
Abstract
Purpose
To elucidate the genetic cause of intellectual deficiency and/or congenital malformations in two parental reciprocal translocation carriers and provide appropriate strategies of assisted reproductive therapy (ART).
Materials and methods
Two similar couples having a child with global developmental delay/intellectual disability symptoms attended the Reproductive and Genetic Hospital of CITIC-Xiangya (Changsha, China) in 2017 and 2019, respectively, in order to determine the cause(s) of the conditions affecting their child and to seek ART to have a healthy baby. Both of the healthy couples were not of consanguineous marriage, denied exposure to toxicants, and had no adverse life history. This study was approved by the Institutional Ethics Committee of the Reproductive & Genetic Hospital of CITIC-Xiangya, and written informed consent was obtained from the parents. Genetic diagnoses were performed by karyotype analysis, breakpoint mapping analysis of chromosomal translocation(s), single-nucleotide polymorphism (SNP) microarray analysis, and whole-exome sequencing (WES) for the two children and different appropriate reproductive strategies were performed in the two families.
Results
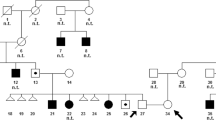
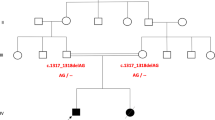
Karyotype analysis revealed that both patients carried parental reciprocal translocations [46,XY,t(7;16)(p13;q24)pat and 46,XY,t(13;17)(q12.3;p11.2)pat, respectively]. Follow-up breakpoint mapping analysis showed no interruption of associated genes, and SNP microarray analysis identified no significant copy number variations (CNVs) in the two patients. Moreover, WES results revealed that patients 1 and 2 harbored candidate compound heterozygous mutations of MCOLN1 [c.195G>C (p.K65N) and c.1061G>A (p.W354*)] and MCPH1 [c.877A>G (p.S293G) and c.1869_1870delAT (p.C624*)], respectively, that were inherited from their parents and not previously reported. Furthermore, the parents of patient 1 obtained 10 embryos during ART cycle, and an embryo of normal karyotype and non-carrier of observed MCOLN1 mutations according to preimplantation genetic testing for structural rearrangement and monogenic defect was successfully transferred, resulting in the birth of a healthy boy. The parents of patient 2 chose to undergo ART with donor sperm to reduce the risk of recurrence.
Conclusions
Systematic genetic diagnosis of two carriers of inherited chromosomal translocations accompanied by clinical phenotypes revealed their cause of disease, which was critical for genetic counseling and further ART for these families.


Similar content being viewed by others

References
Hamerton JL, Canning N, Ray M, Smith S. A cytogenetic survey of 14,069 newborn infants. I. Incidence of chromosome abnormalities. Clin Genet. 1975;8(4):223–43.
Maeda T, Ohno M, Matsunobu A, Yoshihara K, Yabe N. A cytogenetic survey of 14,835 consecutive liveborns. Jinrui Idengaku Zasshi. 1991;36(1):117–29.
Pasinska M, Lazarczyk E, Julga K, Bartnik-Glaska M, Nowakowska B, Haus O. Multiple occurrence of psychomotor retardation and recurrent miscarriages in a family with a submicroscopic reciprocal translocation t(7;17)(p22;p13.2). BMC Med Genet. 2018;11(1):69.
Chen JK, Liu P, Hu LQ, Xie Q, Huang QF, Liu HL. A foetus with 18p11.32-q21.2 duplication and Xp22.33-p11.1 deletion derived from a maternal reciprocal translocation t(X;18)(q13;q21.3). Mol Cytogenet. 2018;11:37.
Paththinige CS, Sirisena ND, Kariyawasam U, Ediriweera RC, Kruszka P, Muenke M, et al. A child with multiple congenital anomalies due to partial trisomy 7q22.1 --> qter resulting from a maternally inherited balanced translocation: a case report and review of literature. BMC Med Genet. 2018;11(1):44.
Larizza L, Finelli P. Developmental disorders with intellectual disability driven by chromatin dysregulation: clinical overlaps and molecular mechanisms. Clin Genet. 2019;95(2):231–40.
Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet. 2010;11:161–87.
Silibello G, Vizziello P, Gallucci M, Selicorni A, Milani D, Ajmone PF, et al. Daily life changes and adaptations investigated in 154 families with a child suffering from a rare disability at a public centre for rare diseases in Northern Italy. Ital J Pediatr. 2016;42(1):76.
McKenzie J, McConkey R. Caring for adults with intellectual disability: the perspectives of family carers in South Africa. J Appl Res Intellect Disabil. 2016;29(6):531–41.
de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367(20):1921–9.
Topper S, Ober C, Das S. Exome sequencing and the genetics of intellectual disability. Clin Genet. 2011;80(2):117–26.
van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annu Rev Genet. 2011;45:81–104.
Kessi M, Xiong J, Wu L, Yang L, He F, Chen C, et al. Rare copy number variations and predictors in children with intellectual disability and epilepsy. Front Neurol. 2018;9:947.
Han JY, Jang JH, Park J, Lee IG. Targeted next-generation sequencing of Korean patients with developmental delay and/or intellectual disability. Front Pediatr. 2018;6:391.
Hu L, Cheng D, Gong F, Lu C, Tan Y, Luo K, et al. Reciprocal translocation carrier diagnosis in preimplantation human embryos. EBioMedicine. 2016;14:139–47.
Tan YQ, Tu C, Meng L, Yuan S, Sjaarda C, Luo A, et al. Loss-of-function mutations in TDRD7 lead to a rare novel syndrome combining congenital cataract and nonobstructive azoospermia in humans. Genet Med. 2019;21(5):1209–17.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Hu L, Wei Y, Luo K, Xie P, Gong F, Xiong B, et al. Clinical outcomes in carriers of complex chromosomal rearrangements: a retrospective analysis of comprehensive chromosome screening results in seven cases. Fertil Steril. 2018;109(3):486–92.
Tan Y, Yin X, Zhang S, Jiang H, Tan K, Li J, et al. Clinical outcome of preimplantation genetic diagnosis and screening using next generation sequencing. Gigascience. 2014;3(1):30.
Schluth-Bolard C, Labalme A, Cordier MP, Till M, Nadeau G, Tevissen H, et al. Breakpoint mapping by next generation sequencing reveals causative gene disruption in patients carrying apparently balanced chromosome rearrangements with intellectual deficiency and/or congenital malformations. J Med Genet. 2013;50(3):144–50.
Moyses-Oliveira M, Di-Battista A, Zamariolli M, Meloni VA, Bragagnolo S, Christofolini DM, et al. Breakpoint mapping at nucleotide resolution in X-autosome balanced translocations associated with clinical phenotypes. Eur J Hum Genet. 2019;27(5):760–71.
Yauy K, Schneider A, Ng BL, Gaillard JB, Sati S, Coubes C, et al. Disruption of chromatin organisation causes MEF2C gene overexpression in intellectual disability: a case report. BMC Med Genet. 2019;12(1):116.
Aristidou C, Theodosiou A, Bak M, Mehrjouy MM, Constantinou E, Alexandrou A, et al. Position effect, cryptic complexity, and direct gene disruption as disease mechanisms in de novo apparently balanced translocation cases. PLoS One. 2018;13(10):e0205298.
Dutta UR, Rao SN, Pidugu VK, Vineeth V S, Bhattacherjee A, Bhowmik AD, et al. Breakpoint mapping of a novel de novo translocation t(X;20)(q11.1;p13) by positional cloning and long read sequencing. Genomics. 2019;111(5):1108–14.
Schluth-Bolard C, Diguet F, Chatron N, Rollat-Farnier PA, Bardel C, Afenjar A, et al. Whole genome paired-end sequencing elucidates functional and phenotypic consequences of balanced chromosomal rearrangement in patients with developmental disorders. J Med Genet. 2019;56(8):526–35.
Dupont JM, Cuisset L, Cartigny M, Le Tessier D, Vasseur C, Rabineau D, et al. Familial reciprocal translocation t(7;16) associated with maternal uniparental disomy 7 in a Silver-Russell patient. Am J Med Genet. 2002;111(4):405–8.
Conroy JM, Grebe TA, Becker LA, Tsuchiya K, Nicholls RD, Buiting K, et al. Balanced translocation 46,XY,t(2;15)(q37.2;q11.2) associated with atypical Prader-Willi syndrome. Am J Hum Genet. 1997;61(2):388–94.
Fukushi D, Yamada K, Suzuki K, Inaba M, Nomura N, Suzuki Y, et al. Clinical and genetic characterization of a patient with SOX5 haploinsufficiency caused by a de novo balanced reciprocal translocation. Gene. 2018;655:65–70.
Cosemans N, Vandenhove L, Maljaars J, Van Esch H, Devriendt K, Baldwin A, et al. ZNF462 and KLF12 are disrupted by a de novo translocation in a patient with syndromic intellectual disability and autism spectrum disorder. Eur J Med Genet. 2018;61(7):376–83.
Xu XJ, Lv F, Liu Y, Wang JY, Song YW, Asan, et al. A cryptic balanced translocation involving COL1A2 gene disruption cause a rare type of osteogenesis imperfecta. Clin Chim Acta. 2016;460:33–9.
Miclea D, Peca L, Cuzmici Z, Pop IV. Genetic testing in patients with global developmental delay / intellectual disabilities. A review. Clujul Med. 2015;88(3):288–92.
Sun M, Goldin E, Stahl S, Falardeau JL, Kennedy JC, Acierno JS Jr, et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum Mol Genet. 2000;9(17):2471–8.
Bargal R, Avidan N, Ben-Asher E, Olender Z, Zeigler M, Frumkin A, et al. Identification of the gene causing mucolipidosis type IV. Nat Genet. 2000;26(1):118–23.
Amir N, Zlotogora J, Bach G. Mucolipidosis type IV: clinical spectrum and natural history. Pediatrics. 1987;79(6):953–9.
Bach G. Mucolipidosis type IV. Mol Genet Metab. 2001;73(3):197–203.
Altarescu G, Sun M, Moore DF, Smith JA, Wiggs EA, Solomon BI, et al. The neurogenetics of mucolipidosis type IV. Neurology. 2002;59(3):306–13.
Zhang X, Li X, Xu H. Phosphoinositide isoforms determine compartment-specific ion channel activity. Proc Natl Acad Sci U S A. 2012;109(28):11384–9.
Li M, Zhang WK, Benvin NM, Zhou X, Su D, Li H, et al. Structural basis of dual Ca(2+)/pH regulation of the endolysosomal TRPML1 channel. Nat Struct Mol Biol. 2017;24(3):205–13.
Vergarajauregui S, Puertollano R. Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic. 2006;7(3):337–53.
Gruber R, Zhou Z, Sukchev M, Joerss T, Frappart PO, Wang ZQ. MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway. Nat Cell Biol. 2011;13(11):1325–34.
Zhou ZW, Tapias A, Bruhn C, Gruber R, Sukchev M, Wang ZQ. DNA damage response in microcephaly development of MCPH1 mouse model. DNA Repair (Amst). 2013;12(8):645–55.
Yang SZ, Lin FT, Lin WC. MCPH1/BRIT1 cooperates with E2F1 in the activation of checkpoint, DNA repair and apoptosis. EMBO Rep. 2008;9(9):907–15.
Liang Y, Gao H, Lin SY, Peng G, Huang X, Zhang P, et al. BRIT1/MCPH1 is essential for mitotic and meiotic recombination DNA repair and maintaining genomic stability in mice. PLoS Genet. 2010;6(1):e1000826.
Trimborn M, Richter R, Sternberg N, Gavvovidis I, Schindler D, Jackson AP, et al. The first missense alteration in the MCPH1 gene causes autosomal recessive microcephaly with an extremely mild cellular and clinical phenotype. Hum Mutat. 2005;26(5):496.
Perche O, Menuet A, Marcos M, Liu L, Paris A, Utami KH, et al. Combined deletion of two Condensin II system genes (NCAPG2 and MCPH1) in a case of severe microcephaly and mental deficiency. Eur J Med Genet. 2013;56(11):635–41.
Wang JK, Li Y, Su B. A common SNP of MCPH1 is associated with cranial volume variation in Chinese population. Hum Mol Genet. 2008;17(9):1329–35.
Farooq M, Baig S, Tommerup N, Kjaer KW. Craniosynostosis-microcephaly with chromosomal breakage and other abnormalities is caused by a truncating MCPH1 mutation and is allelic to premature chromosomal condensation syndrome and primary autosomal recessive microcephaly type 1. Am J Med Genet A. 2010;152A(2):495–7.
Funding
This work was supported by grants from the National Key Research & Developmental Program of China (2018YFC1004900), the National Natural Science Foundation of China (81771645 and 81971447), Hunan Provincial Grant for Innovative Province Construction (2019SK4012), and Research Grant of CITIC-Xiangya (YNXM-201916).
Author information
Authors and Affiliations
Contributions
Dehua Cheng and Shimin Yuan conducted the genetic studies, drafted the initial manuscript, and wrote the manuscript; Liang Hu participated in sequence alignment and performed the initial analyses; Keli Luo and Fei Gong participated in data collection; Duo Yi and Changfu Lu participated in data analysis and FISH experiment; Guangxiu Lu and Ge Lin conceived of the study; Yue-Qiu Tan conceived of the study, helped draft the initial manuscript, and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
This study was approved by the Institutional Ethics Committee of the Reproductive & Genetic Hospital of CITIC-Xiangya, and written informed consent was obtained from the parents.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 17 kb).
Rights and permissions
About this article

Cite this article
Cheng, D., Yuan, S., Hu, L. et al. The genetic cause of intellectual deficiency and/or congenital malformations in two parental reciprocal translocation carriers and implications for assisted reproduction. J Assist Reprod Genet 38, 243–250 (2021). https://doi.org/10.1007/s10815-020-01986-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-020-01986-1