
Abstract
Introduction
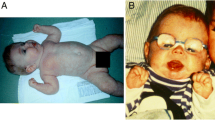
Alpha-1,3-glucosyltransferase congenital disorder of glycosylation (ALG6-CDG) is a congenital disorder of glycosylation. The original patients were described with hypotonia, developmental disability, epilepsy, and increased bleeding tendency.
Methods
Based on Euroglycan database registration, we approached referring clinicians and collected comprehensive data on 41 patients.
Results
We found hypotonia and developmental delay in all ALG6-CDG patients and epilepsy, ataxia, proximal muscle weakness, and, in the majority of cases, failure to thrive. Nine patients developed intractable seizures. Coagulation anomalies were present in <50 % of cases, without spontaneous bleedings. Facial dysmorphism was rare, but seven patients showed missing phalanges and brachydactyly. Cyclic behavioral change, with autistic features and depressive episodes, was one of the most significant complaints. Eleven children died before the age of 4 years due to protein losing enteropathy (PLE), sepsis, or seizures. The oldest patient was a 40 year-old Dutch woman. The most common pathogenic protein alterations were p.A333V and p.I299Del, without any clear genotype–phenotype correlation.
Discussion
ALG6-CDG has been now described in 89 patients, making it the second most common type of CDG. It has a recognizable phenotype and a primary neurologic presentation.


Similar content being viewed by others


Abbreviations
- ALG6:
-
Alpha-1,3-glucosyltransferase
- CDG:
-
Congenital disorder of glycosylation
- TIEF:
-
Transferrin isoelectric focusing
- PLE:
-
Protein-losing enteropathy
References
Al-Owain M, Mohamed S, Kaya N, Zagal A, Matthijs G, Jaeken J (2010) A novel mutation and first report of dilated cardiomyopathy in ALG6-CDG (CDG-Ic): a case report. Orphanet J Rare Dis 5:7
Dercksen M, Crutchley AC, Honey EM, Lippert MM, Matthijs G, Mienie LJ, Schuman HC, Vorster BC, Jaeken J (2013) ALG6-CDG in South Africa: genotype-phenotype description of five novel patients. JIMD Rep 8:17–23
Drijvers JM, Lefeber DJ, de Munnik SA, Pfundt R, van de Leeuw N, Marcelis C, Thiel C, Koerner C, Wevers RA, Morava E (2010) Skeletal dysplasia with brachytelephalangy in a patient with a congenital disorder of glycosylation due to ALG6 gene mutations. Clin Genet 77:507–509
Goreta SS, Dabelic S, Pavlinic D, Lauc G, Dumic J (2012) Frequency determination of α-1,3 glucosyltransferase p.Y131H and p.F304S polymorphisms in the croatian population revealed five novel single nucleotide polymorphisms in the hALG6 gene. Genet Test Mol Biomark 16:50–53
Ichikawa K, Kadoya M, Wada Y, Okamoto N (2013) Congenital disorder of glycosylation type Ic: report of a Japanese case. Brain Dev 35:586–589
Jaeken J, Lefeber D, Matthijs G (2015) Clinical utility gene card for: ALG6 defective congenital disorder of glycosylation. Eur J Hum Genet 23. doi: 10.1038/ejhg.2014.146
Lefeber DJ, Morava E, Jaeken J (2011) How to find and diagnose a CDG due to defective N-glycosylation. J Inherit Metab Dis 34:849–852
Miller BS, Freeze HH, Hoffmann GF, Sarafoglou K (2011) Pubertal development in ALG6 deficiency (congenital disorder of glycosylation type Ic). Mol Genet Metab 103:101–103
Scott K, Gadomski T, Kozicz T, Morava E (2014) Congenital disorders of glycosylation: new defects and still counting. J Inherit Metab Dis 37:609–617
Vuillaumier-Barrot S, Le Bizec C, Durand G, Seta N (2001) The T911C (F304S) substitution in the human ALG6 gene is a common polymorphism and not a causal mutation of CDG-Ic. J Hum Genet 46:547–548
Westphal V, Murch S, Kim S, Srikrishna G, Winchester B, Day R, Freeze HH (2000) Reduced heparan sulfate accumulation in enterocytes contributes to protein-losing enteropathy in a congenital disorder of glycosylation. Am J Pathol 157:1917–1925
Westphal V, Xiao M, Kwok PY, Freeze HH (2003) Identification of a frequent variant in ALG6, the cause of Congenital Disorder of Glycosylation-Ic. Hum Mut 22:420–421
Acknowledgments
The authors are thankful for the Euroglycanet and EURO-CDG networks
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
All authors were compliant and followed the ethical guidelines, according to the requirements of JIMD
Conflict of interest
None.
Additional information
Communicated by: Marc Patterson
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Mutations described in alpha-1,3-glucosyltransferase 6 (ALG6) according to the Exome Aggregation Consortium (ExAC) database. Missense, splice site, and frameshift mutations (178 in total) were ranked by allele frequency. (The 3 or 5′ untranslated region and intronic and silent variants were filtered out.) The three common variants in our patients are highlighted. They rank 9th, 22nd, and 107th for the c.257 + 5G > A, p.A333V, and p.I299Del respectively. (XLSX 40 kb)
Rights and permissions
About this article

Cite this article
Morava, E., Tiemes, V., Thiel, C. et al. ALG6-CDG: a recognizable phenotype with epilepsy, proximal muscle weakness, ataxia and behavioral and limb anomalies. J Inherit Metab Dis 39, 713–723 (2016). https://doi.org/10.1007/s10545-016-9945-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-016-9945-x