
Abstract
Most cancers and in particular carcinomas metastasise via the lymphatics to draining lymph nodes from where they can potentially achieve systemic dissemination by invasion of high endothelial blood venules (HEVs) in the paracortex [1, 2]. Currently however, the mechanisms by which tumours invade and migrate within the lymphatics are incompletely understood, although it seems likely they exploit at least some of the normal physiological mechanisms used by immune cells to access lymphatic capillaries and traffic to draining lymph nodes in the course of immune surveillance, immune modulation and the resolution of inflammation [3, 4]. Typically these include directional guidance via chemotaxis, haptotaxis and durotaxis, adhesion to the vessel surface via receptors including integrins, and junctional re-modelling by MMPs (Matrix MetalloProteinases) and ADAMs (A Disintegrin And Metalloproteinases) [5,6,7]. This short review focusses on a newly emerging mechanism for lymphatic entry that involves the large polysaccharide hyaluronan (HA) and its key lymphatic and immune cell receptors respectively LYVE-1 (Lymphatic Vessel Endothelial receptor) and CD44, and outlines recent work which indicates this axis may also be used by some tumours to aid nodal metastasis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Article
The majority of immune cells that traffic from peripheral tissues to afferent lymphatics effect their entry within the first 200 μm of initial blind-ended capillaries at the distinctive overlapping junctions between the oakleaf shaped endothelial cells [5]. Here, and to a certain extent in adjoining lymphatic pre-collectors the interdigitating membrane flaps of such cells are “buttoned” at their sides by the adherens-junction protein VE-cadherin (Vascular Endothelial cadherin) and tight junction proteins including Claudin-5, ZO-1 (Zonula Occludens-1), ESAM (Endothelial Selective Adhesion Molecule) and JAM-A (Junctional Adhesion Molecule-A), while their tips are lined with PECAM-1 (Platelet Endothelial Cell Adhesion Molecule-1, CD31), and the lymphatic endothelial HA receptor LYVE-1 [5, 8,9,10] to form discrete junctional “portals” of approximately 0.5–1 μm through which individual immune cells can enter by pushing and squeezing [5, 11,12,13,14]. As predicted from its location in such portals, we recently demonstrated that LYVE-1 plays a key role in the entry of antigen presenting Dendritic cells (DCs) and macrophages to initial capillaries by engaging with its ligand HA, anchored in the surface glycocalyx of the incoming immune cells by the closely related leucocyte receptor CD44 [15,16,17]. This role of LYVE-1 has similarities to the one played by CD44 in mediating the exit of T cells from inflamed blood capillaries. There however, the HA glycocalyx is present on the (luminal) endothelium, where it serves to capture activated circulating T cells (which also express CD44 but lack an HA coat) by forming a CD44:HA:CD44 sandwich [18,19,20]. Synthesized primarily by HA synthase-2 (HAS2) in DCs and macrophages, the nascent HA chains associate with CD44 in intracellular vesicles, from which the bound complexes are then exported to the immune cell surface to form a glycocalyx of some 200-300 nm thickness as observed by high resolution (Airyscan) confocal microscopy [17]. Conclusive evidence that LYVE-1:HA interactions can be critical for vessel entry in vivo came from studies tracking the migration of fluorescently labelled DCs in oxazolone hypersensitized skin, which demonstrated that Lyve1 gene deletion or mAb blockade led to a peri-lymphatic accumulation of DCs in the dermis and a consequent delay or inhibition of their trafficking to downstream axial LNs respectively [16]. Likewise, deletion of CD44 or enzymatic depletion of the HA glycocalyx resulted in a marked reduction in DC nodal migration compared to controls. Moreover, the significance of LYVE-1 • HA mediated DC migration for in vivo immune function was underscored by experiments with mice immunised intradermally with influenza virus nucleoprotein or ovalbumin peptide as model vaccines. These showed that both Lyve1 gene deletion and mAb blockade disrupted the generation of DC primed peptide-specific CD4 and CD8 T-cell responses in downstream lymph nodes, confirming that in vivo, the LYVE-1• HA mediated entry of immune cells to lymphatics can be rate-limiting for protective immunity ([16] and reviewed in Johnson et al. [14]). The critical importance of LYVE-1 for immune cell trafficking has been further reinforced by the discovery that the process is controlled by the Circadian clock gene BMAL1 which directly regulates the expression of LYVE-1, CCL21 and CCR7 in lymphatic endothelium to facilitate maximal DC migration during the sleeping hours, when the immune activity of lymph nodes is at its highest [21, 22].
As regards the precise molecular mechanism of HA-mediated entry, we have observed that contact with the DC glycocalyx triggers redistribution of LYVE-1 into ring-like assemblages termed transmigratory cups in the underlying endothelium, owing to their similarity to structures that form around T-cells during exit across blood vascular endothelium [23,24,25]. Coincident with engaging LYVE-1 in these cups, we reported that migrating DCs undergo polarisation to form a uropod at the trailing edge, in which CD44 is concentrated with its bound HA glycocalyx, and a leading edge or lamellipodium that extends to explore the surrounding endothelial surface for potential sites of transit, most likely at tri-cellular junctions [17]. Hence, the adhesive interface between HA and LYVE-1 likely provides a flexible foothold for the DC to attach to the outer surface of a lymphatic capillary while the actomyosin machinery provides the tractive force for transit to the vessel lumen either through amoeboid motility or adhesion to integrins at the leading edge, directionally guided by a haptotactic gradient of the chemokine CCL21 in the perilymphatic matrix via its signaling receptor CCR7 [26]. Additionally, DCs can trigger localised discharge of CCL21 from lymphatic endothelium to guide their own transmigration, through Ca2+ triggered exocytosis of the chemokine from pre-stored depots in trans Golgi vesicles [27, 28]. Indeed, the LYVE-1 HA axis appears to regulate such discharge, insofar as engagement of DCs via LYVE-1 can induce CCL21 secretion from LECs, and release of CCL21 from dermal lymphatic capillaries is disrupted in Lyve1−/− mice. (Johnson and Jackson unpublished). Besides DCs, our ongoing investigations have confirmed that tissue macrophages also utilize their HA glycocalyx to mediate exit from inflamed tissue to afferent lymphatics via LYVE-1, notably during the phase of resolution that enables restoration of normal homeostasis in tissues such as the infarcted heart and peritoneum [29, 30].
Of course, the LYVE-1 HA axis is not the only one to play a role in lymphatic entry. With the exception of DCs and macrophages, most other immune cell populations appear to lack a HA glycocalyx, and must instead initiate lymphatic transit by other mechanisms. In the case of TEM and TREG cells for example, it has been reported that entry involves the chemotactic lipid Sphingosine 1-P along with lymphotoxin (LTα1β3) and its receptor LTβR [31, 32]. Neutrophils, the first responders to tissue injury can adhere to the vessel endothelium via β2 integrins and transmigrate through the combined actions of the matrix metalloproteinases MMP8 and MMP9, the serine protease neutrophil elastase and the arachidonate-derived chemorepellent, 12,hydroxyeicosatetraenoate (12(S)HETE) that together promote junctional retraction [33, 34]. Furthermore, it appears likely that both these populations, which migrate primarily in response to inflammation, can enter dermal lymphatics through downstream collectors which lack LYVE-1 and have conventional tight (zippered) endothelial junctions rather than buttoned junctions. Here, it has been reported that the process of entry is mediated by adhesion between integrins on the immune cells and VCAM-1 (Vascular Endothelial Adhesion Molecule – 1) and ICAM-1 (Intercellular Adhesion Molecule – 1) in the inflamed endothelium [35,36,37,38,39,40]. Notably, it was shown that such integrin dependent transit through downstream collectors is also used by DCs, most likely to accelerate their passage to downstream lymph nodes in inflamed tissues [35]. Indeed integrin-mediated adhesion may even combine with HA-mediated adhesion for DC entry at initial capillaries or pre-collectors, as both ICAM-1 and VCAM-1 have been observed to co-localise with LYVE-1 during transmigration via transmigratory cups [36, 37]. Hence, while the individual mechanisms used to initiate lymphatic entry can differ between immune cell types, they can also vary according to the tissue context. Moreover, the overall process actually involves a series of steps that likely involve contributions from a number of different adhesion receptors and their ligands including ALCAM (Activated Leukocyte Cell Adhesion Molecule), L1CAM, CD99, Mannose receptor, 41-BB/CD137 and CLEVER-1(Combined Lymphatic Endothelial and Vascular Endothelial Receptor − 1), as well as chemokines, chemoattractants and proteinases that together activate signaling pathways leading to the loss of VE-cadherin and junctional retraction [7, 41].
The use of the LYVE-1·HA adhesion axis by DCs and macrophages for lymphatic entry can be reconciled with the unusual mechanics of the binding interaction and its tissue context. Unlike CD44, to which individual HA chains bind through conventional ”sticking” interactions, the HA chains can “slide” through the cleft-like binding site in LYVE-1, a process that likely equips DCs and macrophages for their fluid-like crawling on lymphatic endothelium and subsequent transmigration in the low shear environment of initial lymphatics [10, 42]. Likewise, the absence of an HA glycocalyx from T cells and neutrophils likely reflects the fact they reside mainly in the blood circulation where the firmer binding properties of CD44 enable their extravasation at sites of tissue injury by adhesion to endothelial HA in the face of rapid blood flow.
The HA pericellular coat in tumours - a potential role in nodal metastasis?
Besides being a feature of tissue based immune cell populations as already discussed, the assembly of an HA glycocalyx or pericellular coat is also a property of other less motile cell types such as fibroblasts, chondrocytes and epithelial cells, where the large polyanionic nature of the sugar polymer acts as a barrier modulating cell:cell contact, as well as a means of tissue anchorage via proteoglycans in the extracellular matrix [43]. Importantly, the HA pericellular coat can be radically altered during cell transformation. Notably, its expansion through upregulation of HAS gene expression and HA deposition are hallmarks of epithelial to mesenchymal transition [44], a fundamental process in carcinoma progression by which tumour cells acquire enhanced migratory capacity [45]. Furthermore, elevated levels of both HA and CD44 are common amongst “aggressive” and metastatic cancers and associated with poor survival [46, 47].
There is now growing evidence to suggest this tumour cell HA coat can contribute to metastasis by facilitating lymphatic invasion of either individual tumour cells or tumour emboli via LYVE-1, perhaps in a similar manner to the trafficking of immune cells. Most if not all tumours grow in close proximity to host lymphatics or generate new peritumoral or even intratumoral lymphatics, both of which have LYVE-1 lined endothelial junctions. Moreover, in immunostained paraffin tissue sections of breast cancer, a CD44-associated pericellular HA matrix has been observed on tumour emboli invading such lymphatic vessels [48], while in vitro cultured breast tumour lines have been reported to adhere via HA to LYVE-1 in transfected fibroblasts [49]. Extending upon these observations, our own more recent studies indicate that assembly of a dense CD44 anchored HA glycocalyx is a consistent feature of metastatic MDA MB 231 and MCF-7 breast carcinomas but not poorly metastasizing SKBR3 cells, and that MDA MB 231 and MCF-7 cells both adhere and transit human LEC monolayers via LYVE-1 transmigratory cups similar to those formed by transmigrating DCs (Johnson and Jackson unpublished). Curiously, such findings chime with those from a previous ultrastructural study in mice where detailed electron microscopic imaging suggested that prostate adenocarcinomas, colon carcinomas and B16 melanomas extend filopodia-like protrusions towards lymphatic vessels during invasion, and that the endothelial cells simultaneously extend transmigratory cup-like projections around the transiting tumour cells [50]. Moreover, in further studies with the mouse 4T1 mammary carcinoma line that can spread directly from the first draining lymph nodes to the blood circulation in Balb/c animals by invading HEVs [51, 52] we found the initial invasion of local lymphatics and metastasis to draining inguinal lymph nodes is both reliant on HA and LYVE-1 and severely impaired in Lyve1−/−mice (Johnson LA and Jackson DG unpublished). It is tempting to speculate that subsequent colonisation of these inguinal nodes by 4T1 as well as invasion of blood vessels for systemic dissemination of the tumour involves yet further interactions with LYVE-1, given its abundance in the sinuses that must be traversed to reach the HEVs; however, this remains to be investigated.
In summary, both tissue-based immune cells and metastatic tumours employ similar adhesive interactions between their endogenous HA surface glycocalyx and the endothelial receptor LYVE-1 to enter afferent lymphatics and migrate to downstream lymph nodes - the former to promote immunity and its resolution and the latter for systemic dissemination (Fig. 1). Accordingly, LYVE-1 is a potential target for the therapeutic blockade of both immune based disorders and cancer metastasis. Regarding the latter however, one important caveat is that lymphatic trafficking is pivotal to the generation of anti-tumour immunity and its disruption would therefore be undesirable. Hence, LYVE-1 blockade might be most effective for the treatment of early-stage cancers, when administered after primary tumour resection, prior to nodal dissemination and local immune activation. Further studies will no doubt help to clarify these issues.

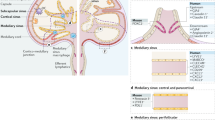
Involvement of LYVE-1 and hyaluronan glycocalyx in entry of immune cells and tumours to initial lymphatics. Pictorial images showing potential similarity between mechanisms used by normal immune cells and tumour cells to enter lymphatic capillaries by means of LYVE-1 and hyaluronan glycocalyx. Panels depict a blind-ended lymphatic in each case containing multiple LYVE-1 lined entry portals (red stippling). Entry of dendritic cells (DC), macrophages (M) and tumour cells (T) involves clustering of the glycocalyx and formation of LYVE-1 enriched transmigratory cups that facilitate transit to the vessel lumen
Data Availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
Zhou H, Lei PJ, Padera TP (2021) Progression of Metastasis through Lymphatic System. Cells 10
Houvenaeghel G, Cohen M, Classe JM, Reyal F, Mazouni C, Chopin N, Martinez A, Darai E, Coutant C, Colombo PE et al (2021) Lymphovascular invasion has a significant prognostic impact in patients with early breast cancer, results from a large, national, multicenter, retrospective cohort study. ESMO Open 6:100316
Tamburini BAJ, Padera TP, Lund AW (2019) Editorial: regulation of Immune function by the lymphatic vasculature. Front Immunol 10:2597
Fujimoto N, Dieterich LC (2021) Mechanisms and Clinical Significance of Tumor Lymphatic Invasion. Cells 10
Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, Vestweber D, Corada M, Molendini C, Dejana E et al (2007) Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med 204:2349–2362
Lammermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Soldner R, Hirsch K, Keller M, Forster R, Critchley DR, Fassler R et al (2008) Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 453:51–55
Jackson DG (2019) Leucocyte trafficking via the lymphatic vasculature - mechanisms and consequences. Front Immunol 10
Yao LC, Baluk P, Srinivasan RS, Oliver G, McDonald DM (2012) Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am J Pathol 180:2561–2575
Banerji S, Ni J, Wang SX, Clasper S, Su J, Tammi R, Jones M, Jackson DG (1999) LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J Cell Biol 144:789–801
Jackson DG (2019) Hyaluronan in the lymphatics: the key role of the hyaluronan receptor LYVE-1 in leucocyte trafficking. Matrix Biol 78–79:219–235
Pflicke H, Sixt M (2009) Preformed portals facilitate dendritic cell entry into afferent lymphatic vessels. J Exp Med 206:2925–2935
Tal O, Lim HY, Gurevich I, Milo I, Shipony Z, Ng LG, Angeli V, Shakhar G (2011) DC mobilization from the skin requires docking to immobilized CCL21 on lymphatic endothelium and intralymphatic crawling. J Exp Med 208:2141–2153
Zhang F, Zarkada G, Yi S, Eichmann A (2020) Lymphatic endothelial cell junctions: Molecular Regulation in Physiology and Diseases. Front Physiol 11:509
Johnson LA, Jackson DG (2021) Hyaluronan and Its Receptors: Key Mediators of Immune Cell Entry and Trafficking in the Lymphatic System. Cells 10
Lawrance W, Banerji S, Day AJ, Bhattacharjee S, Jackson DG (2016) Binding of Hyaluronan to the native lymphatic vessel endothelial receptor LYVE-1 is critically dependent on receptor clustering and Hyaluronan Organization. J Biol Chem 291:8014–8030
Johnson LA, Lawrance W, Roshorn YM, Hanke T, Banerji S, Jackson DG (2017) The lymphatic vessel endothelial receptor LYVE-1 mediates dendritic cell entry to the afferent lymphatics via transmigratory cups that engage the leukocyte hyaluronan glycocalyx. Nat Immunol 18:762–770
Johnson LA, Banerji S, Lagerholm BC, Jackson DG (2021) Dendritic cell entry to lymphatic capillaries is orchestrated by CD44 and the hyaluronan glycocalyx. Life Sci Alliance 4. https://doi.org/10.26508/lsa.202000908
DeGrendele HC, Estess P, Picker LJ, Siegelman MH (1996) CD44 and its ligand hyaluronate mediate rolling under physiologic flow: a novel lymphocyte-endothelial cell primary adhesion pathway. J Exp Med 183:1119–1130
Nandi A, Estess P, Siegelman MH (2000) Hyaluronan anchoring and regulation on the surface of vascular endothelial cells is mediated through the functionally active form of CD44. J Biol Chem 275:14939–14948
Petrey AC, de la Motte CA (2014) Hyaluronan, a crucial regulator of inflammation. Front Immunol 5:101
Holtkamp SJ, Ince LM, Barnoud C, Schmitt MT, Sinturel F, Pilorz V, Pick R, Jemelin S, Muhlstadt M, Boehncke WH et al (2021) Circadian clocks guide dendritic cells into skin lymphatics. Nat Immunol 22:1375–1381
Wang C, Barnoud C, Cenerenti M, Sun M, Caffa I, Kizil B, Bill R, Liu Y, Pick R, Garnier L et al (2023) Dendritic cells direct circadian anti-tumour immune responses. Nature 614:136–143
Carman CV, Springer TA (2004) A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol 167:377–388
Millan J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ (2006) Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol 8:113–123
Nieminen M, Henttinen T, Merinen M, Marttila-Ichihara F, Eriksson JE, Jalkanen S (2006) Vimentin function in lymphocyte adhesion and transcellular migration. Nat Cell Biol 8:156–162
Weber M, Hauschild R, Schwarz J, Moussion C, de Vries I, Legler DF, Luther SA, Bollenbach T, Sixt M (2013) Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science 339:328–332
Vaahtomeri K, Brown M, Hauschild R, De Vries I, Leithner AF, Mehling M, Kaufmann WA, Sixt M (2017) Locally triggered release of the chemokine CCL21 promotes dendritic cell transmigration across lymphatic endothelia. Cell Rep 19:902–909
Vaahtomeri K, Moussion C, Hauschild R, Sixt M (2021) Shape and function of interstitial chemokine CCL21 gradients are Independent of Heparan Sulfates produced by lymphatic endothelium. Front Immunol 12:630002
Vieira JM, Norman S, Del Villa C, Cahill TJ, Barnette DN, Gunadasa-Rohling M, Johnson LA, Greaves DR, Carr CA, Jackson DG et al (2018) The cardiac lymphatic system stimulates resolution of inflammation following myocardial infarction. J Clin Invest 128:3402–3412
Ravaud C, Ved N, Jackson DG, Vieira JM, Riley PR (2021) Lymphatic clearance of Immune cells in Cardiovascular Disease. Cells 10
Brinkman CC, Iwami D, Hritzo MK, Xiong Y, Ahmad S, Simon T, Hippen KL, Blazar BR, Bromberg JS (2016) Treg engage lymphotoxin beta receptor for afferent lymphatic transendothelial migration. Nat Commun 7:12021
Piao W, Xiong Y, Famulski K, Brinkman CC, Li L, Toney N, Wagner C, Saxena V, Simon T, Bromberg JS (2018) Regulation of T cell afferent lymphatic migration by targeting LTbetaR-mediated non-classical NFkappaB signaling. Nat Commun 9:3020
Hampton HR, Bailey J, Tomura M, Brink R, Chtanova T (2015) Microbe-dependent lymphatic migration of neutrophils modulates lymphocyte proliferation in lymph nodes. Nat Commun 6:7139
Rigby DA, Ferguson DJ, Johnson LA, Jackson DG (2015) Neutrophils rapidly transit inflamed lymphatic vessel endothelium via integrin-dependent proteolysis and lipoxin-induced junctional retraction. J Leukoc Biol 98:897–912
Arasa J, Collado-Diaz V, Kritikos I, Medina-Sanchez JD, Friess MC, Sigmund EC, Schineis P, Hunter MC, Tacconi C, Paterson N et al (2021) Upregulation of VCAM-1 in lymphatic collectors supports dendritic cell entry and rapid migration to lymph nodes in inflammation. J Exp Med 218. https://doi.org/10.1084/jem.20201413
Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG (2006) An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med 203:2763–2777
Teijeira A, Garasa S, Pelaez R, Azpilikueta A, Ochoa C, Marre D, Rodrigues M, Alfaro C, Auba C, Valitutti S et al (2013) Lymphatic endothelium forms integrin-engaging 3D structures during DC transit across inflamed lymphatic vessels. J Invest Dermatol 133:2276–2285
Hunter MC, Teijeira A, Halin C (2016) T cell trafficking through lymphatic vessels. Front Immunol 7:613
Teijeira A, Hunter MC, Russo E, Proulx ST, Frei T, Debes GF, Coles M, Melero I, Detmar M, Rouzaut A et al (2017) T Cell Migration from Inflamed skin to draining Lymph Nodes requires Intralymphatic crawling supported by ICAM-1/LFA-1 interactions. Cell Rep 18:857–865
Vigl B, Aebischer D, Nitschke M, Iolyeva M, Rothlin T, Antsiferova O, Halin C (2011) Tissue inflammation modulates gene expression of lymphatic endothelial cells and dendritic cell migration in a stimulus-dependent manner. Blood 118:205–215
Arasa J, Collado-Diaz V, Halin C (2021) Structure and Immune Function of Afferent Lymphatics and Their Mechanistic Contribution to Dendritic Cell and T Cell Trafficking. Cells 10
Bano F, Banerji S, Howarth M, Jackson D, Richter R (2016) A single molecule assay to probe monovalent and multivalent bonds between hyaluronan and its key leukocyte receptor CD44 under force. Sci Rep 6:34176
Toole BP (2004) Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer 4:528–539
Camenisch TD, Schroeder JA, Bradley J, Klewer SE, McDonald JA (2002) Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2-ErbB3 receptors. Nat Med 8:850–855
Lu W, Kang Y (2019) Epithelial-mesenchymal plasticity in Cancer Progression and Metastasis. Dev Cell 49:361–374
Setala LP, Tammi MI, Tammi RH, Eskelinen MJ, Lipponen PK, Agren UM, Parkkinen J, Alhava EM, Kosma VM (1999) Hyaluronan expression in gastric cancer cells is associated with local and nodal spread and reduced survival rate. Br J Cancer 79:1133–1138
Ropponen K, Tammi M, Parkkinen J, Eskelinen M, Tammi R, Lipponen P, Agren U, Alhava E, Kosma VM (1998) Tumor cell-associated hyaluronan as an unfavorable prognostic factor in colorectal cancer. Cancer Res 58:342–347
Williams CS, Leek RD, Robson AM, Banerji S, Prevo R, Harris AL, Jackson DG (2003) Absence of lymphangiogenesis and intratumoural lymph vessels in human metastatic breast cancer. J Pathol 200:195–206
Du Y, Liu Y, Wang Y, He Y, Yang C, Gao F (2011) LYVE-1 enhances the adhesion of HS-578T cells to COS-7 cells via hyaluronan. Clin Invest Med 34:E45–54
Azzali G (2006) On the transendothelial passage of tumor cell from extravasal matrix into the lumen of absorbing lymphatic vessel. Microvasc Res 72:74–85
Brown M, Assen FP, Leithner A, Abe J, Schachner H, Asfour G, Bago-Horvath Z, Stein JV, Uhrin P, Sixt M et al (2018) Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science 359:1408–1411
Padera TP, Kadambi A, di Tomaso E, Carreira CM, Brown EB, Boucher Y, Choi NC, Mathisen D, Wain J, Mark EJ et al (2018) Lymphatic metastasis in the absence of functional intratumor lymphatics. Science 296:1883–1886
Funding
This work was funded by the UK Medical Research Council under Core Grant MC_UU_00008/2.
Author information
Authors and Affiliations
Contributions
Not applicable. Single author.
Corresponding author
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Presented at the 9th International Congress on Cancer Metastasis through the Lymphovascular System, May 4–6, 2023, in San Francisco, CA. To be published in a Special Issue of Clinical and Experimental Metastasis: Molecular Mechanisms of Cancer Metastasis.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jackson, D.G. Lymphatic trafficking of immune cells and insights for cancer metastasis. Clin Exp Metastasis (2023). https://doi.org/10.1007/s10585-023-10229-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10585-023-10229-3