merck, Eisai, Bayer, Taiho, Novartis, MSD, and AstraZeneca; received research funding from MSD and Paxman Coolers; and has received travel grants from AstraZeneca, Eisai, Roche, and Taiho Pharmaceutical. David Tan has received honoraria and travel support from AstraZeneca, Novartis, Roche, Merck Serono, MSD, Bayer, Genmab, Takeda, Eisai, and Clovis; has advisory activity with Astra Zeneca, MSD, Roche, Bayer, Genmab, Tessa Therapeutics, and Eisai; has received research funding from Astra Zeneca, Karyopharm Therapeutics, Bayer, Roche (Foundation Medicine), National Medical Research Council Singapore, Pangestu Family Foundation Gynaecological Cancer Research Fund, and Cancer Science Institute Singapore; and has stock ownership with Asian Microbiome Library (AMiLi). Ross Soo has advisory activity with Amgen, Astra-Zeneca, Bayer, BMS, Boehringer Ingelheim, Lilly, Merck, Novartis, Pfizer, Roche, Taiho, Takeda, and Yuhan; and has received research funding from AstraZeneca and Boehringer Ingelheim. Joline Lim has advisory activity with Pfizer and Novartis; has received research funding from Synthon; and has received conference support from Novartis, Pfizer, and AstraZeneca. Wei Peng Yong has advisory activity with Abbvie, Amgen, AstraZeneca, BMS, Ipsen, Novartis, and MSD; and has received support on the speaker bureaus for Bayer, Eisai, Lilly, Sanofi, and Taiho. Boon Cher Goh has received research funding from MSD, Adagene, Bayer Healthcare, and Alx Oncology; has advisory activity with Adagene, MSD, and Novartis; and has stock ownership with Merus Therapeutics and Gilead Sciences. Soo Chin Lee has advisory activity with Pfizer, Novartis, Astra Zeneca, ACT Genomics, Eli Lilly, MSD, and Roche; has received research funding from Pfizer, Eisai, Taiho, ACT Genomics, Bayer, and Karyopharm; and has received conference support from Amgen, Pfizer, and Roche. Matilda Lee, Siew Eng Lim, and Ling Zhi Wang declare that they have no conflicts of interest that might be relevant to the contents of this manuscript.
Ethics Approval
This study was approved by the National Healthcare Group Domain Specific Review Board prior to commencement. The procedures used in this study adhere to the tenets of the Declaration of Helsinki.
Consent to Participate
Informed consent was obtained from all individual participants included in the study.
Consent for Publication
Consent was obtained from all individual participants included in the study.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
Authors’ Contributions
Soo Chin Lee and Boon Cher Goh contributed to the study conception and design. Matilda Lee, Andrea Wong, Raghav Sundar, David Tan, Ross Soo, Cheng Ean Chee, Joline Lim, Wei Peng Yong, Siew Eng Lim, Boon Cher Goh, and Soo Chin Lee recruited patients into the trial and contributed to data collection. Lingzhi Wang performed the paclitaxel pharmacokinetic analysis. Matilda Lee, Lingzhi Wang and Soo Chin Lee performed the data analysis and data interpretation. The original draft of the manuscript was prepared by Matilda Lee and Soo Chin Lee, and all authors have commented on, read, and approved the final manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Lee, M.X., Wong, A.L.A., Ow, S. et al. Phase Ib Dose-Finding Study of Varlitinib Combined with Weekly Paclitaxel With or Without Carboplatin ± Trastuzumab in Advanced Solid Tumors. Targ Oncol 17, 141–151 (2022). https://doi.org/10.1007/s11523-022-00867-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-022-00867-0