
Abstract
The transitions to multicellularity mark the most pivotal and distinctive events in life’s history on Earth. Although several transitions to “simple” multicellularity (SM) have been recorded in both bacterial and eukaryotic clades, transitions to complex multicellularity (CM) have only happened a few times in eukaryotes. A large number of cell types (associated with large body size), increased energy consumption per gene expressed, and an increment of non-protein-coding DNA positively correlate with CM. These three factors can indeed be understood as the causes and consequences of the regulation of gene expression. Here, we discuss how a vast expansion of non-protein-coding RNA (ncRNAs) regulators rather than large numbers of novel protein regulators can easily contribute to the emergence of CM. We also propose that the evolutionary advantage of RNA-based gene regulation derives from the robustness of the RNA structure that makes it easy to combine genetic drift with functional exploration. We describe a model which aims to explain how the evolutionary dynamic of ncRNAs becomes dominated by the accessibility of advantageous mutations to innovate regulation in complex multicellular organisms. The information and models discussed here outline the hypothesis that pervasive ncRNA-based regulatory systems, only capable of being expanded and explored in higher eukaryotes, are prerequisite to complex multicellularity. Thereby, regulatory RNA molecules in Eukarya have allowed intensification of morphological complexity by stabilizing critical phenotypes and controlling developmental precision. Although the origin of RNA on early Earth is still controversial, it is becoming clear that once RNA emerged into a protocellular system, its relevance within the evolution of biological systems has been greater than we previously thought.
Similar content being viewed by others


Introduction
For the first 2.5 billion years (Ga) of life on Earth, the size of most species was generally much smaller than 1 mm, and rarely was this exceeded (Carroll 2001). Prokaryotes, with few exceptions, have remained unicellular organisms optimizing their size (Carroll 2001), gene and protein content (Ochman and Davalos 2006) as well as the flexibility of their protein-based gene regulation (Lozada-Chavez et al. 2006) in order to maximize metabolisms that drive Earth's biogeochemical cycles (DeLong et al. 2010). Plants, animals, fungi, and protozoan seaweeds, on the other hand, are multicellular organisms that dominate the terrestrial landscapes and the oceans since one billion years ago with a remarkable diversity in genotypic and phenotypic complexity (King 2004; Knoll 2011). A multicellular organism is a collection of self-organized cells that express different phenotypes, despite having the same genotype, in response to the specialization of tasks to perform a cooperative physiological division of labour within an economic organization.
How Difficult is the Transition to Multicellularity?
At least 25 independent transitions to multicellularity have been recorded during the evolution of cellular complexity on Earth (Bonner 1998; Grosberg and Strathmann 2007; Rokas 2008; Knoll 2011). The transition to multicellularity has been repeatedly promoted from unicellular and colonial ancestors (Bonner 1998; Carroll 2001; Kaiser 2001; Medina et al. 2003; King 2004; Grosberg and Strathmann 2007). In several bacterial and eukaryotic organisms, these transitions are an inducible response to environmental stimuli such as predation and starvation (Bonner 1998; Lurling and Van Donk 1999; Kaiser 2001; Kolter et al. 2001; Kong et al. 2009). Surprisingly, multicellurarity can be reverted back to an unicellular state in several bacterial lineages (Velicer et al. 1998; Kolter et al. 2001). A special case are defectors (i.e., mutant cell lineages that selfishly improve their own fitness and fail to cooperate with the other cell types of the organism) in vertebrates, where non-viral transmissible cancerous cells could in effect become independently evolving unicellular colonies (Banfield et al. 1965; Strathmann 1991; Pearse and Swift 2006; Weiss et al. 2006).
The frequent origination and spread of multicellularity suggests that (1) selection favoring this transition is pervasive across organisms and time, (2) the genetic and developmental obstacles to this transition are relatively “easy” to overcome, and (3) adaptive mechanisms that control defectors and stabilize the transition are widely available in natural populations (Grosberg and Strathmann 2007). According to Grosberg and Strathmann (2007), all these pieces of empirical evidence together support the idea that evolution of muticellularity can be considered itself a significant but minor transition, and that cellular diversity can evolve easily when functionally called for by selective advantages. Nevertheless, multicellularity shows two states that presumably are not just extremes of a continuous spectrum but are fundamentally different: simple or complex (Bonner 1998; King 2004; Rokas 2008; Knoll 2011). Increase of organismal size, diversity of cell types, division of labor and functional specialization are interrelated reflections of multicellular complexity; however, all these factors are differentially represented between simple and complex multicellular organisms, as described below (Table 1). Thus, we here support the hypothesis that transitions to complex multicellularity have required, in addition to the advent of the eukaryotic cell and several other key factors, the development of a pervasive non-protein-coding RNA-based gene regulation. Non-protein-coding RNAs (ncRNAs) and proteins have been used as regulatory molecules in prokaryotes and eukaryotes, and conventional wisdom holds that increased complexity requires more regulators. Nevertheless, we here polarize this major evolutionary transition to the contrary: pervasive non-coding RNA-based regulatory systems are prerequisite to complex multicellularity. We are going to argue (1) why pervasive non-coding RNA-based regulatory systems can only be supported and easily explored in higher eukaryotes, and (2) how a vast expansion of regulatory ncRNAs rather than large numbers of novel protein regulators can easily contribute to the emergence of complex multicelullarity.
Simple multicellular organisms (SMOs) include filaments, balls or sheets of cells that arise either via mitotic division from a single progenitor with the offspring sticking together (aquatic origin) or when several solitary cells aggregate to form a colony (terrestrial origin). They form a coherent and reproducible morphology by cell-cell adhesion, and differentiation of somatic and reproductive cells is common (Bonner 1998; Wolpert and Szathmary 2002; Grosberg and Strathmann 2007). However, complex differentiation patterns and intercellular signaling are limited, so that every cell lies in direct contact with the environment during active metabolism (Knoll 2011). SMOs are found in both multiple eukaryotic lineages (such as chlorophyceae, dictyostelia and oomycetes) and in some eubacterial clades, e.g., cyanobacteria, myxobacteria and actinobacteria (Bonner 1998; Kaiser 2001; Rokas 2008). In fact, the first signs of cell differentiation come from fossils of filamentous and mat-forming cyanobacteria-like organisms that diverged once between 2.4 and 2.1 Ga (Tomitani et al. 2006).
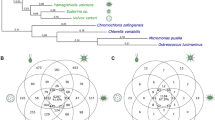
Complex multicellularity, on the other hand, is limited to Eukarya where it arose independently in at least six clades (Fig. 1): once for the eumetazoan animals (King 2004), but multiple times (with possible secondary losses) in embryophytic land plants, florideophyte red algae, stramenopile brown algae (from the order Laminariales), basidiomycete, and ascomycete fungi (Niklas 2000; Medina et al. 2003; Bonner 2004; Grosberg and Strathmann 2007; Rokas 2008; Cock et al. 2010; Knoll 2011). Complex multicellularity arose relatively late in the history of life, probably less than 1000 million years (Ma) ago (Benton and Ayala 2003), and left an extended fossil record during the Ediacaran and Cambrian periods (~600 Ma ago) (Knoll 2011). Complex multicellular organisms (CMOs) show not only evidence of genes involved in cell-cell and cell-matrix adhesion but also a diverse “toolkit” of genes associated with developmental and cell-death programs comprising intercellular signaling, specialization of cell types, and multiple tissue differentiation patterns mediated by complex regulatory networks. This genetic toolkit (including protein-based gene regulation) has been the product of evolutionary innovations, tinkering and expansions of genetic material from ancestral unicellular organisms (King 2004; Bowman and Floyd 2007; King et al. 2008; Rokas 2008; Specht and Bartlett 2009; Cock et al. 2010; Srivastava et al. 2010). Interestingly, the multicellular genetic toolkit corresponds only to a few hundred genes from a few dozen gene families (Bowman and Floyd 2007; Rokas 2008; Erwin 2009; Specht and Bartlett 2009; Knoll 2011) that belong to the 3,000 novel gene families diverged from the last common ancestor of eukaryotes (Koonin et al. 2004).

Multiple origins of CM and C-value and G-value paradoxes among the 100 Eukarya with complete genome sequences. The phylogenetic relationships for higher taxa were obtained from Hedges (2002) and Hampl et al. (2009); main eukaryotic clades with independent origins of CM are underlined and marked with *. Red circles indicate taxa harboring at least one CMO with a complete sequence genome. There is no clear relationship between multicellularity and genome size, although CMOs tend to have larger genomes (black dots). This observation is, however, at least in part biased by the overrepresentation of vertebrates and flowering plants. Comparable proportions of protein-coding genes (bars) are found among unicellular species, SMOs and CMOs. The total number of nuclear protein-coding genes and the non-organellar genome size are depicted in Mega base (Mb) pairs for each genome project (with 7X or greater fold coverage), and were obtained from the reports of their latest publication and updated databases; when necessary, the gene content was corrected by counting the coding sequences for proteins (CDS) from the corresponding annotation of their best gene models
The Eukaryotic Cell as a Source for Complex Multicellularity
Large organismal size, the origin of endosymbiotic energy production, and a “passive” increase of non-protein-coding DNA (ncDNA) in the genome may have predisposed the eukaryotic cell as substrate for complex multicellularity.
Increase of Body Size and Cell Differentiation
Organismal size positively correlates with the number of cell types in CMOs (Valentine et al. 1994; Bonner 1998; Carroll 2001; Bonner 2004; McCarthy and Enquist 2005; DeLong et al. 2010) (Table 1). Once size had increased, the putative advantages of this change would follow (Bonner 1998). Knoll (2011) proposed that large three-dimensional sizes of CMOs could have been enhanced by a positive feedback cycle from the availability of ambient oxygen (pO2). The gradual increase of pO2 in the oceans and atmosphere about 2.5 Ga ago (Holland 2006), and the exposure to pO2 during the transition of life from water to land (Bonner 1998; Hedges et al. 2004; Knoll 2011), would have increased the permissible size of diffusion-limited multicellular organisms. Oxygen may not have started all CMOs, but it would have imposed severe constraints on the evolution of macroscopic organisms with high energy demands. A larger size, in turn, would have allowed larger surface-to-interior gradients of oxygen, nutrients, signaling molecules, bulk transport, as well as the formation of reactive oxygen species (in response to environmental cues) which would have been capable of inducing cell differentiation (Blackstone 2000; Aguirre et al. 2005; Lesser 2006; Knoll 2011). Environmental cues selecting cell differentiation more than just larger organismal size should have been crucial for the transition to CMOs, given that the presence of developmentally differentiated cell types in a colony (or in an organism) is what makes it truly multicellular (Wolpert and Szathmary 2002).
The Acquisition of an Endosymbiotic Energy Production
In general terms, a cell type is a cell with a discrete pattern of gene expression driving a distinct morphological or functional cellular shape in comparison to other cell types with the same organismal genotype. Gene expression of cell types not only needs the involvement of signal(s) to start and maintain differentiation and a considerable diversity of regulatory elements to control expression, but it also needs a powerful source of energy to express both conserved and novel genes in a combinatorial manner. Thus, the massive difference in mean genome size between prokaryotes and eukaryotes is most revealing in terms of the energy available to transcribe and translate their genes (Lane and Martin 2010). Whereas the energetic cost of possessing genes is trivial (~2%), the cost of expressing them as RNA transcripts and proteins is not: protein synthesis consumes most (~75%) of the cell's total energy budget (Wagner 2005; Lane and Martin 2010). For example, if a bacterial genome is increased tenfold in size, it could still be replicated, but there is no current known mechanism (such as the number of regulatory proteins or ribosomes, carbon metabolism, respiratory chain or giant polyploids) that can circumvent the energetic barrier to express ten times as many proteins (Lane and Martin 2010). The origin of eukaryotes, however, entails a bioenergetic innovation that is key to sustain multicellular life. Lane and Martin (2010) elegantly argue that the endosymbiosis that gave rise to mitochondria restructured the distribution of DNA in relation to bioenergetic membranes. By enabling oxidative phosphorylation across a wide area of internal membranes, mitochondrial genes permitted a remarkable 200,000-fold expansion of genome size compared to bacteria. Thereby, mitochondrial power expanded the genotype that an eukaryotic cell could express, inherit, and evolve by four to six orders of magnitude, affording the cell the possibility (but not the necessity) of becoming complex.
A ncRNA-Based Regulatory Network is Hidden Within ncDNA
Large-scale tandem and block duplications have been extensively reported among eukaryotes, hinting at polyploidy in their ancestry (Gregory 2005). Furthermore, fundamental changes in gene structure, such as the advent of introns, allowed the expansion of the eukaryotic proteome by alternative splicing (Nilsen and Graveley 2010). In contrast to prokaryotes (Konstantinidis and Tiedje 2004), however, there is no clear relationship between eukaryotic complexity and either genome size (C-value) or the number of protein-coding genes (G-value) (Fig. 1). In higher eukaryotes, indeed, the vast majority of nuclear DNA is non-protein-coding (ncDNA) (Lynch and Conery 2003; Mattick et al. 2007; Lynch et al. 2011). Intragenic ncDNA is either intronic sequence or untranslated region (UTR), whereas intergenic ncDNA is composed (in different proportions across species) of repetitive transposable elements from class I (e.g., LINEs, SINEs and LTRs) and class II (e.g., MITEs), simple sequence repeats as well as segmental and pseudogene duplications (Gregory 2005; Lynch et al. 2011). The progressive expansion of ncDNA in higher eukaryotic organisms is thought to be a consequence of the reduced efficiency of selection acting against the passive accumulation of “mutationally hazardous” DNA in taxa experiencing elevated magnitudes of random genetic drift (Lynch et al. 2011). This effect may be explained because of reduced effective population sizes (invertebrates and vascular plants ~105–106, and vertebrates ~104–105; whilst prokaryotes ~108, and unicellular eukaryotes and fungi ~107) (Lynch and Conery 2003; Wagner 2005), reduced recombination in large genomes, and a mutational bias toward insertions of large segments of DNA (Lynch 2006; Lynch et al. 2011). The extent and ways by which these ncDNA components contribute to the phenotype of eukaryotic species are still being elucidated (Kazazian 2004; Gregory 2005). Nevertheless, some ncDNA components retain several regulatory elements (in particular cis-regulatory ones), and a considerable proportion of them encodes for a huge diversity of ncRNAs (i.e., functional RNA molecules that are not translated into a protein), at least a substantial fraction of which is thought to have regulatory functions.
Certainly, regulatory ncRNAs were first found in prokaryotes and, for instance, they currently represent ~2% of the total number of genes in the unicellular Escherichia coli, which represents almost half of the genes encoding for protein regulators (~5%) (Storz and Waters 2009). There is increasing evidence of (non-coding) antisense transcripts in bacteria for which regulatory functions are at least suspected (Sharma et al. 2010). More than 100 types of regulatory ncRNAs have been identified throughout the bacterial kingdom, and riboswitches are one of the best known examples. A particular class of riboswitches, namely those responding to coenzyme thiamine pyrophosphate, has also been identified in some eukaryotic lineages (Cheah et al. 2007; Bocobza and Aharoni 2008); however, it exhibits an uncertain evolutionary origin in Eukarya (Sudarsan et al. 2003; Bocobza and Aharoni 2008). With the exception of small nucleolar RNAs (snoRNA) present in Archaea and Eukarya, there is no homologous relationship between the regulatory ncRNAs found in prokaryotes and eukaryotes. Furthermore, the physiological role of bacterial regulatory ncRNAs has been evolutionary driven to mediate rapid responses to changing environmental conditions by modulating specific metabolic pathways or stress cues, like pathogenesis and SOS response (Repoila and Darfeuille 2009; Storz and Waters 2009).
ncRNAs Dominate the Genotype and Phenotype of Complex Multicellular Organisms
In contrast to prokaryotes, regulatory ncRNAs are encoded basically everywhere in the eukaryotic genome, and in particular they cover the ncDNA regions which dominate the genotypes of CMOs. Regulatory ncRNAs are transcribed from pseudogenes, they are also produced from protein-coding loci both by alternative splicing and as independent transcripts (e.g., anti-sense RNAs, enhancer RNAs, intronic transcripts, UTR associated RNAs, repetitive elements), and from their own “intergenic” genes (Kim et al. 2009; Ponting et al. 2009; Voinnet 2009; Ren 2010; Cabili et al. 2011). It appears generally accepted by now that eukaryotic ncDNA is pervasively transcribed (Deng et al. 2006; Berretta and Morillon 2009; Kapranov et al. 2010; Clark et al. 2011; Tisseur et al. 2011), but see van Bakel et al. (2010, 2011) for a dissenting opinion. Accordingly, it has been extensively reported that regulatory ncRNAs show a differential, widespread and complex transcription that gives rise to a considerable number of functional ncRNA families (Table 2) (Gingeras et al. 2007; Jacquier 2009).
Different classes of small and large ncRNAs have been reported as regulating a larger number of both species-specific and deeply conserved cellular processes in tissue identity and stem cell self-renewal and differentiation through well defined mechanisms within the major eukaryotic clades (Millar and Waterhouse 2005; Lin and Gangaraju 2009; Ponting et al. 2009; Arendt et al. 2010; Bartel and Nodine 2010) (Fig. 2). Furthermore, ncRNAs have been shown to regulate almost every level of gene expression, including the activation and repression of homeotic genes and the targeting of chromatin-remodeling complexes (i.e., epigenesis) (Mattick et al. 2008; Chu et al. 2011; Pauli et al. 2011). Indeed, recent transcriptome analyses and different experimental approaches in cell development provide strong evidence that perturbations in ncRNA regulation are involved in complex developmental disorders, including cancers and neurological diseases in mammals; for review see Costa (2005); Spector and Prasanth (2007). Nevertheless, the loss of ncRNA function rarely results in a lethal phenotype. One of the counter-examples is the elimination of microRNA-1-2 (a miRNA expressed in skeletal muscle in vertebrates), which results in a lethal phenotype with defects to cardiac morphogenesis, electrical conduction, and cell cycle control in mouse (Zhao et al. 2007). With the exception again in mouse (Bernstein et al. 2003), however, the genetic inactivation of the central ncRNA processing enzyme Dicer in vertebrates does not dramatically affect cell differentiation and gene expression patterning (Cobb et al. 2005; Giraldez et al. 2005; Harfe et al. 2005). All these lines of evidence in conjunction with the evolutionary trends of these regulators (described below) support the hypothesis that regulatory ncRNAs are primarily moderators of complex multicellular phenotypes on Earth.

Regulatory functions of ncRNAs are involved in all the stages of the central dogma of biology. Genetic information flows from DNA (the genotype) to RNA (with thick black lines). RNA (the phenotype) then decodes proteins (from a mRNA) and/or ncRNAs (from a transcript or a mRNA). Regulatory ncRNAs control gene expression (shown in dashed lines), whereas structural ncRNAs (e.g., ribosomal and transfer RNAs) are involved in protein synthesis (shown in thin doubled-line). ncRNA symbology: mi, micro; nc, non-coding; pi, piwi; rasi, repeat-associated small interfering; si, short interfering; sn, small nuclear; sno, small nucleolar; r, ribosomal; t, transfer; SRP, signal recognition particle; TF, transcription factor. Figure is modified with kind permission from Condorelli and Dimmeler (2008)
Why Can ncRNAs Quickly and Selectively Acquire a Regulatory Functionality in Complex Multicellular Organisms?
The observations that i) energy costs constrain the evolution of gene expression in any organism, ii) ncRNAs could be fixed and even conserved on eukaryotic genomes by nearly neutral forces (perhaps without any advantage or disadvantage for the organism in the beginning), and that iii) possible pervasive transcription of new ncRNAs could perturb existing regulatory networks, raise the question how critical changes of gene regulation have arisen during the evolution of complex multicellularity. Wagner (2005) suggested at least three scenarios that may operate at the same time to promote any gene expression change. First, changes in messenger RNA (mRNA) or protein half-life may contribute significantly to reduce the cost of controlling the cellular concentration of a gene product, given that half-lives are energetically less constrained than synthesis rates. Second, a substantial influx of regulatory mutations may balance the total energy consumption by increasing the synthesis of some gene products and decreasing the synthesis of others. Finally, substantial changes in mRNA and protein synthesis rates can only go to fixation in a large population (like in bacteria and unicellular eukaryotes) when they provide an advantage sufficiently large to overcome the cost and thus the effect of selection opposing it. Ohta’s Near Neutral Theory shows, however, that slightly deleterious mutations can be fixed by genetic drift (Ohta 1992). This effect allows to transcripts without an adaptive effect to persist and spread in a population despite its small energetic cost, and to accumulate mutations. The mutation-selection balance is shifted, furthermore, in favour of genetic drift in small populations, as we expect for the higher multicellular organisms. Indeed, apparently non-functional pseudogenes (i.e., degenerate copies of functional protein-coding transcripts) continue to be expressed in measurable numbers from the human genome (Zheng et al. 2007). New potential regulatory changes can then open the possibility (but again not the necessity) to become positively selected by exploring and canalizing functional phenotypic novelties.
ncRNA-based regulatory networks have shown their evolutionary potential to guide new critical phenotypes. First, ncRNAs may allow the cell to exert faster control of gene expression and thus to improve adaptation to environmental conditions in a specific space and time through low energetic costs. A major distinction between protein- and RNA-based gene regulations can be found in their mode of action. Transcription factors (TFs) recognize sequence-specific cis-motifs in the regulatory upstream regions of target genes (i.e., the promoter), and regulate transcription when bound to their promoters. After transcription, in contrast, microRNAs (miRNA) recognize complementary cis-acting sites in the 3' UTR of the mRNA, and ultimately control the protein translation of the mRNA by promoting its degradation once bound to a target site (Millar and Waterhouse 2005) (Table 2). On the basis of common sequence motifs and structural features, snoRNAs bind to a transcript via base complementarity to regulate alternative splicing at sequences subject to RNA editing (Kishore and Stamm 2006). Additionally, a large number of different functions have been found for long ncRNAs (lncRNA) which often do not appear to show dependence on particular secondary structures. The control of mRNA populations by lncRNA includes the organization of nuclear bodies, a direct involvement in epigenetic mechanisms and transcriptional regulation as well as the inhibition of several receptors (Mattick et al. 2009; Spector et al. 2009; Chu et al. 2011; Kim and Sung 2011). Furthermore, snoRNAs and lncRNA can be also processed into small regulatory RNAs similar to miRNAs (Spector et al. 2009; Scott and Ono 2011). Therefore, precision in genic output (as measured by the variation in number and half-life of mRNA molecules) is often achieved by ncRNAs rather than by protein regulators. ncRNAs could confer robustness (i.e., invariance of the resulting phenotype in the face of perturbation) to regulatory networks by preventing unwanted ectopic protein molecules, buffering fluctuations in expression levels or reducing transcriptional noise (Hornstein and Shomron 2006).
Unlike protein-coding sequences, moreover, ncRNAs do not necessarily exhibit a conserved sequence to perform their functions. In most cases, indeed, the biogenesis or function of the RNA molecule is only possible if the molecule folds into a characteristic two- and three-dimensional structure via formation of intra-molecular base pairs into “stems” or “helices” (Fig. 3b). The disruption of these paired regions through mutations in the primary sequence may result in conformational changes of the structure and can compromise the function of the RNA molecule; however, compensatory mutations that replace one type of base pair by another one in the paired regions of the molecule can restore its functional conformation (Fig. 3c). Under the influence of various selection forces, such as purifying (speed up the loss of mutant alleles), stabilizing (stabilize the frequency of an allele in a population), or positive (promote faster fixation of an advantageous alleles in a population), a RNA molecule may accumulate nucleotide double-substitutions (i.e., covariations) to maintain a structural-functional class or may store mutations to explore a new one.

Comparison of the 5′ stems of the regulatory 7SK small nuclear RNAs. a) Phylogenetic distribution of 7SK candidate sequences in Arthropoda. Figure is adapted with kind permission of Gruber et al. (2008). A black dot indicates a match in the genomic sequence; the hexagons refer to partial ESTs. Aligned blocks are shown in black, gray bars indicate gaps in the alignment, and missing sequence data adjacent to EST regions appear white. b) Simplified cartoon model of the functional secondary structure of 7SK RNA (based on Peterlin et al. 2012) showing the position of the M3 stem located at the 5’ stem region. The position of the M3 stem region is shown by a blue block at the top of the alignment in a) highlighting the highly conserved motif GAUC-GAUC. c) Structural models for the M3 stems in several eumetazoan clades. The consensus structural models for Drosophilidae, Neoptera and Arthropoda are based on the alignment in a). Conserved regions are colored in red; two and three compensatory mutations are shown in ochre and green. Lower case letters denote a deletion in some sequences. Helices are highlighted by a gray background. The open rectangle at the M3 stem for Drosophilidae shows the highly conserved motif GAUC-GAUC. Hairpin loops (with variable size and no clear consensus folds) are drawn as dashed ellipses. Figure is reproduced with kind permission of Mosig et al. (2009)
The effect of mutations on structured RNAs is quite well understood. On the one hand, RNAs are robust against mutations in the sense that a large fraction of mutations does not affect the folding (Fig. 3c). These neutral mutants form extended neutral networks in sequence space on which drift leads to a diffusion-like evolutionary dynamics. Another large fraction of mutations leads to dramatic structural changes, so that any frequent structure is realized in the vicinity of any arbitrary chosen sequence, an effect termed shape space covering (Schuster et al. 1994). Taken together, these two competing effects imply that drift leads to a rapid exploration of novel variants at the fringes of the neutral networks (Huynen 1996; Huynen et al. 1996). In small populations, slightly detrimental mutations at ncDNA regions can also be fixed by genetic drift (Ohta 1973), so that the small energy cost of transcribing non-functional ncRNAs does not lead to their rapid elimination from the genome. Furthermore, second order effects, such as an increased probability to produce offspring with a detrimental mutation in a gene that was fitness-neutral in the parent, cannot be selected because drift dominates the very small effective fitness effect. The first order effect of an advantageous mutation is readily selectable, however. Thus, non-functional transcripts can “wait” for rare advantageous mutations to place them under stabilizing selection. The evolutionary dynamics thereby becomes dominated by the accessibility of advantageous mutations (Fontana and Schuster 1998). This structural accessibility of the RNA molecule could explain in part the structural and functional convergence of the regulatory miRNA-like RNA class in plants, animals and fungi, which is in congruency with an independent origin of their multicellular development (Liu et al. 2010; Carrington et al. 2011).
Evolution and Selection of Regulatory ncRNAs in Eukaryotes: The Case of microRNAs
Functional regulatory ncRNAs, even those under strong stabilizing selection, may exhibit rapid evolution at the sequence level. Thus, some subclasses are clearly under purifying selection acting predominantly on the secondary structure in order to keep their regulatory function (Ponting et al. 2007; Pain et al. 2008; Amar et al. 2009; Marques and Ponting 2009; Ponting et al. 2009; Gerstein et al. 2011). Furthermore, the genetic structure involved in the transcription and function of some regulatory ncRNAs, such as introns and splice sites, is also subject of stabilizing selection (Hiller et al. 2009; Rose et al. 2011). Hence several models have been suggested to describe how selection may be driving the birth-and-death evolutionary dynamics of some ncRNAs (Hornstein and Shomron 2006; Rajewsky and Chen 2007; Lai et al. 2008; Peterson et al. 2009; Carrington et al. 2011).
In particular, comparative analyses of miRNA homologs in diverse animal and plant species have revealed both long-term maintenance and taxa-specific occurrence of miRNA families (Carrington et al. 2007; Donoghue et al. 2008; Wang et al. 2008; Carrington et al. 2011). In animals, it has been proposed that new miRNAs could evolve by point mutations from existing hairpin structures in the genome that are transcribed at low levels and in specific cell types. This process is followed by selection against inadequate miRNA/mRNA pairing and expression modifications, and simultaneously is preserving many of the neutral or advantageous targets that increase the expression of the miRNA (without being highly deleterious to the organism) (Rajewsky and Chen 2007; Lai et al. 2008; Peterson et al. 2009; Carrington et al. 2011). Thus, animal miRNAs and fungal miRNA-like RNAs (milRNAs) have exploited the robustness of the hairpin structure both to maintain the regulatory function and to diversify their activity by targeting imperfectly complementary sequences (Lai et al. 2008; Liu et al. 2010). Because miRNA/target pairing is almost perfectly complementary and extends over twice as many nucleotides in plants, when compared to animals and fungi, the evolution of new plant miRNAs has been proposed to be driven by inverted duplication of target gene sequences (i.e., pre-miRNA-encoding regions) (Carrington et al. 2004). Thus, newly formed expressed RNA hairpins may experience successive selective sweeps until they reach an evolutionarily optimized form, allowing a one-to-one miRNA/target relationship to be established, and then be incorporated into new or existing regulatory networks (de Meaux et al. 2008; Carrington et al. 2011).
Both models have been supported by experiments showing that unlike highly conserved-ancient plant and animal miRNAs, evolutionary young miRNAs are typically expressed at low levels, processed imprecisely, lack targets, and display patterns of neutral variation (Rajewsky and Chen 2007; de Meaux et al. 2008; Lu et al. 2008; Carrington et al. 2011). These trends suggest that young MIRNA loci tend to evolve neutrally, possibly helping to maintain negative pleiotropic effects at low levels, until compensatory and advantageous mutations have emerged (Rajewsky and Chen 2007; Carrington et al. 2011). Exceptional evidence for evolutionary optimization of young pre-miRNA stem-loop structures has been recently presented for two miRNA-encoding loci in Arabidopsis thaliana (de Meaux et al. 2008) as well as for five miRNA-encoding loci in Drosophila melanogaster (Lu et al. 2008). In both works, the authors demonstrated that pervasive variation occurs at miRNA-encoding loci, and that the structural variation among alleles suggests non-random evolution of a thermoresistant substructure in the miRNA precursor, which presumably impacts the processing of the mature form. In A. thaliana, for instance, miR856 shows a weak signature of a selective sweep and miR824 displays signs of balancing selection; whereas the polymorphism pattern of miR310/311/312/313 in D. melanogaster is indicative of hitchhiking under positive selection.
Detailed knowledge of evolutionary trends of regulatory non-coding transcripts is currently restricted to the much better understood microRNAs. Even though some genome projects have estimated the proportion and conservation of some ncRNA families, their extension and evolutionary patterns on a large phylogenetic scale are still being determined. Part of the methodological problem lies in the characterization of ncRNA evolution by large inhomogeneous variations in both sequence and structure (Mosig et al. 2009) (Fig. 3a and c), which hampers their discovery and functional description. The assessment of a large compendium of ncRNAs in basal eukaryotes would help us to understand the origin and evolution of ncRNA-based gene regulation in Eukarya. Interestingly, both genomic and functional similarities among regulatory ncRNAs might be the result of convergent evolution due to duplication and transposition mechanisms, similarly processing enzymes, and comparable selective pressures across species, but it might also reflect either the divergent evolution from a common RNA ancestor or the transition from one ncRNA type to another (Scott and Ono 2011).
Why and How Can a Vast Expansion of RNA Regulators Contribute to the Emergence of Complex Multicellularity Rather than Large Numbers of Novel Protein Regulators?
Many families of regulatory proteins that contribute to the complexity of developmental programs have been conserved, expanded and innovated throughout the evolution of eukaryotes. They are, however, under more stringent constraints than regulatory ncRNAs. Novel transcription factors, for instance, require the independent acquisition of DNA binding sites in all target promoters, while additional paralogs are forced to rapidly diverge from an existing regulatory module to avoid deleterious effects at the time of acquiring a new target or domain of expression (e.g., in a new tissue) (Rajewsky and Chen 2007). Furthermore, for a new protein to come under positive selection, the mRNA must be transcribed (easy with pervasive transcription) and the messenger must be translated (maybe also easy, but protein synthesis is energy intensive). Hence a neutral coding gene should be selected against more strongly than a neutral non-translated transcript. Indeed, protein-coding genes can lose their coding capacity and hang around long enough as pseudogenes to become regulatory ncRNAs (Yano et al. 2004; Guo et al. 2009). One of the best documented examples is the regulatory ncRNA Xist, which is exclusively present in eutherian mammals and its regulatory role is involved in the initiation process of X chromosome inactivation (Table 2). Xist evolved by pseudogenization of the protein-coding gene Lnx3, which is conserved in all vertebrate classes, except Eutheria. Both Xist and Lnx3 are thus present in a mutually exclusive manner at a syntenic locus in all vertebrates (Duret et al. 2006).
Conversely, multiple aspects of ncRNA function and evolution, such as a) leaky repression of transcripts to allow “fine-tuning” of gene expression, b) reduction of phenotypic variation by buffering genetic noise or avoiding unwanted ectopic molecules, c) a presumably regulatory increase in lineages of CMOs over geologic time, and d) the rarely dramatic impact in a phenotype due to loss of function, suggest that regulatory ncRNAs act as key players in canalizing genetic programs (Hornstein and Shomron 2006; Peterson et al. 2009). In this context, canalization can be visualized as the process of formation of virtual phenotypic (relatively) invariant canals in which developmental programs flow (Hornstein and Shomron 2006). If the main function of ncRNA regulation is to stabilize gene expression levels, then almost paradoxically, ncRNAs may increase the heritability of a phenotypic trait through natural selection by decreasing its expression variability about the mean, i.e., making robust the phenotype (a result) by canalizing the genetic program (a cause). Hence, regulatory ncRNAs could be the evolutionary innovatory instrument to canalize development such that phenotypic variation decreases over geologic time at the cost of increasing developmental precision and consequently enhancing the morphological complexity (Peterson et al. 2009).
Here, we suggest a model that explains why novel RNA regulators with advantageous functions are readily accessible in multicellular organisms. Consider a gene that is expressed at some homogeneous level x in an organism. The expression level x will be selected to optimize the organism’s fitness. Now suppose that the organism has two cell types. Figure 4 shows the fitness landscape as function of the expression levels in the two cell types f(x 1 , x 2 ). As long as the regulation of the gene is identical in both cell types, only the points on the median line (x 1 = x 2 ) are accessible. If a regulatory ncRNA is innovated in one of the two cell types (regulator + in the Fig. 4), then it can modify the expression levels of the gene. Evolution can now adjust the organism's fitness to optimize f(x 1 , x 2 ) without the constraint that x 1 = x 2 . Mathematically, we know that max f(x 1 , x 2 ) ≥ max f(x, x) simply because we now optimize over a larger domain. In general, the optimum for f(x 1 , x 2 ) will be reached for some point x 1 ≠ x 2 so that the fitness difference f(x 1 , x 2 ) – max f(x, x) will generically be strictly positive for a non-empty set A of expression values x 1 and x 2 . If the new regulatory ncRNA takes the system into this set A with a small energetic cost (S cost ), then it has a “window of opportunity” to be placed under stabilizing selection. Thus, this new regulatory ncRNA could make the phenotypic traits influenced by the regulated gene much more “evolvable”. If the organism’s fitness depends on the heritability of those phenotypic traits and, accordingly, also on the expression levels of that gene, then the new regulatory ncRNA becomes subject to positive selection (S*) to optimize f(x 1 ,x 2 ) (relationship shown in Fig. 4 as S cost + S*). If the fitness depends continuously on the expression levels, moreover, set A touches the maximal value on the median line and hence is reachable from (x, x) by either a relative increase or a relative decrease of x 1 over x 2 . In summary, for every novel RNA regulatory molecule, there is a “window of opportunity” for being placed under stabilizing selection if it can be used to disentangle expression patterns of other functionally relevant genes in different cell types. We argue that this mechanism makes advantageous innovations easily accessible in differentiated multicellular organisms. The resulting refinement in regulation in turn could lead to the innovation of further cell types, opening the same “window of opportunity” again for the incorporation of further regulators. Since ncRNAs are much easier to generate from genomic DNA and energetically cheaper to produce than functional proteins in eukaryotes, we conclude that complex multicellular organisms should be able to rapidly accumulate ncRNA regulators –as we observe indeed in evolutionary history.

Model for the acquisition of ncRNA regulators in CMOs. The figure shows the fitness landscape of gene a as a function of its expression levels in two cell types f(x 1 ,x 2 ). As long as the regulation of the gene a is identical in both cell types, only the points on the median line (x 1 = x 2 ) are accessible. If a regulatory ncRNA is innovated (regulator +) in one of the cell types, then it can modify the expression levels of gene a in that compartment. If the new regulatory ncRNA takes the system into a new set A of expression values with a small energetic cost (Scost), then it has a “window of opportunity” to be placed under stabilizing selection. Thus, this new regulatory ncRNA could make the phenotypic traits influenced by the regulated gene a much more “evolvable”. If the organism’s fitness depends on the heritability of those traits and, accordingly, on the expression levels of gene a, then the new regulatory ncRNA becomes subject to positive selection (S*) to optimize f(x 1 ,x 2 ). The resulting refinement in regulation in turn could lead to the innovation of further cell types, opening the same “window of opportunity” again for the incorporation of further regulators. See text for a detailed description
This avenue to increasing cellular complexity is much less attractive in single-cell organisms, and in particular prokaryotes with their small size, in which gene products are distributed much more homogeneously. The mechanism of Fig. 4 critically depends on the availability of at least two cell types with the same genetic background and the same ancestral pattern of gene expression from the parental cell, a situation simply not available either in unicellular organisms or in SMOs organized in colonies during certain life stages. Solitary cells thus do not generically provide an opportunity for the invasion of novel regulators, at least not when they are already well-adapted to their environment. The smaller size and hence the expected larger effective population size, in concert with strong dependence of genome size on energy constraints, also implies a much larger selective pressure against non-functional transcripts. Accordingly, prokaryotes should also have a much smaller repertoire of nearly neutral transcripts that persists for much shorter time scales, and thus have a much smaller chance to stumble across a functional improvement or innovation. All these constraints have placed protein regulators, instead of ncRNAs, to “easily” fulfill the selective advantages to dominate the regulation of phenotypes in prokaryotes. Global protein regulators, acting in conjunction with cis-regulatory elements, protein and RNA co-regulators, and specific environmental signals, control the morphogenesis and metabolic complexity of bacterial species (Martinez-Antonio and Collado-Vides 2003; Lozada-Chavez et al. 2008). Accordingly, bacterial protein regulators are highly flexible in switching their mode of action between global and local targeting and have a propensity to spawn homologs with similar function at short evolutionary time scales (Babu et al. 2006; Lozada-Chavez et al. 2006, 2008). Furthermore, the total number of transcription factors positively correlates with genome size and life style (Cases et al. 2003; Babu et al. 2006). Therefore, each bacterial species has evolved its own set of transcription factors, suggesting that the emergence of distinct repertoires of protein regulators is a crucial step for the adaptation to new environments and to control the bacterial phenotypic variance.
Role of RNA Throughout Evolution of Life on Earth
RNA is one of the three major biopolymers that manages the fluxes of biological information of life on Earth. The “RNA world” hypothesis proposes that the biochemical, structural, and catalytic properties of RNA once were sufficient to encode the genotype, to decode the phenotype, and to drive evolution in the early state of life on Earth. As a corollary, RNA has been replaced to a large extent by DNA and proteins on the way to contemporary life. Its capabilities, however, have been preserved in the core of the most fundamental and highly conserved biological processes across the three domains of life (Joyce 2002). Furthermore, the pervasive regulatory role of RNA in the three domains of life, particularly in eukaryotes, has been recently recognized (Fig. 2).
We suggest here that non-coding RNA-based genetic regulation is a prerequisite for the emergence of multicellular complexity. The evolution of complex multicellularity is based upon the successive innovation and refinement of cell and tissue types, requiring a flexible layer of compartment-specific gene regulation. We have argued here that novel regulators that refine gene expression patterns are easily selected for, and that non-coding transcripts are entirely capable of fulfilling such roles at all levels of cellular regulation. Since they are less costly than peptides, non-functional transcripts are available as evolutionary raw material for much longer time spans, and preferably in smaller populations where genetic drift is stronger. Thus, non-coding transcripts have much more time and much more wide-spread opportunities to find an adaptable role. Functional ncRNAs, therefore, are integrated into the genomes of complex multicellular organisms at a faster pace than paralogous proteins. Consequently, regulatory ncRNAs become the driving force behind increasingly complex modes of regulation. Accordingly, it would not be hard to expect that the total number of regulatory ncRNAs could be higher than the total number of protein regulators in CMOs. If so, regulatory ncRNAs could substantially contribute to the theorical and practical definition of simple and complex multicellularity. The origin of RNA on early Earth is still controversial (Robertson and Joyce 2010). Nevertheless, it is becoming clear that once RNA emerged into a protocellular system, its relevance within the central dogma of biology has been greater than we have thought.
References
Aguirre J, Rios-Momberg M, Hewitt D et al (2005) Reactive oxygen species and development in microbial eukaryotes. Trends Microbiol 13:111–118
Amar L, Chen CL, Zhou H et al (2009) Genome-wide evolutionary analysis of the noncoding RNA genes and noncoding DNA of Paramecium tetraurelia. RNA 15:503–514
Arendt D, Christodoulou F, Raible F et al (2010) Ancient animal microRNAs and the evolution of tissue identity. Nature 463:1084–U1105
Babu MM, Teichmann SA, Aravind L (2006) Evolutionary dynamics of prokaryotic transcriptional regulatory networks. J Mol Biol 358:614–633
Banfield W, Woke PA, MacKay CM, Cooper HL (1965) Mosquito transmission of a reticulum cell sarcoma of hamsters. Science 148:1239–1240
Bartel DP, Nodine MD (2010) MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Genes Dev 24:2678–2692
Bell G, Mooers AO (1997) Size and complexity among multicellular organisms. Biol J Linn Soc 60:345–363
Benton MJ, Ayala FJ (2003) Dating the tree of life. Science 300:1698–1700
Bernstein E, Kim SY, Carmell MA et al (2003) Dicer is essential for mouse development. Nat Genet 35:215–217
Berretta J, Morillon A (2009) Pervasive transcription constitutes a new level of eukaryotic genome regulation. Embo Reports 10:973–982
Bistis GN, Perkins David D, Read Nick D (2003) Different cell types in Neurospora crassa. Fungal Genetics Newsletter 50:17–19
Blackstone NW (2000) Redox control and the evolution of multicellularity. Bioessays 22:947–953
Bocobza SE, Aharoni A (2008) Switching the light on plant riboswitches. Trends Plant Sci 13:526–533
Bonner JT (1998) The origins of multicellularity. Integr Biol 1:28–36
Bonner JT (2004) Perspective: the size-complexity rule. Evolution 58:1883–1890
Bowman JL, Floyd SK (2007) The ancestral developmental tool kit of land plants. Int J Plant Sci 168:1–35
Cabili MN, Trapnell C, Goff L et al (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25:1915–1927
Carrington JC, Allen E, Xie ZX et al (2004) Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat Genet 36:1282–1290
Carrington JC, Fahlgren N, Howell MD et al (2007) High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One 2:e219
Carrington JC, Cuperus JT, Fahlgren N (2011) Evolution and functional diversification of MIRNA genes. Plant Cell 23:431–442
Carroll SB (2001) Chance and necessity: the evolution of morphological complexity and diversity. Nature 409:1102–1109
Cases I, de Lorenzo V, Ouzounis CA (2003) Transcription regulation and environmental adaptation in bacteria. Trends Microbiol 11:248–253
Cheah MT, Wachter A, Sudarsan N et al (2007) Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature 447:497–500
Chu C, Qu K, Zhong FL et al (2011) Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Molecular Cell 44:667–678
Clark MB, Amaral PP, Schlesinger FJ et al (2011) The reality of pervasive transcription. Plos Biology 9:e1000625
Cobb BS, Nesterova TB, Thompson E et al (2005) T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med 201:1367–1373
Cock JM, Sterck L, Rouze P et al (2010) The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature 465:617–621
Condorelli G, Dimmeler S (2008) MicroRNAs: components of an integrated system controlling cardiac development, physiology, and disease pathogenesis. Cardiovasc Res 79:551–552
Costa FF (2005) Non-coding RNAs: new players in eukaryotic biology. Gene 357:83–94
de Meaux J, Hu JY, Tartler U et al (2008) Structurally different alleles of the ath-MIR824 microRNA precursor are maintained at high frequency in Arabidopsis thaliana. Proc Natl Acad Sci U S A 105:8994–8999
DeLong JP, Okie JG, Moses ME et al (2010) Shifts in metabolic scaling, production, and efficiency across major evolutionary transitions of life. Proc Natl Acad Sci U S A 107:12941–12945
Deng XW, Li L, Wang XF et al (2006) Genome-wide transcription analyses in rice using tiling microarrays. Nat Genet 38:124–129
Donoghue PCJ, Heimberg AM, Sempere LF et al (2008) MicroRNAs and the advent of vertebrate morphological complexity. Proc Natl Acad Sci U S A 105:2946–2950
Duret L, Chureau C, Samain S et al (2006) The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science 312:1653–1655
Erwin DH (2009) Early origin of the bilaterian developmental toolkit. Phil Trans Roy Soc B Biol Sci 364:2253–2261
Fontana W, Schuster P (1998) Continuity in evolution: on the nature of transitions. Science 280:1451–1455
Gerstein MB, Mu XJ, Lu ZJ et al (2011) Analysis of genomic variation in non-coding elements using population-scale sequencing data from the 1000 Genomes Project. Nucleic Acids Res 39:7058–7076
Gingeras TR, Kapranov P, Cheng J et al (2007) RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316:1484–1488
Giraldez AJ, Cinalli RM, Glasner ME et al (2005) MicroRNAs regulate brain morphogenesis in zebrafish. Science 308:833–838
Gregory TR (2005) Synergy between sequence and size in large-scale genomics. Nat Rev Genet 6:699–708
Grosberg RK, Strathmann RR (2007) The evolution of multicellularity: a minor major transition? Annu Rev Ecol Evol Syst 38:621–654
Gruber AR, Kilgus C, Mosig A et al (2008) Arthropod 7SK RNA. Mol Biol Evol 25:1923–1930
Guo XY, Zhang ZL, Gerstein MB et al (2009) Small RNAs originated from pseudogenes: cis- or trans-Acting? Plos Comput Biol 5:e1000449
Hampl V, Hug L, Leigh JW et al (2009) Phylogenomic analyses support the monophyly of Excavata and resolve relationships among eukaryotic "supergroups". Proc Natl Acad Sci U S A 106:3859–3864
Harfe BD, McManus MT, Mansfield JH et al (2005) The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc Natl Acad Sci U S A 102:10898–10903
Hedges SB (2002) The origin and evolution of model organisms. Nat Rev Genet 3:838–849
Hedges SB, Blair JE, Venturi ML et al (2004) A molecular timescale of eukaryote evolution and the rise of complex multicellular life. BMC Evol Biol 4:2
Hiller M, Findeiss S, Lein S et al (2009) Conserved introns reveal novel transcripts in Drosophila melanogaster. Genome Res 19:1289–1300
Holland HD (2006) The oxygenation of the atmosphere and oceans. Phil Trans Roy Soc B Biol Sci 361:903–915
Hornstein E, Shomron N (2006) Canalization of development by microRNAs. Nat Genet 38:S20–S24
Huynen MA (1996) Exploring phenotype space through neutral evolution. J Mol Evol 43:165–169
Huynen MA, Stadler PF, Fontana W (1996) Smoothness within ruggedness: the role of neutrality in adaptation. Proc Natl Acad Sci U S A 93:397–401
Jacquier A (2009) The complex eukaryotic transcriptome: unexpected pervasive transcription and novel small RNAs. Nat Rev Genet 10:833–844
Joyce GF (2002) The antiquity of RNA-based evolution. Nature 418:214–221
Kaiser D (2001) Building a multicellular organism. Annu Rev Genet 35:103–123
Kapranov P, St Laurent G, Raz T et al (2010) The majority of total nuclear-encoded non-ribosomal RNA in a human cell is 'dark matter' un-annotated RNA. BMC Biol 8:149
Kazazian HH (2004) Mobile elements: drivers of genome evolution. Science 303:1626–1632
Kim, ED and Sung, S (2011) Long noncoding RNA: unveiling hidden layer of gene regulatory networks. Trends Plant Sci (in press)
Kim VN, Han J, Siomi MC (2009) Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10:126–139
King N (2004) The unicellular ancestry of animal development. Dev Cell 7:313–325
King N, Westbrook MJ, Young SL et al (2008) The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature 451:783–788
Kishore S, Stamm S (2006) Regulation of alternative splicing by snoRNAs. Cold Spring Harb Symp Quant Biol 71:329–334
Knoll AH (2011) The multiple origins of complex multicellularity. Annu Rev Earth Planet Sci 39:217–239
Kolter R, Branda SS, Gonzalez-Pastor JE et al (2001) Fruiting body formation by Bacillus subtilis. Proc Natl Acad Sci U S A 98:11621–11626
Kong FX, Yang Z, Yang Z et al (2009) Benefits and costs of the grazer-induced colony formation in Microcystis aeruginosa. Ann Limnol-Int J Lim 45:203–208
Konstantinidis KT, Tiedje JM (2004) Trends between gene content and genome size in prokaryotic species with larger genomes. Proc Natl Acad Sci U S A 101:3160–3165
Koonin EV, Fedorova ND, Jackson JD et al (2004) A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol 5:R7
Lai EC, Liu N, Okamura K et al (2008) The evolution and functional diversification of animal microRNA genes. Cell Res 18:985–996
Lane N, Martin W (2010) The energetics of genome complexity. Nature 467:929–934
Lesser MP (2006) Oxidative stress in marine environments: biochemistry and physiological ecology. Annu Rev Physiol 68:253–278
Lin HF, Gangaraju VK (2009) MicroRNAs: key regulators of stem cells. Nat Rev Mol Cell Biol 10:116–125
Liu Y, Lee HC, Li LD et al (2010) Diverse pathways generate microRNA-like RNAs and dicer-independent small interfering RNAs in fungi. Molecular Cell 38:803–814
Lozada-Chavez I, Janga SC, Collado-Vides J (2006) Bacterial regulatory networks are extremely flexible in evolution. Nucleic Acids Res 34:3434–3445
Lozada-Chavez I, Angarica VE, Collado-Vides J et al (2008) The role of DNA-binding specificity in the evolution of bacterial regulatory networks. J Mol Biol 379:627–643
Lu J, Fu YG, Kumar S et al (2008) Adaptive evolution of newly emerged micro-RNA genes in Drosophila. Mol Biol Evol 25:929–938
Lurling M, Van Donk E (1999) Grazer-induced colony formation in Scenedesmus acutus (Chlorophyceae): ecomorph expression at different temperatures. J Phycol 35:1120–1126
Lynch M (2006) The origins of eukaryotic gene structure. Mol Biol Evol 23:450–468
Lynch M, Conery JS (2003) The origins of genome complexity. Science 302:1401–1404
Lynch M, Bobay LM, Catania F et al (2011) The repatterning of eukaryotic genomes by random genetic drift. Annu Rev Genomics Hum Genet 12:347–366
Marques AC, Ponting CP (2009) Catalogues of mammalian long noncoding RNAs: modest conservation and incompleteness. Genome Biol 10:R124
Martinez-Antonio A, Collado-Vides J (2003) Identifying global regulators in transcriptional regulatory networks in bacteria. Curr Opin Microbiol 6:482–489
Mattick JS, Taft RJ, Pheasant M (2007) The relationship between non-protein-coding DNA and eukaryotic complexity. Bioessays 29:288–299
Mattick JS, Amaral PP, Dinger ME et al (2008) The eukaryotic genome as an RNA machine. Science 319:1787–1789
Mattick JS, Mercer TR, Dinger ME (2009) Long non-coding RNAs: insights into functions. Nat Rev Genet 10:155–159
McCarthy MC, Enquist BJ (2005) Organismal size, metabolism and the evolution of complexity in metazoans. Evol Ecol Res 7:681–696
Medina M, Collins AG, Taylor JW, Valentine JW, Lipps JH, Amaral-Zettler L, Sogin ML (2003) Phylogeny of Opisthokonta and the evolution of multicellularity and complexity in Fungi and Metazoa. Int J Astrobiol 2:203–211
Millar AA, Waterhouse PM (2005) Plant and animal microRNAs: similarities and differences. Funct Integr Genomics 5:129–135
Mosig A, Zhu L, Stadler PF (2009) Customized strategies for discovering distant ncRNA homologs. Brief Funct Genomic Proteomic 8:451–460
Niklas KJ (2000) The evolution of plant body plans - A biomechanical perspective. Ann Bot 85:411–438
Nilsen TW, Graveley BR (2010) Expansion of the eukaryotic proteome by alternative splicing. Nature 463:457–463
Ochman H, Davalos LM (2006) The nature and dynamics of bacterial genomes. Science 311:1730–1733
Ohta T (1973) Slightly deleterious mutant substitutions in evolution. Nature 246:96–98
Ohta T (1992) The nearly neutral theory of molecular evolution. Annu Rev Ecol Syst 23:263–286
Pain A, Mourier T, Carret C et al (2008) Genome-wide discovery and verification of novel structured RNAs in Plasmodium falciparum. Genome Res 18:281–292
Pauli A, Rinn JL, Schier AF (2011) Non-coding RNAs as regulators of embryogenesis. Nat Rev Genet 12:136–149
Pearse AM, Swift K (2006) Transmission of devil facial-tumour disease - An uncanny similarity in the karyotype of these malignant tumours means that they could be infective. Nature 439:549–549
Peterlin BM, Brogie JE, Price DH (2012) 7SK snRNA: a noncoding RNA that plays a major role in regulating eukaryotic transcription. Wiley Interdiscip Rev RNA 3:92–103
Peterson KJ, Dietrich MR, McPeek MA (2009) MicroRNAs and metazoan macroevolution: insights into canalization, complexity, and the Cambrian explosion. Bioessays 31:736–747
Ponting CP, Ponjavic J, Lunter G (2007) Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res 17:556–565
Ponting CP, Oliver PL, Reik W (2009) Evolution and functions of long noncoding RNAs. Cell 136:629–641
Rajewsky N, Chen K (2007) The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet 8:93–103
Ren B (2010) Enhancers make non-coding RNA. Nature 465:173–174
Repoila F, Darfeuille F (2009) Small regulatory non-coding RNAs in bacteria: physiology and mechanistic aspects. Biology of the Cell 101:117–131
Robertson MP, Joyce GF (2010) The origins of the RNA world. Cold Spring Harb Perspect Biol. doi:10.1101/cshperspect.a003608
Rokas A (2008) The origins of multicellularity and the early history of the genetic toolkit for animal development. Annu Rev Genet 42:235–251
Rose D, Hiller M, Schutt K et al (2011) Computational discovery of human coding and non-coding transcripts with conserved splice sites. Bioinformatics 27:1894–1900
Roush S, Slack FJ (2008) The let-7 family of microRNAs. Trends Cell Biol 18:505–516
Schuster P, Fontana W, Stadler PF et al (1994) From sequences to shapes and back: a case study in RNA secondary structures. Proc Biol Sci 255:279–284
Scott MS, Ono M (2011) From snoRNA to miRNA: dual function regulatory non-coding RNAs. Biochimie 93:1987–1992
Sharma CM, Hoffmann S, Darfeuille F et al (2010) The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464:250–255
Specht CD, Bartlett ME (2009) Flower evolution: the origin and subsequent diversification of the angiosperm flower. Annu Rev Ecol Evol Syst 40:217–243
Spector DL, Prasanth KV (2007) Eukaryotic regulatory RNAs: an answer to the 'genome complexity' conundrum. Genes Dev 21:11–42
Spector DL, Wilusz JE, Sunwoo H (2009) Long noncoding RNAs: functional surprises from the RNA world. Genes Dev 23:1494–1504
Srivastava M, Simakov O, Chapman J et al (2010) The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 466:720–U723
Storz G, Waters LS (2009) Regulatory RNAs in Bacteria. Cell 136:615–628
Strathmann R (1991) From metazoan to protist via competition among cell lineages. Evol Theor 10:67–70
Sudarsan N, Barrick JE, Breaker RR (2003) Metabolite-binding RNA domains are present in the genes of eukaryotes. RNA 9:644–647
Tisseur M, Kwapisz M, Morillon A (2011) Pervasive transcription - Lessons from yeast. Biochimie 93:1889–1896
Tomitani A, Knoll AH, Cavanaugh CM et al (2006) The evolutionary diversification of cyanobacteria: molecular-phylogenetic and paleontological perspectives. Proc Natl Acad Sci U S A 103:5442–5447
Valentine JW, Collins AG, Meyer CP (1994) Morphological complexity increase in metazoans. Paleobiology 20:131–142
van Bakel H, Nislow C, Blencowe BJ et al (2010) Most "Dark Matter'' transcripts are associated with known genes. Plos Biology 8:e1000371
van Bakel H, Nislow C, Blencowe BJ et al (2011) Response to "The reality of pervasive transcription". Plos Biology 9:e1001102
Velicer GJ, Kroos L, Lenski RE (1998) Loss of social behaviors by Myxococcus xanthus during evolution in an unstructured habitat. Proc Natl Acad Sci U S A 95:12376–12380
Voinnet O (2009) Origin, biogenesis, and activity of plant MicroRNAs. Cell 136:669–687
Wagner A (2005) Energy constraints on the evolution of gene expression. Mol Biol Evol 22:1365–1374
Wang SM, Lu J, Shen Y et al (2008) The birth and death of microRNA genes in Drosophila. Nat Genet 40:351–355
Weiss RA, Murgia C, Pritchard JK et al (2006) Clonal origin and evolution of a transmissible cancer. Cell 126:477–487
Wolpert L, Szathmary E (2002) Multicellularity: evolution and the egg. Nature 420:745–745
Yano Y, Saito R, Yoshida N et al (2004) A new role for expressed pseudogenes as ncRNA: regulation of mRNA stability of its homologous coding gene. J Mol Med-Jmm 82:414–422
Zhao Y, Ransom JF, Li A et al (2007) Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 129:303–317
Zheng DY, Frankish A, Baertsch R et al (2007) Pseudogenes in the ENCODE regions: consensus annotation, analysis of transcription, and evolution. Genome Res 17:839–851
Acknowledgements
IL-C is funded by the fellowship 185993 from the National Council of Science and Technology of Mexico. We thank Christian Arnold and an anonymous referee for valuable comments and suggestions. We dedicate this article to the memory of Lynn Margulis, whose work has allowed us to go forward on the understanding of the origin and evolution of complex life on Earth.
Author information
Authors and Affiliations
Corresponding author
Additional information
"Dedicated to Lynn Margulis, 1938-2011"
Rights and permissions
About this article
Cite this article
Lozada-Chávez, I., Stadler, P.F. & Prohaska, S.J. “Hypothesis for the Modern RNA World”: A pervasive Non-coding RNA-Based Genetic Regulation is a Prerequisite for the Emergence of Multicellular Complexity. Orig Life Evol Biosph 41, 587–607 (2011). https://doi.org/10.1007/s11084-011-9262-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11084-011-9262-1