
Abstract
The clinical development of cancer drugs is rapidly moving from empirical “one drug fits all” or development-by-tumor-type approaches towards more personalized treatment models. A deeper understanding of cancer and the immune system, novel technologies, and powerful analytics have fueled an increase in precision oncology approaches integrating the molecular profiles of the tumor with the clinical profile of the patient. While this approach has been successful for targeted therapies, the complex mode of action of immunotherapies will likely require integration of clinical profiling with more comprehensive profiling of the tumor, of the tumor microenvironment, and of the immune system of the patient. Integration of precision oncology into clinical research for immunotherapies is viewed as a means to better select patients in the early clinical phase of drug development to (1) maximize the benefit-to-risk ratio for the patient, (2) generate early proof of concept and proof of relevance for the investigational drug, and (3) inform on how to best combine or sequence the therapeutic with other drugs. Here we discuss the upsides and challenges of incorporating precision immuno-oncology into early-phase clinical trials.
Similar content being viewed by others


1 Introduction
The approval of immune checkpoint inhibitors [ICIs; anti-PD-1, anti-PD-L1, and anti-CTLA-4 monoclonal antibodies (mAb)] for the treatment of malignant diseases for which few therapeutic options existed has created a paradigm shift in oncology and highlighted the therapeutic benefits that can be afforded by anti-cancer immunity. However, despite having revolutionized the approach to cancer treatments, ICIs still do not provide a long-term benefit to the majority of cancer patients [1, 2]. Between 10 and 30% of patients with stage IV melanoma or advanced non-small-cell lung cancer (NSCLC) are long-term survivors on immunotherapies [3,4,5], while much fewer with other tumor types are expected to be alive at 5 years. Nevertheless, the unprecedented rates of long-lasting clinical responses observed with ICIs in some patients have led to an avalanche of monotherapy and combination therapy approaches to cancer immunotherapy being investigated. In 2018, there were 3394 immuno-oncology agents in development encompassing 417 targets [6, 7]. They include novel ICIs, immune checkpoint agonists of co-stimulatory receptors, cytokines, cytokine blockers, other immunomodulators including activators of the innate immune system, T-cell engagers including CD3-targeted bispecific antibodies (bsAbs), neoantigen-based vaccines, oncolytic viruses, and adoptive transfer of tumor-specific effector cells.
A greater understanding of the complex tumor—tumor microenvironment—immune system interactions has fueled the rapid clinical advances of immunotherapies. The immune system is a notably complex bionetwork comprised of a multitude of highly diversified and functionalized cells, soluble mediators, and organs all interacting and collaborating as a dynamic but organized system to guard human health [8, 9]. Most cancers do not consist of a homogeneous cancer cell population but are comprised of a diverse collection of cells harboring distinct genetic make-up as a result of mutational or epigenetic changes [10, 11]. Finally, the response rate in phase 1 trials for anticancer drugs hovers around 15–20% [12], which is not optimal for patients, clinicians, or drug developers. In this context, there is a growing need to conduct optimized early-phase clinical trials specifically tailored to evaluate the safety and efficacy of novel and combination immunotherapies for which the traditional “one size fits all”, “all-comer”, or “single-analyte biomarker” approaches are unlikely to yield transformational information. Here we discuss the benefits and challenges of incorporating precision medicine, an approach to cancer treatment that accounts for variability in the genes, environment, and lifestyle of each person [13], into early-phase cancer clinical trials.
2 Current Enrollment Paradigm for Immunotherapy Early-Phase Clinical Trials
Enrollment in early clinical trials commonly utilizes eligibility criteria based on clinical considerations to protect trial participants and avoid noise in the safety data [14]. Protocol development and patient selection for immunotherapy trials remain extremely conservative and are mostly based on what was established for cytotoxic or targeted therapies, notwithstanding that they may not be optimal for innovative Investigational Immune-Products (IIPs). Most of the IIP phase 1 trials are concurrently continuing to select patients based on standard criteria: (1) normal organ function; (2) prior lines of therapies; (3) prior autoimmune disorder or autoimmune-mediated toxicity; (4) co-medication; (5) performance status; (6) QTC value; (7) brain metastasis; (8) co-morbidities; and (9) tumor type. Very few trials have incorporated even standard immuno-markers such as tumor mutational burden (TMB), microsatellite instability (MSI), loss of major-histocompatibility complex (MHC) class I or, for anti-PD-(L)1 mAbs, PD-L1 status. Most early-phase studies for ICIs have applied standard inclusion and exclusion criteria, sometime with the addition of a single biomarker linked to the drug mechanism of action. For example, KEYNOTE-001, a phase 1 study of the anti-PD-1 mAb pembrolizumab, was based on tumor site of origin and disease state; assessment of PD-L1 expression in the tumor by immunohistochemistry (PD-L1 is one of the ligands of PD-1) was only performed in a subset of patients [15, 16]. A survey of the inclusion and exclusion criteria for recruiting and not-yet-recruiting phase 1 (or phase 1/2) cancer immunotherapy trials on clinicaltrial.gov (accessed 13 June 2019) suggested that this approach remains the norm [see, e.g.: NCT02817633 (for anti-TIM-3 mAb alone or in combination with an anti-PD-1 mAb or anti-PD-1 mAb plus an anti-LAG-3 mAb), NCT03708328 (bsAb targeting PD-1 and TIM-3), NCT03889275 (oncolytic virus in combination with anti-PD-L1 mAb), NCT02947165 (anti-TGF-beta mAb in combination with anti-PD-1 mAb), NCT03809624 [bsAb targeting PD-L1 and 4-1BB), NCT02904226 (anti-ICOS mAb alone or in combination with anti-PD-1 mAb or anti-CTLA-4 mAb)]. In order to better select patients prior to enroll an IIP phase 1 trial, we suggest considering pre-selection of patients based on immuno-genomic markers or profiles to obtain a better match between the new agent and the patient/tumor characteristics (Fig. 1).

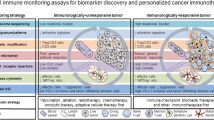
Implementation of precision oncology for cancer investigational immune products (IIP in clinical phase 1 studies). Prospective phase 1 trial participants will undergo clinical profiling, and tumor molecular profiling, tumor immune profiling, and/or patient immune profiling based on the biological/clinical hypothesis for a particular IIP. If the patient is deemed eligible based on clinical considerations, the patient will be assigned, based on an actionable integrating profile to either the immunotherapy (IIP trial, a targeted therapy, standard of care/Physician’s Choice Therapy (PCT) or best supportive care (BSC). If the patient’s profiling is not readily actionable or if the patient is equally eligible for more than one possible treatment, the patient’s potential participation in the IIP trial should be determined by the Molecular Tumor Board. *A list of US Food and Drug Administration (FDA)-approved targeted therapies against cancer molecular drivers/actionable targets can be found at https://www.fda.gov/medical-devices/vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-vitro-and-imaging-tools
3 Next-Generation Immunotherapy Early Clinical Trial Design
In contrast to the traditional “one-size-fits-all” approach to clinical trial design for chemotherapy and the more recent immunotherapy “all-comer” studies, which incorporate multiple expansion cohorts [17, 18], a precision oncology approach to an early immunotherapy trial would instead enroll patients after integrating clinical and immunological information derived from each patient with his/her tumor immuno-genomic landscape. This would afford the patient a maximized chance for treatment efficacy and thus an opportunity to increase the chances of the drug’s success in early clinical trials.
Recent technology and computing advancements have increased the feasibility of deep, comprehensive profiling of clinical trial participants. Molecular profiling of tumors is entering mainstream clinical practice [19,20,21] as an increasing number of clinicians use next-generation sequencing (NGS)-based tests to identify targetable cancer mutations and matching treatments. In addition, more and more groups are using NGS in retrospective analysis of clinical trials to discover novel biomarkers predictive of response, to gain further insights on the drug mode of action and its clinical development, and to gather insights for the development of novel therapeutics. To date, precision oncology has been used mostly in early clinical trials to match targeted therapies to tumor molecular aberrations where it has led to higher rates of response and longer time-to-treatment failure [22]. In this setting, the full tumor mutational and immunological landscapes, as well as cellular heterogeneity, are often not taken into consideration, though they may confer de novo resistance to the therapy.
The emergence of immunotherapies and their rapid integration into clinical standard of care has added another level of complexity to the implementation of precision oncology in early-phase clinical trials. As immunotherapies harness the ability of the immune system to detect and destroy cancer cells, precision oncology incorporation in early immunotherapy clinical trials is likely to demand the integration of tumor genomics, with information derived from the tumor microenvironment, assessment of infiltration of immune cells into the tumor, and the immune system. The nature of the tumor micro-environment, the potential infiltration of immune cells in the tumor, and the overall immune fitness of the patient are all thought to play a role in response to immunotherapy [23, 24]. Immuno-profiling of tumors and assessing the overall fitness and responsiveness of the patient’s immune system could also be used to optimize patient stratification in immunotherapy trials, to inform on combination strategies, and to help with the prediction of immune-related serious adverse events (irSAEs). Immuno-profiling can also provide valuable insights into the mechanisms of action of immunotherapies. Current methodologies used for the molecular profiling of tumors and immuno-profiling (blood and tumors) are presented in Tables 1 and 2, respectively.
4 Practical Considerations for the Implementation of Comprehensive Patient Profiling
Integrating clinical, molecular, and immunological profiling into early clinical trials can help identify patients whose cancer has the right profile for a given clinical program and are therefore most likely to respond, monitor their response to the investigational drug(s), and identify patients at an increased risk for irSAEs. Comprehensive profiling also allows retrospective analyses to be performed against a greater number of variables than the one utilized for enrollment to inform about further determinants of response to therapy. However, this does not come without its own set of challenges, on top of the very real risk of having too stringent criteria that would not allow for effective trial enrollment due to a heightened screen failure rate.
The first step is the identification of possible biomarkers of interest for the study, often based on the known pharmacological and toxicological properties of the experimental therapeutic, and a biological or clinical hypothesis. However, incorporation of biomarkers into an early-phase clinical study design can pose unique technical, logistic, and regulatory challenges that need to be addressed so that quality and regulatory requirements are met. These include sample collection, assay type, analytics, and level of qualification/validation, turn-around-time, and cost. The evaluation of phase 1 sites by sponsors or CROs should incorporate their ability to connect with relevant platforms, technology, and analytics to be able to achieve the proposed patient selection.
Sample collection considerations: (1) Source of tissue; (2) collection method (taking into consideration invasiveness of the method); (3) amount of specimen to be collected (should be minimized yet allow for possible retesting including as part of additional or bridging studies); (4) sample processing, preservation, and storage (methods should preserve analytes’ integrity as much as possible, and must be feasible and practical in a clinical setting); (5) shipping conditions to the testing laboratory. If formalin-fixed paraffin-embedded (FFPE) tissue specimens are used, there is a need to address how tumor purity will be determined for assays that require a minimum percent of tumor content.
Assay considerations: (1) Define intended use; (2) select best suited assay technology and data analytics (as there can be significant differences or trade-offs between offerings); (3) assess need to perform assay in a Clinical Laboratory Improvement Amendments (CLIA) laboratory (e.g., if biomarker will be used to direct patient enrollment); (4) if applicable, determine assay development timeline so that assay is ready by the time the trial starts to enroll; (5) fit-for-purpose qualification or validation (which identifies the most important elements of a qualification/validation program supporting its intended use) should be performed in the central laboratory that will run the assay; (6) analytics need to be carefully assessed especially when the assay is NGS-based as different companies or laboratories can have different algorithms for variant calling and interpretation (e.g., single-nucleotide variants (SNVs), insertion or deletion mutation (indel), fusion, and copy number variation (CNV)), cancer DNA analysis (e.g., TMB, neoantigen burden and ranking) [25], and to appraise RNA expression signatures; (7) advanced analytics and machine learning algorithms might need to be purposely developed to deliver actionable insights; (8) consider impact of intra- and inter-tumor heterogeneity and clonal evolution (e.g., necessity to collect more than one biopsy per tumor, to biopsy more than one tumor site, use of liquid biopsy vs. tissue biopsy or combination of both, sampling at enrollment vs. repeat or longitudinal sampling).
Turn-around-time and cost considerations: (1) Turn-around-time from sample collection to data receipt of more than 14 calendar days may have an adverse effect on patient recruitment and, in fast-progressing disease, the patients themselves; (2) specimen collection, processing, and analysis for complex amalgam of biomarkers can significantly increase early clinical trial cost, especially if longitudinal sampling is performed.
Biomarkers used in clinical cancer research could pave the way to the development of companion diagnostic tests that can be co-approved with the therapeutic agent. The development of companion diagnostics is outside the scope of this article but more information can be found on this topic on the US Food and Drug Administration (FDA) website [26].
5 Conclusion
Given the sheer number of immunotherapies entering clinical trials and the fact that current immunotherapies seem to provide durable benefits in only a minority of patients, there is an increasing need to assess IIP mono and combination therapies with rigor and justification. A major challenge of early clinical development is to generate data in a timely fashion that will assess and hopefully verify the safety and relevance of the therapeutics under development in the current context of standard of care. Most if not all IIPs entering clinical testing are based on a precise biological or clinical hypothesis that can help guide a biomarker-directed enrollment approach. For example, some assets may help overcome resistance or boost the efficacy of existing ICIs, while others may help turn a cold tumor into a hot tumor [27,28,29].
Clinical development of immunotherapies could be expedited using innovative early clinical trial study designs that match patients with several specific genotypes and phenotypes with a specific immunotherapy approach. There is momentum to integrate precision medicine into clinical trials to help ensure that the right treatment gets to the right patients. We suggest an early patient selection based immuno-genomic markers (or profiles) to better triage individuals to standard therapy or palliative care versus consideration for phase 1 trials with a new targeted therapy or IIP. The overall strategy is summarized in Fig. 1. Conducting biomarker-directed early-phase immunotherapy clinical trials can lead to fast assessment of the safety and the clinical relevance of the asset, which will be beneficial to patients and companies alike, and delight the clinical team. This will also help evaluate the benefits of conducting precision oncology clinical research for cancer immunotherapies. With new comprehensive immuno-genomic profiling platforms, the feasibility of this type of testing is becoming increasingly practical in these trials. It is prime time to revisit the phase 1 conservative world and deploy the extensive knowledge acquired in the laboratory to better benefit the patients who, in clinical research, are generally facing a critical prognosis as their disease usually has progressed on conventional therapies or experimental drugs administered as part of a clinical trial. New innovative pilot studies to prospectively evaluate the impact of patient selection for IIP phase 1 studies are warranted.
References
Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–5.
Carbognin L, Pilotto S, Milella M, Vaccaro V, Brunelli M, Caliò A, et al. Differential activity of nivolumab, pembrolizumab and MPDL3280A according to the tumor expression of programmed death-ligand-1 (PD-L1): sensitivity analysis of trials in melanoma, lung and genitourinary cancers. PLoS One. 2015;10:e0130142.
Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann Oncol. 2019;30:582–8.
Garon EB, Hellmann MD, Rizvi NA, Carcereny E, Leighl NB, Ahn M-J, et al. Five-year overall survival for patients with advanced non-small-cell lung cancer treated with pembrolizumab: results from the phase I KEYNOTE-001 study. J Clin Oncol. 2019;JCO.19.00934.
Gettinger S, Horn L, Jackman D, Spigel D, Antonia S, Hellmann M, et al. Five-year follow-up of nivolumab in previously treated advanced non-small-cell lung cancer: results from the CA209-003 study. J Clin Oncol. 2018;36:1675–84.
Kaiser J. Too much of a good thing? Science. 2018;359:1346–7.
Tang J, Pearce L, O’Donnell-Tormey J, Hubbard-Lucey VM. Trends in the global immuno-oncology landscape. Nat Rev Drug Discov. 2018;17:783–4.
Nicholson LB. The immune system. Essays Biochem. 2016;60:275–301.
Rieckmann JC, Geiger R, Hornburg D, Wolf T, Kveler K, Jarrossay D, et al. Social network architecture of human immune cells unveiled by quantitative proteomics. Nat Immunol. 2017;18:583–93.
McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613–28.
Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–13.
Chakiba C, Grellety T, Cousin S, Italiano A. Risks and benefits of phase 1 oncology trials in the era of personalized medicine. Ann Oncol. 2016;27:1372P.
What is precision medicine?—Genetics Home Reference—NIH. https://ghr.nlm.nih.gov/primer/precisionmedicine/definition. Accessed 14 Jun 2019.
CFR—Code of Federal Regulations Title 21. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=56.111. Accessed 14 Jun 2019.
Patnaik A, Kang SP, Rasco D, Papadopoulos KP, Elassaiss-schaap J, Beeram M, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res. 2015;21:4286–94.
Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Keff R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109–17.
Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–7.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, Mcdermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54.
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23.
Hux A, Lewis A, Sachwitz D, Gregory T. Clinical utility of next-generation sequencing in precision oncology. J Am Acad Physician Assist. 2019;32:35–9.
Xie J, Lu X, Wu X, Lin X, Zhang C, Huang X, et al. Capture-based next-generation sequencing reveals multiple actionable mutations in cancer patients failed in traditional testing. Mol Genet Genom Med. 2016;4:262–72.
Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Falchook GS, Fu S, et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center Initiative. Clin Cancer Res. 2012;18:6373–83.
Trujillo JA, Sweis RF, Bao R, Luke JJ. T cell-inflamed versus non-T cell-inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res. 2018;6:990–1000.
Gnjatic S, Bronte V, Brunet LR, Butler MO, Disis ML, Galon J, et al. Identifying baseline immune-related biomarkers to predict clinical outcome of immunotherapy. J Immunother Cancer. 2017;5:1–18.
Tumor Mutational Burden (TMB)|Friends of Cancer Research. https://www.focr.org/tmb. Accessed 14 Jun 2019.
Companion Diagnostics|FDA. https://www.fda.gov/medical-devices/vitro-diagnostics/companion-diagnostics. Accessed 13 Jun 2019.
Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, et al. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol Res. 2015;3:506–17.
Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18:197–218.
Shergold AL, Millar R, Nibbs RJB. Understanding and overcoming the resistance of cancer to PD-1/PD-L1 blockade. Pharmacol Res. 2019;145:104258.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
Open Access fee paid by Personalis, Inc.
Conflict of interest
Xavier Paliard is an employee and shareholder of Personalis, Inc. Olivier Rixe reports no conflicts of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Paliard, X., Rixe, O. Precision Oncology for Cancer Immunotherapies in Early-Phase Clinical Trials. Targ Oncol 14, 631–637 (2019). https://doi.org/10.1007/s11523-019-00678-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-019-00678-w