
Abstract
Sensitivity to lodging of oat varieties has been reduced in the last decades through the introduction of dwarfing genes. However, lodging may still cause significant yield loss, underscoring the need for new oat varieties with higher levels of lodging tolerance. In the present study, we analysed lodging and plant height in a collection of European oat accessions including landraces, old and modern varieties, in order to perform a genome-wide association study (GWAS) for identifying markers associated to lodging tolerance. This collection has been recently genotyped by the Infinium 6K SNP array for oat and SNP data were analysed as continuous intensity ratios, rather than as discrete genotypes (Tumino et al. 2016, Theor Appl Genet 129, pp. 1711–1724). Phenotypes for lodging severity, plant height and growth habit were collected under natural conditions in eight European countries. Plant height correlated to lodging severity as previously observed in many studies, explaining about 30% of lodging variation. GWAS analyses detected six significant associations for lodging and two for plant height. These results indicate that GWAS can successfully be used for identifying markers associated to lodging in oat, even though lodging is a quantitative trait influenced by several plant characteristics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lodging is the failure of plants to maintain an upright position under unfavorable weather conditions (rain and strong wind). When lodging occurs in cereals during or just after anthesis it can cause severe losses in grain yield (Pendleton 1954; Stanca et al. 1979), mainly because of reducing carbon assimilation within the canopy and efficiency in light interception by leaves (Setter et al. 1997). Various types of lodging can be distinguished. Stem buckling at the lower internodes is defined as stem lodging, while buckling of the middle internodes is known as brackling. In addition, plants may lodge as a result of root anchorage failure, which is called root lodging. In oat and barley all types of lodging have been observed, while wheat is known to be insensitive to brackling.
Tolerance to lodging can therefore be affected by several plant characteristics, including height, stem strength and root anchorage (see Berry et al. 2004 for a review). Several studies have shown that height is the main factor affecting lodging tolerance in cereals (e.g. Stanca et al. 1979; Berry and Berry 2015). Shorter plants are less affected by lodging, due to the reduction of the leverage force applied on the base of the stem (Berry et al. 2004). Breeding has reduced stem length in wheat and barley and as a result also the risk of lodging. In oat cultivation, however, lodging can still cause significant yield losses (Marshall et al. 1986). A few dwarfing genes are available in oat that have been successfully used in the last decades by breeders, producing high-yielding varieties with lower lodging risk (Marshall and Murphy 1980; Milach et al. 1997; Morikawa et al. 2007). However, reducing plant height too much can have a detrimental effect on yield potential (Flintham et al. 1997). Knowledge about genetic factors controlling stem strength and root anchorage in cereals and their effect on lodging tolerance is scarce, probably because these are complex traits that may be affected by many factors (e.g. stem diameter, stem wall thickness, cell wall composition, root spread, root depth, root diameter, root number), requiring time-consuming phenotyping to be dissected and fully understood. Berry and Berry (2015) studied the relationship between natural lodging and lodging-related traits in two doubled haploid wheat populations. The strongest correlation was observed between lodging and plant height, followed by stem diameter and stem material strength. They also found a positive correlation between stem diameter and stem wall thickness and a negative correlation between stem diameter and stem material strength. The rice gene ABERRANT PANICLE ORGANIZATION 1 (APO1), encoding for an F-box-protein involved in controlling rachis branch number of the panicle, has been shown to be responsible for increasing stem diameter (Ookawa et al. 2010), providing a first clue of the genetic basis of stem strength in cereals. Root anchorage has been investigated more in maize than in other cereals, and several QTLs and candidate genes have been identified affecting root morphology and tolerance to root lodging (Bruce et al. 2001; Landi et al. 2007; Landi et al. 2010). However, the physiological mechanisms and genetic factors affecting root lodging are still largely unclear.
In oat many QTLs for plant height have been identified in several studies by using bi-parental populations (Siripoonwiwat et al. 1996; Holland et al. 1997; De Koeyer et al. 2004; Wooten et al. 2009), but only few QTL analyses have addressed oat lodging (De Koeyer et al. 2004; Tanhuanpää et al. 2012).
The recent development of new genomic tools, notably the 6K Infinium SNP array (Tinker et al. 2014) and the dense hexaploid oat consensus map (12,000 markers) based on 12 bi-parental populations (Chaffin et al. 2016; Yan et al. 2016), have made it possible to map oat traits at a high resolution (Esvelt Klos et al. 2016). Winkler et al. (2016) used the array for a genome-wide association study (GWAS) in a diverse panel of oat accessions from the USDA National Small Grains Collection, assembled to represent a gene pool relatively unaffected by 20th century breeding activities, but failed to find significant associations for lodging tolerance. In the present work we used this map and SNP array for a GWAS for lodging tolerance in a new European oat collection, established by the AVEQ project (http://aveq.jki.bund.de/aveq). Genetic diversity of the AVEQ collection and GWAS for frost tolerance were addressed by Tumino et al. (2016) using continuous SNP intensity ratios, providing a framework for the present GWAS study. Objectives of this work are (1) to study the phenotypic variation of lodging and height in the AVEQ collection and their relationship, (2) to test the effectiveness of GWAS for finding loci associated with a complex trait such as lodging, (3) to provide new genetic markers to be used in breeding programs aimed to improve lodging tolerance in oat.
Materials and methods
Plant materials and DNA preparation
The diverse collection analyzed in the present study is a subset of the European oat collection of about 600 accessions established within the AVEQ project (http://aveq.jki.bund.de/aveq). Selection of the accessions aimed to maximize the diversity with respect to country of origin, year of registration and type of cultivation (winter or spring). It includes 126 winter and spring A. sativa accessions from 27 European countries, including breeding lines, old and modern cultivars and a few landraces. For DNA extraction leaf tissues were collected and pooled at second-leaf stage from 10 plants per accession. Genomic DNA was extracted using a CTAB-based buffer followed by chloroform extraction.
Phenotypic and genotypic data
During the AVEQ project the oat accessions were evaluated in 8 European countries (France, Italy, Estonia, Czech Republic, Poland, Bulgaria, Romania and Germany). The collection was divided into two sets, which were phenotyped in 2008 (set AVEQ08, 75 accessions) and 2009 (set AVEQ09, 62 accessions). Eleven standard cultivars were included in both sets. The accessions were phenotyped for lodging at mature and immature (from anthesis to late milk stage; IBPGR 6.1.5) stage, for plant height and growth habit (i.e. tendency of the culm to grow prostrate or erect at a juvenile stage; scored according the UPOV rating) in spring-sown field trials. At each location, each accession was planted in single 2 × 1 m plots and standard cultivars were replicated in five plots.
The present study is based on 3567 SNP markers from the Infinium 6K Oat Array (Tinker et al. 2014). SNP data curation is described in detail in Tumino et al. (2016). Briefly, SNP fluorescence signals were used to calculate intensity ratios according to the formula X/(X + Y), where X and Y are normalized hybridization intensities for alleles A and B. We used these ratios as continuous marker data, instead of defining clusters and calling discrete genotypes as usually performed by GenomeStudio (Illumina Inc., San Diego, CA). The GenomeStudio Polyploid Clustering Module enables detecting signal clusters, but time-consuming manual curation may be required to adjust clusters and check uncertain assignments. This is particularly true for allopolyploids. For the 6K Oat Array, almost 20% of the polymorphic markers assayed in a diverse set of 595 mostly American oat varieties detected more than two clusters (Tinker et al. 2014), which indicates hybridisation to homoeologous loci (Wang et al. 2014), assuming accessions to be pure and nearly homozygous.
The SNP quality filtering process aimed to remove monomorphic markers and single data points with extremely high or low total signal intensity values and resulted in a dataset of 126 samples and 3567 SNPs with fewer than 25% missing values.
Data analysis
Population structure was determined by a Principal Component Analysis (PCA) on a correlation matrix based on SNP hybridization ratios, conducted in R (R Core Team 2014) using the function prcomp (package stats). Before calculation all missing values were replaced by the mean ratio per marker. The genetic structure of the collection is described in detail in Tumino et al. (2016).
To estimate accession adjusted means, phenotypic data were analyzed by using an ANOVA model including accession and location as fixed factors. We decided to analyze the two sets of accessions (AVEQ08 and AVEQ09) separately, because of the small overlap of accessions between them. Locations in which low or no variation in lodging score was observed were not included in the model (Fig. 2). Best linear unbiased estimations (BLUEs) for the accession effects were used to perform association mapping.
Three alternative models for genome-wide association studies (GWAS) were fitted in GenStat (VSN International 2014). These three models differed in the way they account for population structure. The first model does not account for population structure. It was a simple linear model including marker effects as a fixed factor: y = μ + m + ε; where y is the trait of interest, μ is the overall mean, m is the vector of marker effects and ε is the vector of residuals. Random variables are underlined. A second linear mixed model corrected for kinship between accessions, including marker as a fixed factor and genotype as a random factor: y = μ + m + a + ε; where a is the vector of accession random effects. The covariance between accessions was structured by a kinship matrix, calculated according to Tumino et al. (2016). A third fixed effects model corrected for population structure using the scores of n principal components: y = μ + p n + m + ε; where p is a sum vector of n principal components scores. The p-values obtained from each association analysis were plotted versus the expected p-values under the null hypothesis (Q–Q plot), to check that they have a uniform distribution (no association). Departure of p-values from the uniform distribution was used as indication that the presence of population structure may increase the type I error rate. −logP-values from the association analyses were visualized as Manhattan plots using the map positions reported in Chaffin et al. (2016). A significance threshold adjusted for multiple testing was calculated by the method described in Li and Ji (2005), separately for AVEQ08 (−logP = 3.365) and AVEQ09 (−logP = 3.246), with α = 0.05.
In addition, to compare QTL profiles between AVEQ08 and AVEQ09 we considered a liberal threshold of −logP = 2 and a sliding window of 10 cM. For each region consistent across the two sets the highest −logP was reported (Figs. 5c, 6c).
Markers with –logP ≥2 were checked for the presence of outlier genotypes (intensity ratios), which could affect significance of phenotype-genotype associations, thus increasing the risk of false positives. This check replaces the filtering against markers with a low minor allele frequency (MAF), which cannot be calculated using continuous intensity ratios. To identify outliers (extreme values of low frequency) we focused our check on the first ten and the last ten percentiles of marker ratios. Discontinuity in these values is an indication of the presence of outliers. We defined discontinuity when the difference between consecutive percentiles was greater than 5% of the range of variation of ratios. Markers containing outliers were excluded from the results.
Results
Population structure
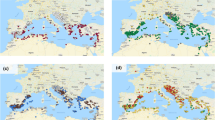
The overall population structure of the AVEQ subset analysed by Tumino et al. (2016) indicated the presence of two genetic groups mainly based on geography and cultivation type (spring versus winter types). One group originating from Continental Europe (group B) contained mainly spring types, which are susceptible to frost, while the other group, with accessions from Atlantic and Mediterranean Europe (group A), was more diverse and included both frost-susceptible and frost-tolerant accessions. However, this collection can be considered weakly structured, as the first two principal components explained only 27% of the total genetic variation (Tumino et al. 2016). Here we performed a PCA within each of the two sets of accession (AVEQ08 and AVEQ09, Fig. 1), in order to support a comparison of the GWAS results between the two sets. These two PCAs showed a similar pattern for the first three principal components. The first principal component differentiated group A (right side quadrants) from group B (left side quadrants) in both sets, explaining 20.5 and 22.9% of the total variance in AVEQ08 and AVEQ09 respectively. The next two PCA components differentiated Central European (upper left quadrants) from Eastern European accessions (lower left quadrants) in group B, and common oats (A. sativa sub. sativa) from red oats (A. sativa sub. byzantina) in group A (but this AVEQ subset included only few red oat accessions). In AVEQ09 the axis that separated common oats from red oats (PC2) explained more than the one separating Central and Eastern European accessions (PC3) (7 vs. 4%) (Fig. 1). In AVEQ08 both these principal components explained around 6% of the variation and their order was reversed (i.e. PC2 in AVEQ08 roughly corresponds to PC3 in AVEQ09 and v.v.) (Fig. 1). A principal component analysis of the whole population produced similar results as when AVEQ08 and AVEQ09 were separately plotted (Online Resource 1). Lodging-susceptible accessions were found both in group A and in group B, although within group B they mainly originated from Eastern Europe (Fig. 1a, b, lower left quadrants).

Principal component analyses based on 3567 SNP hybridization intensity ratios for the two sets of accessions. The scatter plots of PC1 versus PC2 and PC1 versus PC3 are showed for AVEQ08 (a, c) and AVEQ09 (b, d). The standard cultivars are represented by squares. Colours varying from blue to red indicate the level of lodging tolerance (blue for tolerant and red for susceptible). The first principal component differentiated group A (positive scores) from group B (negative scores) as described in Tumino et al. (2016). (Color figure online)
Phenotypic variation
A large variation among accessions for lodging score was observed in eleven locations, while in six locations the lodging scores were very low, as the weather conditions had been not severe, making it impossible to discriminate among the accessions (Fig. 2). The median lodging score per accession covered the full range from 0 to 9 in AVEQ08 and almost the full range (from 0 to 7) in AVEQ09 (Fig. 3, Online Resource 2a, b). The accession factor was highly significant in both sets (ANOVA, p < 0.001). Lodging score measured at an immature stage was very similar to lodging at maturity, with a lower range of variation ranging from 0 to 6 and from 0 to 5 in AVEQ08 and AVE09 respectively (Online Resource 2c, d). This observation indicated that the most sensitive accessions were already lodged to a less severe degree at an immature stage. The accession effect in both sets was still highly significant (p < 0.001).

Phenotypic variation of lodging score per location. The box represents the first, second and third quartiles. The whiskers length is the interquartile range multiplied by 1.5. The data points were plotted over the boxes. Locations tagged by an asterisk were not included in the model because of low phenotypic variation observed

Lodging score variation per accession. The box represents the first, second and third quartiles. The whiskers length is the interquartile range multiplied by 1.5
Plant height ranged from 70 to 130 cm in AVEQ08 and from 65 to 135 cm in AVEQ09, with a continuous variation across the range (Online Resource 2e, f). Although plant height is known to be highly affected by environmental factors, particularly by sowing date in our study, the accession effect on height was very significant (p < 0.001) in both sets.
Regarding growth habit a low variation was observed with most of the accessions showing an erect habit (Online Resource 2g, h).
Diagnostic plots of residuals for all traits indicated no substantial deviation from the assumption of normality and homogeneity of variance (not shown).
Correlation between lodging tolerance and plant height
To check correlation between lodging and plant height, both lodging and plant height BLUEs were corrected for structure using a fixed effect model including only population structure as a factor (using the Ward’s clustering membership as reported in Tumino et al. 2016). Then, BLUEs adjusted for structure were used for calculating Pearson’s correlation.
Pearson’s correlation corrected for population structure indicated that lodging scored at maturity correlated with plant height, with r = 0.53 and r = 0.60 for AVEQ08 and AVEQ09 respectively. About 30% of variation in lodging scores can be explained by plant height in both the sets (Fig. 4). Uncorrected Pearson’s correlation coefficients were high (0.74 and 0.72 for AVEQ08 and AVEQ09 respectively), indicating that part of the covariation between lodging and height could be due to population structure.

Correlation plots between lodging and plant height BLUEs adjusted for population structure, for AVEQ08 (a) and AVEQ09 (b). The solid line represents a linear fit of the data
Genome-wide association analysis for lodging and plant height
For the lodging data at mature growth stage, the GWAS model that corrected for population structure by using a kinship matrix outperformed the other models, as determined using Q–Q plots (not shown). The improvement of the Q–Q plots of this model compared to the model that does not correct for population structure is shown in Online Resource 3. Therefore, only the GWAS results obtained by this model are presented here. GWAS analyses for lodging at immature stage and growth habit are not presented because lodging phenotypic data at immature stage were very similar to data at maturity and growth habit data appeared not well balanced, as some phenotypic classes were over-represented (Online Resource 2g, h), increasing the risk of spurious associations due to rare extreme values.
Two QTLs for lodging were detected in AVEQ08 in Mrg24 (five significant markers in the range 57–60 cM) and in Mrg21 (one marker at 194 cM) (Fig. 5a). In AVEQ09 five markers were positive in various linkage groups, one of them coinciding with the QTL found in Mrg24 for AVEQ08 (Fig. 5b). Five regions were consistent across the two sets (Fig. 5c).

GWAS results for lodging. a Manhattan plot illustrating p-values from the GWAS analysis in AVEQ08. b Manhattan plot illustrating p-values from the GWAS analysis in AVEQ09. c Consistency across AVEQ08 and AVEQ09. Regions with p-values above a mild threshold of −logP = 2 in both sets were detected by using a sliding window of 10 cM. For these regions the highest −logP value is reported (solid line). The numbers at the top of the plot indicate the consensus chromosomes as named in Chaffin et al. (2016)
Regarding plant height, two significant markers have been found in the bottom part of Mrg01, one for AVEQ08 and one for AVEQ09, possibly detecting the same QTL (Fig. 6a, b). Another significant marker mapped in Mrg13 at 58 cM in AVEQ08 (Fig. 6b). Four regions were consistent across the two mapping sets, using the sliding window comparison approach (Fig. 6c).

GWAS results for plant height. a Manhattan plot illustrating p-values from the GWAS analysis in AVEQ08. b Manhattan plot illustrating p-values from the GWAS analysis in AVEQ09. c Consistency across AVEQ08 and AVEQ09. Regions with p-values above a mild threshold of −logP = 2 in both sets were detected by using a sliding window of 10 cM. For these regions the highest −logP value is reported (solid line). The numbers at the top of the plot indicate the consensus chromosomes as named in Chaffin et al. (2016)
Discussion
The use of continuous marker intensity ratios
In the present study we used genotypes from bulks of ten plants per accession, in order to take into account potential within-accession diversity such as expected in landraces or old traditional varieties. For an association analysis, the bulking approach may be convenient, especially when phenotypes from genebank accessions are already available. In fact, those phenotypes would be average measures of bulks of potentially heterogeneous accession and therefore should be associated to an average measure of bulk genotypes, not to an individual genotype taken from this bulk. However, SNP array data of heterogeneous bulked samples cannot be interpreted as discrete genotypes. To circumvent the need to (artificially and inappropriately) convert continuous signals into discrete genotypes for heterogeneous samples, we developed a method that accepts continuous SNP signal ratios (Tumino et al. 2016), which represent within-bulk allele frequency estimations. The higher the number of heterogeneous accessions, the more advantageous will be the use of continuous ratios. One concern may be that the use of continuous ratios for those SNPs that clearly cluster into discrete classes, would reduce the statistical power of a GWAS analysis. To study this concern we have compared, for the positive markers identified in our analysis, the GWAS p-values obtained using continuous ratios with those obtained by discrete genotypes (Online Resource 4). Similar p-values were obtained by the two methods (Online Resource 4), supporting the validity of continuous scoring. Three out of 19 markers were discarded by the Illumina quality filters as discrete scores could not be assigned, indicating that continuous scoring is able to use information from more markers. Phenotypes (BLUE values) and genotypes (normalized X and Y signals) used to perform the association analyses presented here can be found as supplementary files (Online Resource 5 and Online Resource 6, respectively).
Continuous intensity ratios have also been successfully used to analyse homogeneous DNA samples of grape, in order to optimize the use of those SNPs that do not pass Illumina quality filters, but still contain information regarding marker-trait association (Myles et al. 2015).
Population structure
We performed a GWAS analysis for lodging tolerance and plant height in a diverse population of European hexaploid oats. The geographical origin of the accessions in the AVEQ collection reflects partly the breeding station from which the accessions originate, and also the type of cultivation that is possible, as the group B (Tumino et al. 2016) includes almost exclusively frost-susceptible spring-type oats. The Eastern Europe group B included many of the lodging-susceptible accessions, emphasising the need to correct for population structure in the GWAS model. Since we compared GWAS results of two sets of accessions, knowledge on population structure and correlation with traits is critical for interpreting the results. For this reason, we determined the population structure within subsets AVEQ08 (75 accessions) and AVEQ09 (62 accessions) separately. The structure was similar in both and comparable to that of the whole set (Tumino et al. 2016), with the first three principal components representing geographical origin of accessions, growth type (winter/spring) and probably the difference between common and red oats. The importance of correctly accounting for population structure was confirmed by the abundance of low p-values for uncorrected models as observed in the Q–Q plots (Online Resource 3).
Lodging tolerance and plant height
In this study several significant marker associations for lodging and plant height have been detected. Some of them mapped in regions previously identified by other studies (Table 1). Since we performed GWAS in two different sets of accessions, we based the interpretation of our results on (i) significance adjusted for multiple testing, based on the method of Li and Ji (2005) and (ii) consistency across the two mapping sets, using a sliding window analysis, taking advantage of the fact that the two mapping sets had very similar population structure. The underlying idea is that consistency across two independent sets of accessions can support statistical testing in single sets. Moreover, we compared 10 cM bins instead of single markers, as the presence of heterogeneous alleles with different frequencies in the two sets might lead to more diffuse peaks of association (Atwell et al. 2010).
Lodging scores measured at maturity correlated with plant height, with a structure-corrected Pearson’s coefficient of 0.53 and 0.60 for AVEQ08 and AVEQ09 respectively (Fig. 4). This finding is in agreement with previous studies about lodging-related traits in oat (De Koeyer et al. 2004), barley (Stanca et al. 1979) and wheat (Berry and Berry 2015) that showed that plant height is the main factor affecting risk of lodging. As a consequence, QTLs for lodging often co-localized with QTLs for plant height (e.g. De Koeyer et al. 2004). In our study, on Mrg23 we detected QTLs for lodging and height within 10 cM from each other (Table 1). Marker associations for lodging on Mrg01 (at 74 cM) and Mrg24 (57–60 cM) mapped close to two QTLs for plant height (at 78 cM and 85 cM respectively) previously identified by Holland et al. (1997) in the Kanota × Ogle (KO) bi-parental population. Previous studies using bi-parental populations identified QTLs for lodging in Mrg01, Mrg05 and Mrg21. In particular, three loci have been detected in Mrg21 around 190 cM, in the range 150–180 cM (De Koeyer et al. 2004) and around 130 cM (Tanhuanpää et al. 2012), where we also detected two associations (Table 1). Interestingly, associations to hullessness have also been found around 178 cM (Tumino et al. 2016) and 194 cM (De Koeyer et al. 2004). Our findings support therefore the suggestion that the N1 allele (controlling hullessness) may also affect lodging as it plays a role in lignin deposition in spikelets and possibly also in stems (De Koeyer et al. 2004; Ougham et al. 1996). Further studies will be needed to exclude that this co-location of QTLs is due to two independent genes in the same chromosomal region. For the other QTLs for lodging reported by De Koeyer et al. (2004) on Mrg01 and Mrg05 a precise position comparison with our results was not possible as shared markers were lacking.
Associations for plant height were detected in five linkage groups (Table 1). The QTLs in Mrg01 and Mrg11 mapped in regions previously associated to height using bi-parental population (Table 1; Holland et al. 1997; De Koeyer et al. 2004; Wooten et al. 2009). QTL mapping for plant height is complicated by pleiotropic effects of genes controlling heading date (vernalization requirement and photoperiod sensitivity), as they affect duration of vegetative stage and therefore height of plant. In fact, many studies reported co-localization of QTLs for height with those for heading date or vernalization (Holland et al. 1997; De Koeyer et al. 2004; Wooten et al. 2009; Winkler et al. 2016). We also have found that the associations with plant height in Mrg01 and Mrg13 co-mapped with QTLs for heading date previously identified in this collection (Tumino et al. 2016) and an association with plant height in Mrg11 co-mapped with a QTL for frost tolerance (Tumino et al. 2016).
Several of the QTLs associated to height in our analysis do not co-localize with QTLs for lodging. This could be related to the statistical power of the analysis. Lodging occurs under unfavourable weather conditions, therefore it was measured only in some environments, while plant height was measured in all environments. A biological reason could be the fact that genes controlling heading date pleiotropically affect also other factors with contrasting effects on lodging. For instance, with increased duration of the vegetative stage, the negative effect of taller plants on lodging tolerance could be partly compensated by a larger root system.
In general, alleles responsible for reducing plant height will reduce also the risk of lodging, as they diminish the bending stress at the base of the culm under unfavourable weather conditions (strong wind and rain). During the Green Revolution, reduction of plant height was one of the main breeding targets for cereals, leading to an increase in grain yield. However, this process seems to have some physiological limitations, and further reduction of plant height may negatively affect yield potential (Flintham et al. 1997; Berry et al. 2015). Therefore, QTL that affect lodging without affecting height can be of great value in further reducing lodging resistance. In our study the most robust association for lodging in Mrg24 did not co-localize with any QTL for plant height, and as such it may represent a valuable marker for breeding programs targeted to enhance lodging tolerance in oat. Further studies will be needed to dissect the genetic basis determining lodging tolerance in oat, addressing QTL mapping for traits such as culm diameter, culm cell wall composition or root anchorage.
References
Atwell S, Huang YS, Vilhjálmsson BJ, Willems G, Horton M, Li Y, Meng D, Platt A et al (2010) Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465:627–631. doi:10.1038/nature08800
Berry PM, Berry ST (2015) Understanding the genetic control of lodging-associated plant characters in winter wheat (Triticum aestivum L.). Euphytica 205:671–689
Berry PM, Sterling M, Spink JH, Baker CJ, Sylvester-Bradley R, Mooney S, Tams A, Ennos AR (2004) Understanding and reducing lodging in cereals. Adv Agron 84:215–269
Berry PM, Kendall S, Rutterford Z, Orford S, Griffiths S (2015) Historical analysis of the effects of breeding on the height of winter wheat (Triticum aestivum) and consequences for lodging. Euphytica 203:375–383. doi:10.1007/s10681-014-1286-y
Bruce W, Desbons P, Crasta O, Folkerts O (2001) Gene expression profiling of two related maize inbred lines with contrasting root-lodging traits. J Exp Bot 52:459–468
Chaffin AS, Huang YF, Smith S, Bekele WA, Babiker E, Gnanesh BN et al (2016) A consensus map in cultivated hexaploid oat reveals conserved grass synteny with substantial subgenome rearrangement. Plant Genome. doi:10.3835/plantgenome2015.10.0102
De Koeyer DL, Tinker NA, Wight CP, Deyl J, Burrows VD, O’Donoughue LS, Lybaert A, Molnar SJ, Armstrong KC, Fedak G, Wesenberg DM, Rossnagel BG, McElroy AR (2004) A molecular linkage map with associated QTLs from a hulless x covered spring oat population. Theor Appl Genet 108:1285–1298. doi:10.1007/s00122-003-1556-x
Esvelt Klos K, Huang YF, Bekele WA, Obert DE, Babiker E, Beattie AD, Bjørnstad Å, Bonman JM, Carson ML, Chao S, Gnanesh BN et al (2016) Population genomics related to adaptation in elite oat germplasm. Plant Genome. doi:10.3835/plantgenome2015.10.0103
Flintham JE, Borner A, Worland AJ, Gale MD (1997) Optimizing wheat grain yield: effects of Rht (gibberellin insensitive) dwarfing genes. J Agric Sci 128:11–25
Holland JB, Moser HS, O’Donoghue LS, Lee M (1997) QTLs and epistasis associated with vernalization responses in oat. Crop Sci 37:1306–1316
Landi P, Sanguineti MC, Liu C, Li Y, Wang TY, Giuliani S, Bellotti M, Salvi S, Tuberosa R (2007) Root-ABA1 QTL affects root lodging, grain yield, and other agronomic traits in maize grown under well-watered and water-stressed conditions. J Exp Bot 58:319–326. doi:10.1093/jxb/erl161
Landi P, Giuliani S, Salvi S, Ferri M, Tuberosa R, Sanguineti MC (2010) Characterization of root-yield-1.06, a major constitutive QTL for root and agronomic traits in maize across water regimes. J Exp Bot 61(3553):3562. doi:10.1093/jxb/erq192
Li J, Ji L (2005) Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity 95:221–227
Marshall HG, Murphy CF (1980) Inheritance of dwarfness in three oat crosses and relationship of height to panicle and culm length. Crop Sci 21:335–338
Marshall HG, Kolb FL, Roth GW (1986) Effects of nitrogen fertilizer rate, seeding rate, and row spacing on semi-dwarf and conventional height spring oat. Crop Sci 27:572–575
Milach S, Rines H, Phillips R (1997) Molecular genetic mapping of dwarfing genes in oat. Theor Appl Genet 95:783–790. doi:10.1007/s001220050626
Morikawa T, Sumiya M, Kuriyama S (2007) Transfer of new dwarfing genes from the weed species Avena fatua into cultivated oat A. byzantina. Plant Breeding 126:30–35. doi:10.1111/j.1439-0523.2007.01309.x
Myles S, Mahanil S, Harriman J, Gardner KM, Franklin JL, Reisch BI, Ramming DW, Owens CL, Li L, Buckler ES, Cadle-Davidson L (2015) Genetic mapping in grapevine using SNP microarray intensity values. Mol Breed 35:88
Ookawa T, Hobo T, Yano M, Murata K, Ando T, Miura H, Asano K, Ochiai Y, Ikeda M, Nishitani R, Ebitani T, Ozaki H, Angeles ER, Hirasawa T, Matsuoka M (2010) New approach for rice improvement using a pleiotropic QTL gene for lodging resistance and yield. Nat Commun 1:132. doi:10.1038/ncomms1132
Ougham HJ, Latipova G, Valentine J (1996) Morphological and biochemical characterization of spikelet development in naked oats (Avena sativa). New Phytol 134:5–12
Pendleton JW (1954) The effect of lodging on spring oat yields and test weights. Agron J 46:265–267
R Core Team (2014) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. http://www.R-project.org/
Setter TL, Laureles EV, Mazaredo AM (1997) Lodging reduces the yield of rice by self-shading and reductions in canopy photosynthesis. Field Crops Res 49:95–106
Siripoonwiwat W, O’Donoghue LS, Wesenberg D, Hoffman DL, Barbosa-Neto JF, Sorrels ME (1996) Chromosomal regions associated with quantitative traits in oat. J Agric Genom 2:1–13
Stanca AM, Jenkins G, Hanson PR (1979) Varietal responses in spring barley to natural and artificial lodging and to a growth regulator. J Agric Sci 93:449–457
Tanhuanpää P, Manninen O, Beattie A, Eckstein P, Scoles G, Rossnagel B, Kiviharju E (2012) An updated doubled haploid oat linkage map and QTL mapping of agronomic and grain quality traits from Canadian field trials. Genome 55:289–301. doi:10.1139/g2012-017
Tinker NA, Chao S, Lazo GR, Oliver RE, Huang Y-F, Poland JA, Jellen EN, Maughan PJ, Kilian A, Jackson EW (2014) A SNP genotyping array for hexaploid oat. Plant Genome. doi:10.3835/plantgenome2014.03.0010
Tumino G, Voorrips RE, Rizza F, Badeck FW, Morcia C, Ghizzoni R, Germeier CU, Paulo MJ, Terzi V, Smulders MJM (2016) Population structure and genome-wide association analysis for frost tolerance in oat using continuous SNP array signal intensity ratios. Theor Appl Genet 129:1711–1724. doi:10.1007/s00122-016-2734-y
VSN International (2014) GenStat for Windows, 17th edn. VSN International, Hemel Hempstead
Wang S, Wong D, Forrest K, Allen A et al (2014) Characterization of polyploid wheat genomic diversity using a high-density 90000 single nucleotide polymorphism array. Plant Biotechnol J 12:787–796. doi:10.1111/pbi.12183
Winkler LR, Michael Bonman J, Chao S, Admassu Yimer B, Bockelman H, Esvelt Klos K (2016) Population structure and genotype-phenotype associations in a collection of oat landraces and historic cultivars. Front Plant Sci 7:1077. doi:10.3389/fpls.2016.01077
Wooten DR, Livingston DP, Lyerly HJ, Holland JB, Jellen EN, Marshall DS, Murphy JP (2009) Quantitative trait loci and epistasis for oat winter-hardiness component traits. Crop Sci 49:1989–1998. doi:10.2135/cropsci2008.10.0612
Yan H, Bekele WA, Wight CP, Peng Y, Langdon T, Latta RG, Fu YB, Diederichsen A, Howarth CJ, Jellen EN, Boyle B, Wei Y, Tinker NA (2016) High-density marker profiling confirms ancestral genomes of Avena species and identifies D-genome chromosomes of hexaploid oat. Theor Appl Genet 129:2133–2149. doi:10.1007/s00122-016-2762-7
Acknowledgements
The authors wish to thank Fabio Reggiani, Donata Pagani and Nadia Faccini for their technical assistance in plant phenotyping. This work was done within AVEQ (AGRI GEN RES 061, EU funded) and FAO-RGV (Italian Ministry of Agriculture funded) projects. Data analysis and writing the paper was supported in part by the Polyploids Project (TKI H263, BO-26.03-002-001). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10681_2017_1939_MOESM1_ESM.pdf
OR01. PCA of the whole population (AVEQ08 plus AVEQ09) based on 3567 SNPs. The scores of accessions belonging to AVEQ08 (a) and AVEQ09 (b) were separately plotted. Standard cultivars are represented by squares and the colour scale indicates the lodging tolerance of the accessions (blue for tolerant and red for susceptible). Supplementary material 1 (PDF 11 kb)
10681_2017_1939_MOESM2_ESM.pdf
OR02. BLUE values for lodging score, lodging score at immature stage, plant height and growth habit in AVEQ08 (a, c, e, g, respectively) and AVEQ09 (b, d, f, h, respectively). Lower values for lodging score mean less lodging, lower values for growth habit mean more erect habit. Supplementary material 2 (PDF 12 kb)
10681_2017_1939_MOESM3_ESM.pdf
OR03. Expected versus observed quantiles of p-values from association analyses of lodging tolerance and plant height for AVEQ08 and AVEQ09. The black and the red curves represent the p-value distribution obtained using simple association (no correction for population structure) and the p-value distribution from the model including a kinship matrix, respectively. Supplementary material 3 (PDF 173 kb)
10681_2017_1939_MOESM4_ESM.pdf
OR04. Comparison of continuous intensity ratios versus discrete calls. For markers with significant –logP values in our analysis, we also called discrete genotypes using GenomeStudio (Illumina Inc., San Diego, CA) with the standard Illumina procedure. Then, continuous ratios and discrete genotypes were included in our GWAS model in order to compare the p-values obtained by using the two methods. The resulting p-values are reported. Markers that did not pass the Illumina quality filter are indicated by NA. Supplementary material 4 (PDF 275 kb)
10681_2017_1939_MOESM5_ESM.xlsx
OR05. Phenotypes. BLUE values per accession for all the phenotypes analysed in this paper. Supplementary material 5 (XLSX 16 kb)
10681_2017_1939_MOESM6_ESM.csv
OR06. Genotypes. X and Y signals normalized by the standard procedure implemented in GenomeStudio (Illumina Inc., San Diego, CA). Supplementary material 6 (CSV 13332 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tumino, G., Voorrips, R.E., Morcia, C. et al. Genome-wide association analysis for lodging tolerance and plant height in a diverse European hexaploid oat collection. Euphytica 213, 163 (2017). https://doi.org/10.1007/s10681-017-1939-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-017-1939-8