
Abstract
Objective
Dynamic contrast enhanced (DCE)-MRI is currently not generally used for intraocular masses as lesions are small, have an inhomogeneous T1 and the eye is prone to motion. The aim of this paper is to address these eye-specific challenges, enabling accurate ocular DCE-MRI.
Materials & methods
DCE-MRI of 19 uveal melanoma (UM) patients was acquired using a fat-suppressed 3D spoiled gradient echo sequence with TWIST (time-resolved angiography with stochastic trajectories sequence). The analysis consisted of a two-step registration method to correct for both head and eye motion. A T1 map was calculated to convert signal intensities to concentrations. Subsequently, the Tofts model was fitted voxel wise to obtain Ktrans and ve.
Results
Registration significantly improved the concentration curve quality (p < 0.001). The T1 of melanotic lesions was significantly lower than amelanotic lesions (888 ms vs 1350 ms, p = 0.03). The average achieved B1+ in the lesions was 91%. The average Ktrans was 0.46 min−1 (range 0.13–1.0) and the average ve was 0.22 (range 0.10–0.51).
Conclusion
Using this eye-specific analysis, DCE of intraocular masses is possible which might aid in the diagnosis, prognosis and follow-up of UM.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Most intraocular lesions are benign, such as choroidal neavi, haemangiomas and leiomyomas, but also various malignant intraocular masses exist. Although uveal melanoma (UM) is relatively rare, with an incidence of four to six per million in central Europe [1], it is the most common primary intraocular tumour. It mostly originates from the choroid (90%), but can also originate from the iris or ciliary body [2, 3]. Other malignant ocular lesions include mainly metastases from other tumour sites or even more rare lesions such as retinoblastoma [4, 5]. As the prognosis and treatment of benign lesions, UM and other malignant ocular lesions differ, it is important to have an accurate diagnosis [2, 5]. For the differentiation between these different lesions the ophthalmologist primarily relies on fundoscopic, fluorescent angiography and ultrasound imaging (Fig. 1a–d) [6]. However, in some patients, this differentiation is quite challenging, especially for amelanotic melanomas, or lesions behind the iris.

Conventional ophthalmic imaging and MRI of patient 13. A–D Conventional ophthalmic imaging of an UM. The fundus photo (A) shows a pigmented lesion with lipofuscin (arrow). The lesion is enhancing with pinpoints (C, arrow) on the fluorescent angiography (FAG, B, C). On ultrasound (US, D), the lesion (dagger) has an intermediate reflectivity, while the retinal detachment (double dagger) has a low reflectivity and the dimensions of the lesion are measured. On MRI, an enhancing lesion (dagger) with associated retinal detachment (double dagger) is visible (E–G). In contrast to the FAG, the change of the signal intensity after contrast administration can be visualized in DCE-MRI (H)
In the last decade, advances in ocular MRI, such as dedicated receive coils [7] and dedicated acquisition strategies [8] have resulted in different new clinical applications of MRI for ocular conditions [9]. MRI offers a superior evaluation of the extend of eye lid tumours [10], can be instrumental in the diagnosis and assessment of disease progression in orbital disease involving extra-ocular muscles [11], provide insight into ocular complaints such as negative dysphotopsia [12] and allows for a more accurate assessment of tumour dimensions for radiotherapy therapy planning [13, 14]. Furthermore, diffusion-weighted imaging is emerging as a promising early marker of therapy response after ocular proton-beam therapy [15], while quantitative dynamic contrast-enhanced MRI (DCE-MRI) could assist in the differential diagnosis of intraocular masses and monitoring of treatment response of UM [16]. The use of DCE-MRI of ocular masses is often limited to the evaluation of the time intensity curve (TIC, Fig. 1f–h) [16,17,18,19]. However, Wei et al. [17] and Kamrava et al. [20] have shown contradicting results on the relation between tumour permeability and metastatic risk. Kamrava et al. [20] found a higher Ktrans in UM patients with monosomy 3, a subset of UM patients who have a strong increased risk of developing metastatic disease [21]. Conversely Wei et al. [17] showed a decreased Ktrans (a lower peak signal intensity) in patients with metastatic disease. Although these papers are an improvement compared to the current clinical practice and other research where some eye-related challenges were not addressed such as small lesion size, eye motion and difference in melanin content.
The limited use of quantitative DCE-MRI might be due to the eye-specific challenges of MR imaging in general. One of the main challenges of DCE-MRI of intraocular masses is the small size of the eye, containing even smaller lesions, generally with a thickness of less than 5 mm. Furthermore, the eye is prone to movement, which in combination with the small lesion size leads to a mismatch of the tumour location between timepoints [18]. Finally, intraocular lesions can be pigmented (large amount of melanin), unpigmented (no melanin) or partially pigmented [22]. This varying degree of pigmentation results in a large variability in pre-contrast longitudinal relaxation time (T1) [23], which directly affects the quantification of concentration of the, generally T1-based, contrast agent. However, recent improvements in ocular MR-imaging protocols such as the use of a surface coil for receiving the signal and implementation of time-resolved angiography with stochastic trajectories sequence [24] allow for the acquisition of DCE-MRI with sufficient temporal and spatial resolution to perform DCE-MRI [18]. The aim of this paper is to overcome eye-specific challenges in the DCE-MRI analysis of intraocular masses.
Methods
Study population
Nineteen patients diagnosed with UM were included for this study. Nine patients were scanned as part of a prospective study and were recruited randomly. This study has been approved by the local ethics committee and subjects were scanned after written informed consent. The data from the remaining ten patients were selected from UM patients with a tumour prominence > 3 mm who received an MRI as part of clinical care. This retrospective inclusion of data was approved by the local ethics committee. The patients were on average 63 years old (range 30–82 years), 68% (n = 13) were male and had a BWI of 26.6 ± 4.5. The lesions had an average prominence of 7.8 mm and an average largest basal diameter of 14.5 mm on ultrasound. The American Joint Committee on Cancer staging [25] ranged from T1 to T4 with most patients in the T3 (n = 8) and T4 (n = 6) stage. Most were primary tumours (18/19) but in one case (UM15), the patient had a large reoccurrence. The primary tumour was treated with ruthenium plaque therapy and was located at the other side of the eye. A detailed description of the cohort of patients can be found in Table 1.
MRI protocol
All patients were scanned before treatment on a 3T MR scanner (Ingenia, Philips Healthcare, the Netherlands) using the protocol described by Ferreira et al. [18] and a 47 mm diameter surface receive coil covering the affected eye (Fig. 2). The scan parameters of the relevant sequences are listed in Table 2. Patients were instructed not to wear eye makeup and the affected eye was taped shut. In the last six patients, a wet gauze was placed on top of the eye to minimize susceptibility artefacts. Patients were immobilized as much as possible using a radiotherapy head support (MaxSupport™ wide shaped, red variant, 117,000 HSSETW, Medeo, Schöftland, Switzerland) (Fig. 2).

Clinical setup for ocular MRI. A The patient was scanned using a 47 mm surface receive coil (arrow). This coil was positioned over the affected eye. The head was supported by a radiotherapy head support (arrowhead). B Clarification of the positioning of the eye coil on a phantom
The dynamic time series were acquired using a fat-suppressed 3D spoiled gradient echo sequence with a spatial resolution of 1.25 × 1.5 × 1.5 mm3. A bolus of 0.1 mmol/kg gadolinium (DOTAREM; Guerbet, Roissy CdG Cedex, France) was administered 6 s after the start of the scan followed by a 20 ml injection of isotonic saline, using a power injector with an injection rate of 2 ml/s. The first eight patients were scanned with a flip angle of 5° to match the then used 7 Tesla protocol, where the flip angle was limited to 5° due to SAR restrictions. The flip angle was increased to 13° for subsequent patients as this provided a more optimal contrast for the contrast agent concentrations in our patients as theoretically the optimal flip angle for our spoiled gradient echo sequence is between 13° and 16° assuming a T1 between 600 and 1500 ms [26]. Time-resolved angiography with stochastic trajectories sequence (TWIST) [24], with a central size of 25% and a peripheral density of 20%, was implemented to increase the temporal resolution to 2 s per dynamic scan to reduce motion artefacts per dynamic image. To determine the baseline lesion T1, a 3D spoiled gradient echo flip angle series, with flip angles of 2°, 5°, 9° and 15°, was acquired before the dynamic scan with the same field of view (FOV). Additionally, a B1+-map was acquired using the DREAM sequence [27]. Finally a post-contrast 3DT1-weighted scan (3DT1gd) was acquired for anatomical reference.
DCE analysis
The analysis of the DCE data consisted of four steps. First, the images of the dynamic scan, flip angle series, B1+-map and 3DT1gd scan were registered to the dynamic dataset and the lesion was segmented on the 3DT1gd image. Second, a T1 map was calculated using the flip angle series and B1+-map, which was subsequently used to calculate the gadolinium concentration for each lesion voxel in the dynamic scan. Finally, pharmacokinetic modelling was applied using the Tofts model [28].
Registration
All timepoints of the dynamic scan were rigidly registered to the 50th of the 125 timepoints in two steps using Elastix 4.9.0 [29]. The 50th timepoint was chosen as it had an intensity comparable to most timepoints, which was beneficial for automatic image registration. The first step consisted of registration of the full FOV to correct for head motion. Subsequently an eye-mask was created using an in-house build Mevislab network (3.0.2, MeVis Medical Solutions AG, Bremen, Germany [30]). The eye-mask was used for a masked registration to reduce eye motion between timepoints. Additionally, the variable flip angle series, B1+-map and 3DT1gd scan were rigidly registered to the 50th timepoint using masked registration.
Segmentation
A lesion mask was created by manually segmenting the UM on the 3DT1gd images using ITK-SNAP [31]. Elastix was used to translate this mask to the registered dynamic scan, using the earlier obtained transformation matrix. Subsequently, the voxels within this mask were selected for the pharmacokinetic analysis.
T1-mapping
The pre-contrast T1 value of each voxel was obtained from the flip angle series in Matlab (version R2019b, MathWorks, Natick, Massachusetts, USA) as described by Fram et al. and Gupta et al. [32, 33]. The flip angles were corrected according to the median achieved B1+ of the lesion. Subsequently, a masked 3D median filter with 26-connected components was applied to the T1 map to remove potential outliers within the lesion. Voxels outside the lesion were excluded from the filter, as the vitreous has significantly higher T1 values than the lesion [34]. The UM were classified as melanotic, amelanotic or mixed based on description of the tumour in the medical status by an ophthalmologist.
Pharmacokinetic modelling
The signal intensities from the DCE images were converted to concentration time profiles, using the relations described by Tofts [28], assuming a gadolinium relaxivity of 3.4 L mmol−1 s−1 [35]. For each voxel, the peak concentration was defined as the 95th percentile of the concentration over time.
Voxel-by-voxel pharmacokinetic modelling (PKM) was performed using nonlinear least squares fitting of the standard Tofts model using in-house build scripts in Matlab. First, the bolus arrival time (BAT) was determined for each patient by fitting the PKM for the first 40 time points of the median lesion concentration curve for 25 different BATs. The BAT with the lowest residuals was selected. The automatic demined BAT was visually correct in 50% of the patients. In the remaining patients, the BAT was shifted with one timepoint in eight cases and two timepoints in three cases. Subsequently, the PKM was fitted to the full dynamic concentration curve (C(t)) for each voxel within the lesion to obtain the Ktrans (vascular permeability [min−1]) and ve (extravascular extracellular space per volume of tissue [unitless]). As no major feeding arteries were in the field of view of the DCE scan, an earlier derived population arterial input function (AIF) was used, which was derived from the carotid arteries in ten brain cancer patients (supplementary Fig. 1).
Evaluation of the registration
The effect of the registration was evaluated by comparing the concentration curves before and after registration of 15 randomly selected lesion voxels for all patients. The curves from all patients were randomized and presented unannotated to prevent a potential bias. The observer scored each curve as being sufficient or insufficient for automatic fitting and scored which of the two curves had the best quality or whether the quality was the same based on the amount of visual variance/spikes and motion artefacts in the concentration curve. Observer 1 (MJ) scored all 285 curves and observer 2 (JWB) scored a random subset of 50 to validate the scores. The scores were evaluated using a two-sided Wilcoxon signed rank test.
Evaluation of the error propagation
The effect of the precision error of the B1+, T1 and registration on the pharmacokinetic were assessed. First, in two additional UM patients, the flip angle series and B1+ map were acquired twice to determine the repeatability of the B1+ and T1 measurements. Second, the effect precision errors in B1+, T1 and registration on Ktrans and ve was assessed to determine the sensitivity of small inaccuracies of the different analysis steps on the final DCE parameters. To this end, the results of different intermediate steps were manually modified in two different patients: a patient with a medium-sized amelanotic tumour with a B1+ of 82% (patient 7) and with a large melanotic tumour and a B1+ of 96% (patient 10). The effect of a precision error in B1+ measurement was assessed by increasing and decreasing the measured B1+ with 2 and 5%. The sensitivity to T1 changes was determined by changing the measured T1 for all voxels by 30 ms, the measured precision error for the T1 mapping, or 60 ms, and 2 and 5% of the average tumour T1, respectively. Finally, the effect of imprecise registration was estimated by artificially shifting the images of individual timepoints. Regular eye motion was assessed by a one voxel shift during 2 timepoints after 175 s, while the most unfavorable case was assessed by the same shift but exactly after bolus arrival. For all cases, the median Ktrans and ve of the tumor were compared with the original analyses.
Statistical analysis
The impact of the different analysis steps on the final PKM was evaluated. The Ktrans values with and without registration and with and without B1+-correction were visualized. The effect of T1-mapping was evaluated by comparing the Ktrans values based on concentration data with the average T1 of all patients with the model in which data from the individual T1 map was used. The difference in Ktrans between melanotic and amelanotic lesions for a population T1 and individual T1 was tested using unpaired t tests.
The reported PKM values in this paper are the median of the voxels within the lesion mask as the values are not normally distributed. The error bars shown in the figures are the 25th and 75th percentile (IQR). The reported unpaired t tests were calculated using Matlab. A p value of 0.05 or smaller was considered statistically significant.
Results
Registration
The centre of the eye moved up to 3.0 mm (average 1.3 mm) during the 4 min acquisition of the dynamic time series with rotations of up to 20° (average 6°) with respect to the first time point.
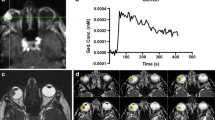
Registration resulted in a significant improvement in the quality of the concentration curves (Z = 8.9, p < 0.001). Figure 3 shows a representative curve before and after registration. Eye motion can result in changes in the enhancement and concentration curves, as even small eye motion can result in mismatch between the ROI and actual lesion location, as can be seen in Fig. 3. Without registration, there were spikes (Fig. 3, asterisk) in the contrast agent concentration, which were caused by motion, most likely eye blinks or a different gaze angle. In this case, most of the outflow was no longer apparent after registration of the eye, although still some residual motion artefacts can be seen in the concentration curve. Note that eye motion was not resolved with the unmasked registration.

Registration. A, B Concentration time curves of a representative patient (18) before and after registration. Before registration motion artefacts were present (asterisk); furthermore, unmasked registration was not able to fully resolve the motion. A movie of the dynamic scan of patient 18 visualizing the motion in the scan can be found in the supplementary materials. C Registration changes the Ktrans of the UM patients with an average of 0.06 min−1
Before registration, 40% (n = 113/285) of the curves were scored as insufficient quality to perform a fit compared to 15% (n = 43/285) after registration. In 55% of the curves (n = 157/285), the curve was scored as improved, in 36% (n = 102/285) of the curves, no clear effect of the registration was observed, while in 9% (n = 26/285), the curve was scored as deteriorated after registration, although in 15/26 of these curves, the curves received the same quality, indicating a minimal difference. No significant difference in scoring was found between the scoring of observer 1 and 2 (p = 0.09).
The reported increase in quality of the concentration curves after registration resulted in a change of the PKM parameter values. The maximum absolute change in Ktrans was 0.25 min−1 with an average absolute change of 0.06 min−1 (Fig. 3c; Table 3).
B1 + and T1-mapping
The median lesion T1 per patient showed a wide range from 522 to 1509 ms as is shown in Fig. 4b. The average T1 of all patients was 1122 ms, this value was used as population T1 for subsequent comparisons on the effect of T1 on the PKM. Amelanotic lesions had an average T1 of 1350 ms, while the average T1 of 888 ms for the melanotic lesions was significantly lower (p = 0.03). The average T1 of the mixed lesions was 1193 ms.

T1-mapping. A Slice of UM patient 10 showing an inhomogeneous T1 of the lesion. The retinal detachment (Asterix) is excluded from the analysis. B T1 values of amelanotic, mixed, melanotic and non-UM lesions (median and IQR). The three melanotic lesions with a higher T1 are small lesions most likely suffering from partial volume effects. The T1 between amelanotic and melanotic lesions is significantly different (p = 0.03). No significant difference was found between mixed lesions and either amelanotic or melanotic lesions. B, C. B1+-correction change the Ktrans with an average of 0.11 min−1. D When a population T1 was used the Ktrans mainly resembled the amount of pigmentation with a low Ktrans for melanotic lesions. When the actual T1 was used the Ktrans changed with an average of 0.15 min−1
The average achieved B1+ in the lesion was 91% (range 77–104%). Correction of the flip angles resulted in an average absolute change in T1 of 208 ms and an absolute change in Ktrans of 0.11 min−1, (Fig. 4c; Table 3).
When a population average T1 was used the Ktrans appeared to mainly resemble the amount of pigmentation, with the melanotic showing a significant lower Ktrans compared to amelanotic lesions (p < 0.01, Fig. 4d). However, when the actual T1 was included in the analysis, the Ktrans changed with 0.15 min−1 on average (Fig. 4d; Table 3) and the bias was resolved as no systematic difference was found between the Ktrans of melanotic and amelanotic lesions (p = 0.37).
The strong effect of the melanin concentration on the perfusion quantification can be seen in a mixed lesion with both a melanotic and amelanotic lobe, (Fig. 5). On the TIC, the amelanotic part of the lesion appeared to be enhancing stronger than the melanotic part, 225% vs 150%. The melanotic lobe has, however, an almost 1000 ms shorter T1 than the amelanotic lobe, on average 494 ms vs 1464 ms, respectively. When the T1 was included in the conversation to concentration, a very similar concentration was found in both lesions, although still a higher peak concentration was measured in the early timepoints of the amelanotic lobe.

The effect of T1 on the signal intensity. Bilobar lesion with both an amelanotic and a melanotic lobe. The amelanotic part of the lesion appears to be enhancing stronger than the melanotic part. When, however, the actual T1 was included in the calculation of the contrast agent concentration, a very similar concentration was found in both lesions, although still a distinct difference was present in the early timepoints
Pharmacokinetic parameters
When all corrections were applied, a wide range of Ktrans values was observed (Fig. 6c). The median Ktrans per lesion ranged from 0.13 to 1.0 min−1 with a mean of 0.46 min−1. The median ve was 0.22 on average with a range from 0.10 to 0.51.

Pharmacokinetic modelling results. A Concentration curve and fit of two voxels of patient 10 showing the inhomogeneity of the lesion. B A sagittal slice of UM patient 10 showing Ktrans and ve maps showing an inhomogeneous values of Ktrans and ve in the lesion. C, D The median and IQR of the Ktrans and ve in the lesion. All patients except one UM had a ve lower than 0.3
Within lesions, a wide distribution of Ktrans and ve was observed. The maximum IQR (75th–25th percentile) of the Ktrans was 0.99 min−1 and the average IQR was 0.40 min−1. For the ve, the maximum IQR was 0.83 and the average IQR was 0.18.
Error propagation
The average precision error of the achieved B1+ and T1 was 0.2% and 30 ms, respectively. Major inaccuracies (5%) in the measurement of the achieved B1+ can result in a difference up to 12% in the determined Ktrans and 19% in the determined ve (Table 4). Differences in the T1 measurement (30 ms) can lead to changes in Ktrans and ve up to 5% (Table 4). Imprecisions in the registration during the second half of the acquisition had a minimal effect on the outcomes of Ktrans and ve (< 1%), while a similar imprecisions directly after contrast uptake resulted in 3% change in Ktrans.
Discussion
Recent developments in ocular MR imaging allow for the acquisition of DCE images with sufficient temporal and spatial resolution to perform DCE-MRI in the eye, as the currently achievable isotropic spatial resolution of ~ 1 mm is sufficient to assess the smaller intraocular lesions, while the 2 s temporal resolution yields more than sufficient time points to determine the inflow characteristics of the lesion [18]. With this improved protocol, DCE-MRI was performed in 19 intraocular lesions and the effect of eye-specific challenges on the quantification was investigated.
As the eye can rotate within the head, registering the complete FOV of all time points is not sufficient to correct for eye motion. Therefore, a dedicated registration method was developed to mitigate the effect of gazing variations on the measured concentration curve. These corrections resulted in absolute changes in Ktrans up to 0.25 min−1. Although rigid registration of the complete Field-of-View has been proposed to correct for motion of intraocular lesions [20], we argue that is not sufficient, since the eye moves within the head. We showed that masked-registration improved the quality of the curves significantly, however, still the quality of 15% of the concentration curves was scored as insufficient to perform an automatic fit on. Smaller lesions had a higher percentage of insufficient quality voxels indicating that our registration cannot fully resolve effects of motion at the edge of the lesion. In 4% (n = 10/285) of the curves, across different subjects and tumour sizes, registration seemed to have deteriorated the quality at some timepoints, although the deterioration was minor compared to the improvement by the registration in the other voxels. This mostly occurred at the edge of the tumour and might be attributed to partial volume effects, although inhomogeneities in the tumour might play a role as well. A MRI protocol with an increased resolution might, therefore, also be favourable to decrease the effect of these residual errors in the registration. Although cued blinking might be implemented to reduce motion during acquisition, the resulting twofold reduction in temporal resolution will likely not be beneficial for the pharmacokinetic modelling [7, 8]. It might be beneficial to register all scans to the higher resolution 3DT1gd instead of one of the dynamic timepoints.
Only one paper [20] was found that used T1 mapping for the quantitative DCE analysis of ocular lesions. We found a significant difference in T1 between melanotic and amelanotic lesions and a strong effect on the quantification indicating that T1 mapping is a crucial step in DCE quantification. We, therefore, recommend to include the actual T1 in the analysis of DCE of ocular lesions to prevent the bias introduced by the amount of melanin in the tumour. The effect of melanin can be seen clearly in patients with a bilobar lesion, as shown in Fig. 5. A similar effect was observed between lesions, where amelanotic lesions appears to enhance more than melanotic lesions. This was, however, primarily the result of its longer T1 and not of an increased contrast agent concentration.
Finally, our results indicate that registration and B1+correction are important steps in the quantitative analysis of ocular DCE, but these steps affect the pharmacokinetic parameters to a lesser extent than the differences in T1, and are independent of the type of lesion. DCE scans are clinically often evaluated by assessing the Time Intensity Curves (TIC), instead of the actual gadolinium concentration which can be calculated from these intensities [16,17,18,19]. However, by comparing these TICs between lesions, the potential differences in T1 between these lesions are ignored, which can lead to erroneous interpretation of the data. A higher (maximum) relative enhancement or area under the curve might, for example, be interpreted as an increased perfusion in the lesion, but might actually be the result of a less pigmented lesion which has, therefore, a higher T1. As the overall shape of the TIC is not affected by the scaling effect of the T1, a classification based on the curve pattern can still be useful when only the signal intensity is available. [36, 37]
In line with earlier genetic and pathology studies [38,39,40,41], our results indicate that uveal melanomas are inhomogeneous, making a voxel wise volumetric assessment of ocular lesions preferred over a single 2D ROI analysis. The relatively thick slices of 3 mm used in the study of Kamrava, likely still resulted in representative sample of the lesion, due to averaging in the slice direction, resulting in the observed correlation between monosomy 3 and Ktrans. However, a lower resolution limits the possibility for motion correction and analysis of small intraocular lesions, making a higher resolution with a voxel wise analysis preferred. Overall, ROI-based (semi-)quantitative DCE-MRI analysis without T1 mapping for ocular lesions is not recommended as this would result in a significant bias as most DCE measures are dependent on the baseline T1.
There are areas in which ocular DCE can be further improved. First of all, the flip angle of the DCE-MRI acquisition was 5° for the first eight patients and 13° for subsequent patients. For the determination of reference PKM values of lesions, a standardized protocol should be used for all patients. On the hardware side, a multichannel eye coil [42, 43] might result in multiple advances in ocular DCE. An increased channel count might not only enable an increased temporal or spatial resolution, but also a potential larger FOV which can be used to determine the AIF on a patient-specific level. As with the single loop–coil approach no major blood vessels were available within the FOV, we relied on a population-based AIF. Population-based AIFs are a widely used approach but result a less accurate estimation of especially Ktrans as the AIF is be influenced by body mass and cardiac output [44, 45]. Eye muscles were investigated as reference tissue, but the perfusion of the eye muscles appeared not to be consistent within a single subject and might be also influenced by lesion location and therefore unreliable.
Although the spatial and temporal resolution achieved with our protocol are high compared to other ocular DCE studies, the spatial resolution of 1.25 × 1.5 × 1.5 mm3 is still a limiting factor for small ocular lesions. The smallest lesion included in this study was 32 voxels. A higher resolution would be preferred as this allows for the edge voxels to be removed from the analysis, as the results from these voxels are less reliable due to partial volume effects, not only of the dynamic scan, but also in the T1 mapping. Therefore, for the current resolution, DCE-MRI of small lesions small lesions (i.e. thickness < 2 mm) is likely less accurate. In situations where the conditions are less optimal (e.g. no orbital coil is or only a 1.5T MRI is available), DCE-MRI of intraocular lesions could be performed, however, although the decrease in image resolution would increase the lesion size required for reliable results. Moreover, although the TWIST sequence successfully reduces the motion artefacts by reducing the acquisition time per dynamic, the effective spatial resolution for rapidly changing concentrations, in particular the inflow of the contrast agent, might be lower. Nevertheless, the effect of precision errors in analysis steps, such as T1 determination or registration, on the obtained pharmacokinetic parameters generally showed to be less than 5%. Furthermore, some choices in the proposed analysis pipeline might benefit from a more thorough evaluation to further improve the analysis, such as the optimal reference image for the registration, the potential benefit of applying the median filter to the source FA-series images instead of the resulting T1 map and other PK models, such as the extended Tofts model to include a vascular component [46]. Finally, it is important to assess the reproducibility of DCE-MRI for ocular lesions between visits and centers.
Some of the proposed steps to enable reliably ocular DCE-MRI, such as the eye-specific registration, might be less easily incorporated into clinical practice. However, the inclusion of differences in T1 between lesions in the analysis, which has the strongest effect on the PK measures, is available in various clinical software packages. Although the elementary TIC classification, e.g. a distinction between a washout and plateau curve, is not affected by the T1, other elementary measures, in particular the relative enhancement, are significantly affected. Clinically, this conversion to concentration is particularly important as conversely to UM, the majority of other intraocular lesions are non-pigmented, resulting a biased evaluation. When the pharmacokinetic parameters have been determined in a larger cohort of patients with intraocular lesions, they could aid to differentiate between benign and different malignant intraocular lesions. Although the eye-specific motion correction is currently not available clinically, head motion can still be corrected with regular registration methods. When only this form of registration is available, a careful evaluation of the individual data is needed to screen for motion and to potentially remove motion-corrupted time points. We anticipate that this approach will be clinically sufficient to aid in the differential diagnosis, especially as other information, such as DWI, can be included in the considerations. This research furthermore showed that some UM are inhomogeneous in composition. For subsequent studies to assess these inhomogeneities, and assess their potential relation to genetic factors and the patients’ prognosis [20], full motion correction will be needed, as these inhomogeneities can amplify the effect of eye motion on the final parameters.
Conclusion
Although MRI of eyes is challenging in many aspects, we showed that quantitative DCE-MRI analysis can be performed for intraocular lesions by increasing the temporal and spatial resolution of the dynamic scan and using dedicated registration and T1 mapping with B1+ correction in the analysis. In the clinic DCE-MRI analysis might aid in the diagnosis, prognosis and follow-up of intraocular masses.
References
Virgili G, Gatta G, Ciccolallo L, Capocaccia R, Biggeri A, Crocetti E, Lutz JM, Paci E (2007) Incidence of uveal melanoma in Europe. Ophthalmology. https://doi.org/10.1016/j.ophtha.2007.01.032
Kaliki S, Shields CL (2017) Uveal melanoma: relatively rare but deadly cancer. Eye. https://doi.org/10.1038/eye.2016.275
Shields CL, Furuta M, Thangappan A, Nagori S, Mashayekhi A, Lally DR, Kelly CC, Rudich DS, Nagori AV, Wakade OA, Mehta S, Forte L, Long A, Dellacava EF, Kaplan B, Shields JA (2009) Metastasis of uveal melanoma millimeter-by-millimeter in 8033 consecutive eyes. Arch Ophthalmol 127:989–998
Fabian ID, Sagoo MS (2018) Understanding retinoblastoma: epidemiology and genetics. Community eye Heal 31:7
Cohen VML (2013) Ocular metastases. Eye. https://doi.org/10.1038/eye.2012.252
Shields CL, Furuta M, Berman EL, Zahler JD, Hoberman DM, Dinh DH, Mashayekhi A, Shields JA (2009) Choroidal nevus transformation into melanoma: analysis of 2514 consecutive cases. Arch Ophthalmol. https://doi.org/10.1001/archophthalmol.2009.151
Beenakker JWM, van Rijn GA, Luyten GPM, Webb AG (2013) High-resolution MRI of uveal melanoma using a microcoil phased array at 7 T. NMR Biomed. https://doi.org/10.1002/nbm.3041
Berkowitz BA, McDonald C, Ito Y, Tofts PS, Latif Z, Gross J (2001) Measuring the human retinal oxygenation response to a hyperoxic challenge using MRI: eliminating blinking artifacts and demonstrating proof of concept. Magn Reson Med 46:412–416
Niendorf T, Beenakker JWM, Langner S, Erb-Eigner K, Bach Cuadra M, Beller E, Millward JM, Niendorf TM, Stachs O (2021) Ophthalmic magnetic resonance imaging: where are we (Heading To)? Curr Eye Res. https://doi.org/10.1080/02713683.2021.1874021
Ferreira T, Pinheiro C, Saraiva P, Jaarsma-Coes M, Duinen S, Genders S, Marinkovic M, Beenakker J-W (2020) MR and CT imaging of the normal eyelid and its application in eyelid tumors. Cancers (Basel) 12:658
Keene KR, van Vught L, van de Velde NM, Ciggaar IA, Notting IC, Genders SW, Verschuuren JJGM, Tannemaat MR, Kan HE, Beenakker J-WM (2021) The feasibility of quantitative MRI of extra-ocular muscles in myasthenia gravis and Graves’ orbitopathy. NMR Biomed 34:e4407–e4407
van Vught L, Dekker CE, Stoel BC, Luyten GPM, Beenakker JM (2021) Evaluation of intraocular lens position and retinal shape in negative dysphotopsia using high-resolution magnetic resonance imaging. J Cataract Refract Surg 47(8):1032–1038. https://doi.org/10.1097/j.jcrs.0000000000000576
Beenakker JWM, Ferreira TA, Soemarwoto KP, Genders SW, Teeuwisse WM, Webb AG, Luyten GPM (2016) Clinical evaluation of ultra-high-field MRI for three-dimensional visualisation of tumour size in uveal melanoma patients, with direct relevance to treatment planning. Magn Reson Mater Phys Biol Med 29:571–577
Fleury E, Trnková P, Erdal E, Hassan M, Stoel B, Jaarma-Coes M, Luyten G, Herault J, Webb A, Beenakker J (2021) 3D MRI-based treatment planning approach for non-invasive ocular proton therapy. Med Phys 48(3):1315–1326. https://doi.org/10.1002/mp.14665
Foti PV, Longo A, Reibaldi M, Russo A, Privitera G, Spatola C, Raffaele L, Salamone V, Farina R, Palmucci S, Musumeci A, Caltabiano R, Ragusa M, Mariotti C, Avitabile T, Milone P, Ettorre GC (2017) Uveal melanoma: quantitative evaluation of diffusion-weighted MR imaging in the response assessment after proton-beam therapy, long-term follow-up. Radiol Med. https://doi.org/10.1007/s11547-016-0697-3
Jiang X, Asbach P, Willerding G, Dulce M, Xu K, Taupitz M, Hamm B, Erb-Eigner K (2015) Dynamic contrast-enhanced MRI of ocular melanoma. Melanoma Res. https://doi.org/10.1097/CMR.0000000000000142
Wei W, Jia G, Von Tengg-Kobligk H, Heverhagen JT, Abdel-Rahman M, Wei L, Christoforidis JB, Davidorf F, Knopp MV (2017) Dynamic contrast-enhanced magnetic resonance imaging of ocular melanoma as a tool to predict metastatic potential. J Comput Assist Tomogr. https://doi.org/10.1097/RCT.0000000000000598
Ferreira TA, Fonk LG, Jaarsma-Coes MG, van Haren GGR, Marinkovic M, Beenakker JWM (2019) MRI of uveal melanoma. Cancers (Basel) 11:1–20
Rodjan F, De Graaf P, Van Der Valk P, Moll AC, Kuijer JPA, Knol DL, Castelijns JA, Pouwels PJW (2012) Retinoblastoma: value of dynamic contrast-enhanced MR imaging and correlation with tumor angiogenesis. Am J Neuroradiol. https://doi.org/10.3174/ajnr.A3119
Kamrava M, Sepahdari AR, Leu K, Wang PC, Roberts K, Demanes DJ, McCannel T, Ellingson BM (2015) Quantitative multiparametric MRI in uveal melanoma: increased tumor permeability may predict monosomy 3. Neuroradiology 57:833–840
Chang MY, Rao NP, Burgess BL, Johnson L, Mccannel TA (2013) Heterogeneity of monosomy 3 in fine needle aspiration biopsy of choroidal melanoma. Mol Vis 19:1892–900
Chien JL, Sioufi K, Surakiatchanukul T, Shields JA, Shields CL (2017) Choroidal nevus: a review of prevalence, features, genetics, risks, and outcomes. Curr Opin Ophthalmol. https://doi.org/10.1097/ICU.0000000000000361
Enochs WS, Petherick P, Bogdanova A, Mohr U, Weissleder R (1997) Paramagnetic metal scavenging by melanin: MR imaging. Radiology. https://doi.org/10.1148/radiology.204.2.9240529
Tudorica LA, Oh KY, Roy N, Kettler MD, Chen Y, Hemmingson SL, Afzal A, Grinstead JW, Laub G, Li X, Huang W (2012) A feasible high spatiotemporal resolution breast DCE-MRI protocol for clinical settings. Magn Reson Imaging. https://doi.org/10.1016/j.mri.2012.04.009
Baron ED, Di Nicola M, Shields CL (2018) Updated ajcc classification for posterior uveal melanoma: a case example of a patient with choroidal melanoma is discussed in light of the latest edition of this cancer staging manual. Retin Today 30-34
De Naeyer D, Verhulst J, Ceelen W, Segers P, De Deene Y, Verdonck P (2011) Flip angle optimization for dynamic contrast-enhanced MRI-studies with spoiled gradient echo pulse sequences. Phys Med Biol. https://doi.org/10.1088/0031-9155/56/16/019
Nehrke K, Börnert P (2012) DREAM-a novel approach for robust, ultrafast, multislice B1 mapping. Magn Reson Med. https://doi.org/10.1002/mrm.24158
Tofts PS (2010) T1-weighted DCE imaging concepts: modelling, acquisition and analysis. Signal 500:400
Klein S, Staring M, Murphy K, Viergever MA, Pluim JPW (2010) Elastix: a toolbox for intensity-based medical image registration. IEEE Trans Med Imaging 29:196–205
Ritter F, Boskamp T, Homeyer A, Laue H, Schwier M, Link F, Peitgen H-O (2011) Medical image analysis: a visual approach. IEEE Pulse 2:60–70
Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, Gerig G (2006) User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage 31:1116–1128
Fram EK, Herfkens RJ, Johnson GA, Glover GH, Karis JP, Shimakawa A, Perkins TG, Pelc NJ (1987) Rapid calculation of T1 using variable flip angle gradient refocused imaging. Magn Reson Imaging. https://doi.org/10.1016/0730-725X(87)90021-X
Gupta RK (1977) A new look at the method of variable nutation angle for the measurement of spin-lattice relaxation times using fourier transform NMR. J Magn Reson. https://doi.org/10.1016/0022-2364(77)90138-X
Koolstra K, Beenakker JWM, Koken P, Webb A, Börnert P (2019) Cartesian MR fingerprinting in the eye at 7T using compressed sensing and matrix completion-based reconstructions. Magn Reson Med. https://doi.org/10.1002/mrm.27594
Geneesmiddeleninformatiebank, Samenvatting van de productkenmerken: Dotarem 0,5 mmol/ml. https://www.geneesmiddeleninformatiebank.nl/smpc/h27039_smpc.pdf. Accessed 30 Sep 2021
Ro SR, Asbach P, Siebert E, Bertelmann E, Hamm B, Erb-Eigner K (2016) Characterization of orbital masses by multiparametric MRI. Eur J Radiol. https://doi.org/10.1016/j.ejrad.2015.11.041
Yuan Y, Kuai XP, Chen XS, Tao XF (2013) Assessment of dynamic contrast-enhanced magnetic resonance imaging in the differentiation of malignant from benign orbital masses. Eur J Radiol. https://doi.org/10.1016/j.ejrad.2013.03.001
Griewank KG, Murali R (2013) Pathology and genetics of uveal melanoma. Pathol J RCPA 45:18–27
Maat W, Jordanova ES, van Zelderen-Bhola SL, Barthen ER, Wessels HW, Schalij-Delfos NE, Jager MJ (2007) The heterogeneous distribution of monosomy 3 in uveal melanomas: implications for prognostication based on fine-needle aspiration biopsies. Arch Pathol Lab Med 131:91–96
Miller AK, Benage MJ, Wilson DJ, Skalet AH (2017) Uveal melanoma with histopathologic intratumoral heterogeneity associated with gene expression profile discordance. Ocul Oncol Pathol 3:156–160
McLean IW, Foster WD, Zimmerman LE, Gamel JW (1983) Modifications of Callender’s classification of uveal melanoma at the Armed Forces Institute of Pathology. Am J Ophthalmol 96:502–509
Graessl A, Muhle M, Schwerter M, Rieger J, Oezerdem C, Santoro D, Lysiak D, Winter L, Hezel F, Waiczies S (2014) Ophthalmic magnetic resonance imaging at 7 T using a 6-channel transceiver radiofrequency coil array in healthy subjects and patients with intraocular masses. Invest Radiol 49:260–270
Glarin RK, Nguyen BN, Cleary JO, Kolbe SC, Ordidge RJ, Bui BV, McKendrick AM, Moffat BA (2021) MR-EYE: high-resolution MRI of the human eye and orbit at ultrahigh field (7T). Magn Reson Imaging Clin N Am 29:103–116
Fedorov A, Fluckiger J, Ayers GD, Li X, Gupta SN, Tempany C, Mulkern R, Yankeelov TE, Fennessy FM (2014) A comparison of two methods for estimating DCE-MRI parameters via individual and cohort based AIFs in prostate cancer: a step towards practical implementation. Magn Reson Imaging. https://doi.org/10.1016/j.mri.2014.01.004
Parker GJM, Roberts C, Macdonald A, Buonaccorsi GA, Cheung S, Buckley DL, Jackson A, Watson Y, Davies K, Jayson GC (2006) Experimentally-derived functional form for a population-averaged high-temporal-resolution arterial input function for dynamic contrast-enhanced MRI. Magn Reson Med. https://doi.org/10.1002/mrm.21066
Sourbron SP, Buckley DL (2011) On the scope and interpretation of the Tofts models for DCE-MRI. Magn Reson Med. https://doi.org/10.1002/mrm.22861
Funding
This work was supported by the Netherlands Organization for Scientific Research (NWO) [protons4vision 14654].
Author information
Authors and Affiliations
Contributions
Study conception and design: MJ, TF, GL, JB. Acquisition of data: MJ, JB. Analysis and interpretation of data: MJ, TF, PH, UH, GL, JB. Drafting of manuscript: MJ, JB. Critical revision: TF, PH, UH, GL.
Corresponding author
Ethics declarations
Conflict of interest
Beenakker and Jaarsma report research support from Philips Healthcare. Ferreira, van Houdt, van der Heide and Luyten declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with animals performed by any of the authors. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Nine patients were scanned as part of a prospective study, which has been approved by the local ethics committee and subjects were scanned after written informed consent [P16-186, METC-LDD]. Ten patients received an MRI as part of clinical care, and their data were included retrospectively with approval of the local ethics committee [G18-047, METC-LDD].
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 (AVI 12854 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jaarsma-Coes, M. ., Ferreira, T.A., van Houdt, P.J. et al. Eye-specific quantitative dynamic contrast-enhanced MRI analysis for patients with intraocular masses. Magn Reson Mater Phy 35, 311–323 (2022). https://doi.org/10.1007/s10334-021-00961-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10334-021-00961-w