
Abstract
To report long-term efficacy of certolizumab pegol (CZP) treatment with and without concomitant DMARDs in patients with psoriatic arthritis (PsA). RAPID-PsA (NCT01087788) was double-blind and placebo-controlled to week 24, dose-blind to week 48, and open-label to week 216. Patients had active PsA with ≥ 1 failed DMARD. At baseline, patients were randomized 1:1:1 to CZP 200 mg every 2 weeks: CZP 400 mg every 4 weeks: placebo. CZP-randomized patients continued their dose into open-label. Observed case efficacy data are reported to week 216 for week 0 CZP-randomized patients (dose combined) with and without baseline DMARD use (DMARD+/DMARD−). Dactylitis (tenderness and ≥ 10% difference in swelling between affected and opposite digits) and enthesitis were measured using Leeds Dactylitis Index (LDI) and Leeds Enthesitis Index (LEI). 273/409 randomized patients received CZP from baseline: 199/273 (72.9%) DMARD+ and 74/273 (27.1%) DMARD− patients. 141/199 (70.9%) DMARD+ and 42/74 (56.8%) DMARD− patients completed Week 216. DMARD+ (79.7%) and 83.3% of DMARD− patients achieved ACR20 response at week 216; 79.2 and 78.1% achieved 75% improvement from baseline in Psoriasis Area and Severity Index (PASI75). High proportions of DMARD+/DMARD− patients with extra-articular manifestations at baseline reported total resolution at week 216; dactylitis 91.4% of DMARD+ and 93.3% of DMARD− patients, enthesitis 74.4% of DMARD+ and 87.5% of DMARD− patients. Long-term improvements in PsA symptoms were observed with CZP monotherapy or concomitant DMARDs, across important psoriatic disease domains, including joint disease, psoriasis, nail disease, dactylitis, and enthesitis.
Trial registration: NCT01087788
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Psoriatic arthritis (PsA) is a chronic, inflammatory disease, affecting up to a third of psoriasis patients [1]. Clinical features of PsA include progressive and erosive joint inflammation and damage, [1], psoriatic skin disease, and extra-articular manifestations including dactylitis and enthesitis [2]. These symptoms are often accompanied by pain, fatigue, and functional impairment [3], with reductions in patient wellbeing and quality of life [4, 5].
The most recent treatment recommendations from the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) advise on optimal therapy choices based on disease activity and appropriate subsequent treatment pathways [6]. The initial treatment recommendations include the conditional use of corticosteroid injections and conventional disease-modifying anti-rheumatic drugs (DMARDs: methotrexate [MTX], sulfasalazine, leflunomide), due to their low costs and universal access. For patients who did not respond adequately to DMARDs, biological DMARDs (biologics: anti-TNF, anti-IL-12/23, anti-IL-17) are recommended, either with or without concomitant DMARD treatment [6].
Certolizumab pegol (CZP) is an Fc-free, PEGylated anti-TNF which has demonstrated rapid clinical improvements in joint and skin disease in patients with PsA [7]. Effects were maintained over 4 years of treatment [8]. Improvements were also observed in extra-articular manifestations including dactylitis, enthesitis, and nail psoriasis [7], which may positively impact patients’ quality of life [9].
CZP can be used to treat PsA alongside DMARDs, or as a monotherapy when treatment with a DMARD is otherwise undesirable [10]. The option of CZP monotherapy treatment is relevant for patients for whom DMARDs are contraindicated such as women who are, or may be planning to become, pregnant. The British Association of Dermatologists recommends that patients with PsA transition to biologic therapies with the aim of stopping the DMARD therapy. Dermatologists may, therefore, be more likely to stop DMARD therapy, compared to rheumatologists [11]. Data are available for the relative efficacy of other anti-TNFs with and without concomitant DMARDs; however similar comparisons in CZP are currently limited [12]. Here, we report the efficacy, safety, and patient-reported outcomes of CZP in patients treated with and without concomitant DMARDs.
Materials and methods
Patients
Patient eligibility criteria have been reported elsewhere [7]. Briefly, patients were ≥ 18 years of age and had a diagnosis of active PsA for at least 6 months according to the Classification Criteria for Psoriatic Arthritis (CASPAR). They must have had active disease: at least 3 tender joints, 3 swollen joints, and at least 1 of the following: erythrocyte sedimentation rate (ESR) ≥ 28 mm/h or C-reactive protein (CRP) > ULN (7.9 mg/L). All patients must have failed ≥ 1 DMARD but were allowed to continue taking certain DMARDs at stable dose levels: leflunomide (≤ 20 mg daily), MTX (≤ 25 mg weekly), or sulfasalazine (≤ 3 g daily). Reasons for stopping DMARD use were not collected for those who had used DMARDs in the past but stopped prior to the first administration of CZP during the study. Use of hydroxychloroquine, azathioprine, cyclosporine, cyclophosphamide, and mycophenolic acid, at any dose, was not permitted within 28 days prior to the baseline visit.
Patients were excluded if they had received > 1 prior anti-TNF, had previously experienced primary failure of an anti-TNF, or had a diagnosis of other non-PsA inflammatory arthritis. Patients with evidence of latent or active tuberculosis (TB) were also excluded, unless prophylactic treatment had commenced ≥ 4 weeks prior to baseline.
All human studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Study design
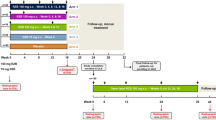
RAPID-PsA (NCT01087788) was a 216-week, multicenter, phase 3 randomized clinical trial of CZP in patients with PsA. The trial was double-blind and placebo-controlled until week 24, dose-blind to week 48, and open-label to week 216.
All patients gave their informed consent prior to their inclusion within the study. Patients with PsA were randomized (1:1:1) at week 0 to CZP (200 mg every 2 weeks [Q2W] or 400 mg every 4 weeks [Q4W]; following 400 mg loading doses at weeks 0, 2, and 4) or placebo. CZP-randomized patients remained on their assigned dose into the dose-blind and open-label stage. Placebo patients were re-randomized (1:1) to CZP (200 mg Q2W or 400 mg Q4W; following 400 mg loading dose) either upon failing to achieve ≥ 10% reduction in tender and swollen joint counts at both weeks 14 and 16 (early escape), or after having completed the 24-week double-blind phase.
No changes to DMARD doses were allowed during the first 48 weeks, after which possible changes were allowed and recorded.
Study procedures and evaluations
Efficacy outcomes
The clinical outcomes measured were disease activity in peripheral joints and skin, health-related quality of life (HRQoL), dactylitis, enthesitis, and nail disease, reported from baseline to week 216. Peripheral joint disease was assessed using swollen and tender joint counts (66 joint assessment and 68 joint assessment, respectively) [3]. Skin disease was assessed in patients with ≥ 3% body surface area (BSA) involvement at baseline, using the Psoriasis Area and Severity Index (PASI), with scores ranging from 0 to 72 [3].
Dactylitis, which refers to swelling of the entire digit, was assessed using the Leeds Dactylitis Index (LDI) in fingers and toes: dactylitic digits were defined as those which exhibited ≥ 10% difference in circumference when compared to the opposite digit and were tender when pressed (“acute” dactylitis) [13]. The scores for each digit were summed to produce a total for the patient; higher scores indicate worse dactylitis [3]. Total resolution was also evaluated: dactylitis was considered to be resolved if no digit was affected.
Enthesitis was assessed using the Leeds Enthesitis Index (LEI), which was specifically developed for patients with PsA, and determines the presence (1) or absence (0) of tenderness in the bilateral epicondyles, medial femoral condyles, and Achilles tendon insertions to give an overall score between 0 and 6 [3, 14]. Total resolution of enthesitis was calculated and presented as the percentage of patients with enthesitis at baseline, achieving LEI = 0.
The modified nail psoriasis severity index (mNAPSI) was calculated for the most affected fingernail at baseline (“target fingernail”) and assessed for three features or groups of features, namely pitting, onycholysis, and oil-drop dyschromia, and nail plate crumbling which were graded for severity on a scale from 0 to 3. The presence or absence of other features, including leukonychia, splinter hemorrhages, hyperkeratosis, and red spots in the lunula, was also assessed (scored 0 or 1), with an overall sum score out of 13 for the target fingernail [15]. Total resolution of nail psoriasis in the target nail was defined as mNAPSI score = 0 for subjects with nail psoriasis at baseline.
Composite measures of disease activity were analyzed, among which week 12 American College of Rheumatologists 20% (ACR20) response was the primary endpoint (reported previously) [7]. Here, we report arthritis joint improvement and involvement using ACR20, 50, and 70 responses and the 28-joint count Disease Activity Score based on C-reactive protein [DAS28(CRP)], though this is not a validated outcome for PsA. Skin response is reported in terms of whether patients achieved 75, 90, or 100% improvement in PASI (PASI75, PASI90, PASI100 responder rates, respectively).
Week 24 change from baseline in total sharp score (mTSS), modified for PsA [16], was the primary radiographic variable to quantify the progression of bone erosions and joint space narrowing. These data have been reported previously [7, 9]. Regular assessments also comprised physician visual analogue scales (VAS) for global disease activity and CRP.
Patient-reported outcomes
Functional health status was assessed using Health Assessment Questionnaire-Disability Index (HAQ-DI). The HAQ-DI is used to emphasize the outcomes of most importance to patients and is based on 20 items, divided into 8 domains: “dressing and grooming,” “arising,” “eating,” “walking,” “hygiene,” “reach,” “grip,” and “common daily activities” [17, 18]. HAQ-DI uses a 4-point scale to assess the degree of difficulty experienced within each domain, summed to provide a score between 0 and 24 which is then divided by the number of categories resulting in a final score between 0 and 3, where 0 = mild limitations and 3 = very severe limitations of physical function [18].
For HAQ-DI, minimal clinically important difference (MCID) was considered to be ≥ 0.35-point decrease from baseline [19].
Safety
Any treatment-emergent adverse events (TEAEs), classified by severity, are reported, in addition to any withdrawals due to TEAEs, drug-related TEAEs, serious TEAEs (including infections and infestations), and deaths. TEAEs and serious TEAEs which occurred after the first CZP administration until 70 days after the last CZP administration were recorded. Serious TEAEs were defined as events that were life-threatening, or those that resulted in death, significant or persistent disability/incapacity, congenital anomaly or birth defect, hospitalization or prolongation of hospitalization, or an important medical event.
Statistical analysis
Efficacy data are presented from baseline to week 216 in patients randomized to CZP at week 0 (randomized set). All data reported within the text are “observed case” values (including only those patients who were assessed at the time point in question) unless otherwise stated; tables and figures report both observed case data and values with imputation for missing data. Imputed categorical data are shown as the percentage of responders out of all randomized patients; imputed quantitative data were calculated using last observation carried forward (LOCF).
All TEAEs were coded according to the Medical Dictionary of Regulatory Activities (MedDRA) version 14.1. Incidences of TEAEs are presented for all patients who received at least one dose of CZP at any stage of the study (the Safety Set). The proportion of the Safety Set that experienced and reported each TEAE, the event rate (ER, per 100 patient-years of exposure) and the incidence rate (IR, incidence of new cases of an event per 100 patient-years of exposure) are reported. Patient-years of exposure was calculated as the sum of individual patient-exposures up until 70 days after patients’ last CZP injection or, in the case of incidence rates, until the patients’ first occurrence of the respective event, when applicable. The most common serious TEAEs, occurring in either of the patient subgroups (DMARD+ or DMARD−), are reported by system organ class (SOC) as well as other TEAEs of interest. Malignancies were identified using the Standardized MedDRA Query (SMQ) “Malignancies.” Based on MedDRA terms and medical review, major adverse cardiovascular events (MACE) were defined as all serious adverse events associated with one or more of the following search criteria: fatal and serious non-fatal myocardial infarction, cerebrovascular events, and congestive heart failure [20].
All statistical analyses are based on the combined data from the CZP 200 mg every 2 weeks (Q2W) and 400 mg every 4 weeks (Q4W) arms, presented separately for the subgroup using concomitant DMARDs at baseline (the DMARD+ group) and the subgroup not using DMARD at baseline (the DMARD− group), irrespective of subsequent changes in DMARD usage.
Data availability
The datasets generated and analyzed during this study are available in anonymized format upon reasonable request via the CSDR platform (www.clinicalstudydatarequest.com).
Results
Patient disposition and baseline characteristics
A total of 409 patients with PsA were randomized, of whom 273 received CZP from week 0 (Fig. 1a). Of the 273 week 0 CZP-randomized patients, 199 (72.9%) were using concomitant DMARDs at baseline and 74 (27.1%) were not. DMARDs at baseline consisted of MTX (174 patients, 63.7%), leflunomide (12 patients, 4.4%), sulfasalazine (12 patients, 4.4%), and hydroxychloroquine (1 patient, 0.4%, despite hydroxychloroquine use at baseline being contraindicated in the study protocol). Baseline demographics and disease characteristics were similar between the two treatment groups, although a higher proportion of patients in the DMARD− group had psoriasis BSA ≥ 3% at baseline (Table 1). The concomitant use of corticosteroids was similar between patients treated with or without concomitant DMARDs at baseline (DMARD+ 24.1%, DMARD− 27.0%) (Table 1).

a RAPID-PsA study design. b Change in DMARD use after initial grouping as DMARD− or DMARD+ at baseline (doses combined). c Discontinuations for all reasons in patients grouped by DMARD use at baseline. a Randomized set. b Safety Set. Comparison of DMARD use at patients’ last visit vs. baseline. c Randomized set. CZP certolizumab pegol, DMARD disease-modifying anti-rheumatic drug, LD loading dose, PsA psoriatic arthritis, Q2W every 2 weeks, Q4W every 4 weeks, SJC swollen joint count, TJC tender joint count, Wk week
In total, over the 4-year duration of the RAPID-PsA study, 207 (75.8%) week 0 CZP-randomized patients took concomitant DMARDs at any time. While the majority of patients did not change their DMARD treatment regimen during the study, 26/199 (13.1%) patients within the DMARD+ group discontinued DMARD treatments, and 5/74 (6.8%) patients within the DMARD− group initiated DMARD use between baseline and their last visit (Fig. 1b). The net reduction in the proportion of patients using DMARDs from baseline to the end of the study was 7.7%.
In total, 183/273 patients completed open-label treatment to week 216: 141/199 (70.9%) of DMARD+ patients and 42/74 (56.8%) of DMARD− patients (Fig. 1c). The most common reasons for discontinuation were adverse events and withdrawal of consent (Supplementary Table 1).
Efficacy
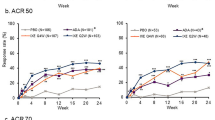
Improvements in joint disease as measured by ACR20 responder rates were observed in 61.7% of DMARD+ and 52.1% of DMARD− patients treated with CZP at week 12, the time point for evaluation of the primary efficacy variable. Among patients assessed at week 216, 79.7% of DMARD+ and 83.3% of DMARD− patients achieved an ACR20 response (Fig. 2a). Sustained improvements in joint disease were also demonstrated by ACR50 and ACR70 responder rates to week 216 in DMARD+ and DMARD− patients (Fig. 2b, c).

Joint disease outcomes in patients with and without concomitant use of DMARDs at baseline. a ACR20 responder rate. b ACR50 response rate. c ACR70 response rate. d mean DAS28 (CRP) scores. Randomized set. Imputed analyses show the percentage of responders out of all randomized patients (for categorical data) or used last observation carried forward (LOCF) imputation (for continuous data). Data points are shown for observed case values at weeks 24, 48, 96, and 216. ACR American College of Rheumatology, ACR20/50/70 American College of Rheumatology 20, 50, and 70% response criteria, CRP C-reactive protein, DAS28 28-joint count Disease Activity Score, DMARD disease-modifying anti-rheumatic drug, OC observed case, LOCF last observation carried forward
A reduction in disease activity from baseline to week 24, as measured by mean change from baseline in DAS28(CRP) [SD], was observed: − 2.07 [1.28] in DMARD+ and − 1.98 [1.25] in DMARD− patients. These reductions were maintained to week 216 in both subgroups of patients (Fig. 2d).
Patients with ≥ 3% BSA affected with psoriasis at baseline showed improvements in PASI as early as week 1, and maximal improvements observed by week 48 were maintained to week 216, both in DMARD+ and DMARD− patients (Supplementary Fig. 1a). PASI75 responses were achieved by 65.7% of DMARD+ patients and 82.2% of DMARD− patients at week 24, with 26.3% of DMARD+ and 24.4% of DMARD− patients also achieving a PASI100 response. These observations were maintained to week 216 for both DMARD+ and DMARD− patients (Fig. 3a, Supplementary Fig. 1b).

Improvements in psoriasis, dactylitis, enthesitis, and functional HRQoL in patients with and without concomitant DMARD use at baseline. a PASI75 responder rate. b Mean Leeds Dactylitis Index (LDI). c Mean Leeds Enthesitis Index (LEI). d HAQ-DI MCID responder rate. Randomized Set. a Patients with ≥ 3% BSA at baseline. b Patients with dactylitis at baseline. c Patients with enthesitis at baseline. d MCID for HAQ-DI was defined as a decrease of ≥ 0.35 points from baseline. Imputed analyses show the percentage of responders out of all randomized patients (for categorical data) or used last observation carried forward (LOCF) imputation (for continuous data). Data points are shown for observed case values at weeks 24, 48, 96, and 216. DMARD disease-modifying anti-rheumatic drug, HAQ-DI Health Assessment Questionnaire-Disability Index, HRQoL health-related quality of life, OC observed case, LDI Leeds Dactylitis Index, LEI Leeds Enthesitis Index, LOCF last observation carried forward, MCID minimal clinically important differences, PASI75 75% reduction of the Psoriasis Area and Severity Index
For patients with dactylitis, enthesitis, or nail psoriasis at baseline, improvements in LDI, LEI, and mNAPSI, respectively, were observed at week 24, and maintained to week 216 in DMARD+ and DMARD− patients (Fig. 3b, c and Table 2). Among CZP-treated patients who remained in the study, total resolution of dactylitis (DMARD+ 91.4%, DMARD− 93.3%), enthesitis (DMARD+ 74.4%, DMARD− 87.5%), and nail psoriasis (DMARD+ 72.3%, DMARD− 67.7%) was achieved at week 216 by high proportions of patients affected at baseline. Even when data were imputed, using LOCF, more than two-thirds of those with dactylitis or enthesitis at baseline achieved total resolution of the respective manifestation of their disease at week 216, both in the DMARD+ and DMARD− groups (Table 2).
Similarly, total resolution of nail psoriasis was achieved by 66.9% of DMARD+ and 57.7% of DMARD– patients affected at baseline, by Week 216 when data were imputed (Table 2).
HAQ-DI MCID (≥ 0.35 decrease from baseline) was achieved by 51.6% of DMARD+ and 59.4% of DMARD– patients at week 24, which increased to 58.7% of DMARD+ patients and 69.0% of DMARD– among patients who were assessed at week 216 (Table 2).
Safety
Total exposure to CZP in RAPID-PsA was 1321 patient-years. In the Safety Set, 279/393 (71.0%) patients were DMARD+ at baseline, while the remaining 114/393 (29.0%) were DMARD−. Similar TEAE event rates of 257.4 per 100 patients-years in DMARD+ patients and 259.3 per 100 patient-years in DMARD− patients were observed in both subgroups of patients (Table 3).
Investigators classified 62.2% of TEAEs as “mild” (DMARD+ 62.3%, DMARD− 61.9%) and 34.9% as “moderate” (DMARD+ 35.0%, DMARD− 34.3%) in severity. The severe TEAEs ER was 6.8 per 100 patient-years for DMARD+ patients and 9.9 per 100 patient-years for DMARD− patients (Table 3). Serious TEAEs were reported for 26.2% DMARD+ and 23.7% DMARD− patients; the serious adverse ER was 11.5 per 100 patient-years for DMARD+ patients and 13.0 per 100 patient-years for DMARD− patients (Table 3). The most commonly reported serious TEAEs, occurred in the MedDRA SOC “infections and infestations,” “musculoskeletal and connective tissue disorders,” and “gastrointestinal disorders.” Serious infections and infestations were reported for 16 (5.7%) DMARD+ patients with an ER of 2.0 per 100 patient-years compared to 7 (6.1%) DMARD− patients with an ER of 3.1 per 100 patient-years (Table 3). The most common serious infection reported was pneumonia, which was reported by two patients each in the DMARD+ and DMARD− subgroups. As reported previously, there were no confirmed cases of active TB [21, 22]. Slightly more DMARD− than DMARD+ patients experienced TEAEs which led to discontinuation of CZP and subsequent withdrawal from the study (Table 3).
MACE were reported for six (2.2%) DMARD+ patients and one (0.9%) DMARD− patient. Malignancies were reported for five (1.8%) DMARD+ patients and two (1.8%) DMARD− patients. In the DMARD+ subgroup of patients, two cases of breast cancer were reported, in addition to one case each of metastatic gastrointestinal cancer, cervix carcinoma stage 0 (non-serious), and lymphoma, while one case each of breast cancer and ovarian cancer were reported in the DMARD− subgroup of patients.
No new safety signals were identified from week 96 to week 216, and no further deaths were reported during this period. Overall, six deaths were reported during the RAPID-PsA study, reported previously [9]. The causes of death included myocardial infarction, cardiac arrest, sudden death, pneumonia and sepsis (reported in the same patient), breast cancer, and lymphoma. Five of these were reported by patients within the DMARD+ subgroup (Table 3).
Discussion
Previously published results from the RAPID-PsA trial have demonstrated sustained improvements in the signs and symptoms of PsA in patients treated with CZP over 4 years [22]. Here, we elaborate on these findings to assess outcomes in patients treated with and without concomitant DMARD therapy. Although for some outcomes we noted a trend towards greater improvements in those patients using concomitant DMARDs, these data demonstrate that patients were able to achieve long-term improvements in their disease, even without additional conventional disease-modifying therapies.
Both groups showed improvements as early as week 1, with maximal improvements seen by week 48 and maintained over 4 years. Improvements were seen not only in joint disease but in every extra-axial domain included in the GRAPPA guidelines, including extra-articular manifestations (enthesitis and dactylitis), which are important for improving patients’ quality of life and well-being and reducing the day-to-day burden of disease [6]. We also saw no difference in the incidence or pattern of adverse events between the groups using CZP with or without conventional DMARDs.
These findings are of particular importance for those patients who are unable or unwilling to take DMARDs such as MTX, for example those who have previously failed to respond, experienced an adverse reaction, have a greater risk of developing an adverse reaction, or may be, or planning to become, pregnant.
The results from RAPID-PsA, reported here, are consistent with published data from the Corrona Registry which showed no significant difference in the time to remission for patients with PsA treated with DMARDs concomitantly with an anti-TNF (etanercept, adalimumab or infliximab) or anti-TNF monotherapy [23].
This, in view of existing treatment recommendations, is of particular interest for dermatologists. The study data should be reassuring for many patients with PsA under the care of dermatology departments, who may only receive anti-TNFs as a monotherapy [11]. The use of DMARDs is also contraindicated in some patients, and therefore treatment options that can provide reductions in disease activity without the need for concomitant therapies are of benefit to these patients. Monotherapy also offers increased convenience for patients and cost-savings for the healthcare system [24]. Women with PsA who are planning a pregnancy are advised to adjust medications, since withdrawal of treatment prior to conception can result in worsening of symptoms [25]. Of particular importance, the British Society for Rheumatology and British Health Professionals in Rheumatology guidelines recommend that MTX and cyclophosphamide use are strictly contraindicated prior to conception and during pregnancy and breastfeeding [25, 26].
Investigation into the use of anti-TNF drugs as monotherapies has previously been conducted in relatively small, non-randomized trials without placebo control, which demonstrated the efficacy of adalimumab, infliximab, and golimumab as monotherapies in patients with PsA [27,28,29]. However, these studies did not include a comparative group of patients treated with combined therapy (anti-TNF and DMARDs) [27,28,29]. For etanercept, post hoc analyses of the combined results from two clinical trials showed comparable efficacy whether the agent was provided as a combined treatment or as a monotherapy at baseline [30,31,32]. Similar to our study, patients treated with adalimumab and concomitant DMARDs demonstrated slightly higher retention rates than those patients on monotherapy, which could explain the trend for improved outcomes in DMARD+ patients [23]. In contrast, the PSUMMIT 2 study of the anti-IL-12/23 p-40 monoclonal antibody, ustekinumab, demonstrated no differences in efficacy with or without concomitant MTX use [33].
Our study has shown sustained improvements in signs and symptoms of PsA whether patients’ treatment was with CZP alone or in combination with DMARDs. These data and the observed long-term efficacy (up to 5.8 years) of CZP suggest that CZP monotherapy is an option that can be considered for women of child-bearing age with chronic PsA [10, 34].
Due to its unique Fc-free structure, CZP showed no to minimal (below 0.1%) placental transfer from mother to baby in a first-of-its-kind clinical study (CRIB study) [35] and minimal (below 0.2%) transfer of CZP from plasma to breast milk [36]. As of August 2018, CZP is the only biologic with clinical trial data in its label supporting potential use in both pregnancy and breastfeeding in chronic rheumatic diseases (axSpA, PsA, RA), as indicated in the CZP SmPC [10, 34, 36]. Given the paucity of data currently available in the published literature, further information in this area will be of great importance moving forward, particularly regarding the use of biologics as monotherapies.
The limitations of the current study included the classification of patients as DMARD+ or DMARD−, since this was according to their DMARD use at baseline, and thus changes in DMARD use throughout the study were not accounted for. However, relatively few patients completely discontinued or initiated DMARD use during the 216-week study. Although the DMARD+ and DMARD− patients were largely similar with respect to the demographic and baseline disease characteristics, it is important to note that patients’ use of DMARDs was not controlled by the study protocol and therefore patients were not randomized into these groups. Formal comparisons of the DMARD+ and DMARD− patient groups were not performed, partly due to potential confounding of some background characteristics which were not assessed during this study. Also, patient withdrawal from the study introduced a risk of bias in the data, a limitation associated with all clinical trials, the impact of which increases in long-term studies such as RAPID-PsA. In order to reduce this risk of bias, observed and imputed data have been reported for this study.
In conclusion, the RAPID-PsA study showed that patients with PsA treated with CZP demonstrated sustained improvements over 4 years in signs and symptoms of their disease across important GRAPPA domains, when taken as a monotherapy, or with concomitant DMARDs. CZP was shown to be efficacious and had a similar safety profile to previous studies investigating CZP, in patients treated with concomitant DMARDs and CZP monotherapy.
Change history
11 October 2018
The above article originally published with an error present in Discussion section and presented correctly in this article. The original article was corrected.
References
Gladman D, Antoni C, Mease P, Clegg D, Nash P (2005) Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis 64(suppl 2):ii14–ii17
Kerschbaumer A, Fenzl KH, Erlacher L, Aletaha D (2016) An overview of psoriatic arthritis–epidemiology, clinical features, pathophysiology and novel treatment targets. Wien Klin Wochenschr:1–5
Mease PJ (2011) Measures of psoriatic arthritis: tender and swollen joint assessment, Psoriasis Area and Severity Index (PASI), Nail Psoriasis Severity Index (NAPSI), Modified Nail Psoriasis Severity Index (mNAPSI), Mander/Newcastle Enthesitis Index (MEI), Leeds Enthesitis Index (LEI), Spondyloarthritis Research Consortium of Canada (SPARCC), Maastricht Ankylosing Spondylitis Enthesis Score (MASES), Leeds Dactylitis Index (LDI), Patient Global for Psoriatic Arthritis, Dermatology Life Quality Index (DLQI), Psoriatic Arthritis Quality of Life (PsAQOL), Functional Assessment of Chronic Illness Therapy–Fatigue (FACIT-F), Psoriatic Arthritis Response Criteria (PsARC), Psoriatic Arthritis Joint Activity Index (PsAJAI), Disease Activity in Psoriatic Arthritis (DAPSA), and Composite Psoriatic Disease Activity Index (CPDAI). Arthritis Care Res (Hoboken) 63(S11):S64–S85
Husted JA, Gladman DD, Farewell VT, Cook RJ (2001) Health-related quality of life of patients with psoriatic arthritis: a comparison with patients with rheumatoid arthritis. Arthritis Care Res (Hoboken) 45(2):151–158
Mease PJ, Menter MA (2006) Quality-of-life issues in psoriasis and psoriatic arthritis: outcome measures and therapies from a dermatological perspective. J Am Acad Dermatol 54(4):685–704. https://doi.org/10.1016/j.jaad.2005.10.008
Coates LC, Kavanaugh A, Mease PJ, Soriano ER, Laura Acosta-Felquer M, Armstrong AW, Bautista-Molano W, Boehncke WH, Campbell W, Cauli A (2016) Group for research and assessment of psoriasis and psoriatic arthritis 2015 treatment recommendations for psoriatic arthritis. Arthritis Rheumatol 68(5):1060–1071
Mease PJ, Fleischmann R, Deodhar AA, Wollenhaupt J, Khraishi M, Kielar D, Woltering F, Stach C, Hoepken B, Arledge T, van der Heijde D (2014) Effect of certolizumab pegol on signs and symptoms in patients with psoriatic arthritis: 24-week results of a phase 3 double-blind randomised placebo-controlled study (RAPID-PsA). Ann Rheum Dis 73(1):48–55. https://doi.org/10.1136/annrheumdis-2013-203696
Mease P, Fleischmann R, Wollenhaupt J, Deodhar A, Gladman D, Hoepken B, Peterson L, van der Heijde D (2016) FRI0472 improvements in joint outcomes of psoriatic arthritis over 4 years of treatment with certolizumab pegol in patients with and without prior anti-TNF exposure. Ann Rheum Dis 75:609
Mease P, Deodhar A, Fleischmann R, Wollenhaupt J, Gladman D, Leszczyński P, Vitek P, Turkiewicz A, Khraishi M, FitzGerald O, Landewé R, de Longueville M, Hoepken B, Peterson L, van der Heijde D (2015) Effect of certolizumab pegol over 96 weeks in patients with psoriatic arthritis with and without prior antitumour necrosis factor exposure. RMD Open 1(1):e000119. https://doi.org/10.1136/rmdopen-2015-000119
EMA (2017) Summary of product characteristics (Cimzia). http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001037/WC500069763.pdf. Accessed 24 Apr 2018
Smith C, Jabbar-Lopez Z, Yiu Z, Bale T, Burden A, Coates L, Cruickshank M, Hadoke T, MacMahon E, Murphy R, Nelson-Piercy C (2017) British Association of Dermatologists guidelines for biologic therapy for psoriasis 2017. Br J Dermatol 177(3):628–636
Gossec L, Smolen J, Ramiro S, De Wit M, Cutolo M, Dougados M, Emery P, Landewé R, Oliver S, Aletaha D (2016) European league against rheumatism (EULAR) recommendations for the management of psoriatic arthritis with pharmacological therapies: 2015 update. Ann Rheum Dis 75:499–510
Ferguson E, Coates L (2014) Optimisation of rheumatology indices: dactylitis and enthesitis in psoriatic arthritis. Clin Exp Rheumatol 32(Suppl 85):113–117
Healy PJ, Helliwell PS (2008) Measuring clinical enthesitis in psoriatic arthritis: assessment of existing measures and development of an instrument specific to psoriatic arthritis. Arthritis Care Res (Hoboken) 59(5):686–691
Cassell SE, Bieber JD, Rich P, Tutuncu ZN, Lee SJ, Kalunian KC, Wu CW, Kavanaugh A (2007) The modified nail psoriasis severity index: validation of an instrument to assess psoriatic nail involvement in patients with psoriatic arthritis. J Rheumatol 34(1):123–129
van der Heijde D, Sharp J, Wassenberg S, Gladman D (2005) Psoriatic arthritis imaging: a review of scoring methods. Ann Rheum Dis 64(suppl 2):ii61–ii64
Bruce B, Fries J (2005) The health assessment questionnaire (HAQ). Clin Exp Rheumatol 23(5):S14
Fries JF, Spitz P, Kraines RG, Holman HR (1980) Measurement of patient outcome in arthritis. Arthritis Rheum (Munch) 23(2):137–145
Mease PJ, Woolley JM, Bitman B, Wang BC, Globe DR, Singh A (2011) Minimally important difference of health assessment questionnaire in psoriatic arthritis: relating thresholds of improvement in functional ability to patient-rated importance and satisfaction. J Rheumatol 38(11):2461–2465. https://doi.org/10.3899/jrheum.110546
Bykerk VP, Cush J, Winthrop K, Calabrese L, Lortholary O, De Longueville M, Van Vollenhoven R, Mariette X (2015) Update on the safety profile of certolizumab pegol in rheumatoid arthritis: an integrated analysis from clinical trials. Ann Rheum Dis 74:96–103
Mease PJ, Fleischmann R, Wollenhaupt J, Deodhar A, Gladman D, Hoepken B, Peterson L, van der Heijde D (2016) FRI0471 certolizumab pegol for the treatment of psoriatic arthritis: 4-year outcomes from the rapid-Psa trial. Ann Rheum Dis 75(Suppl 2):608–609. https://doi.org/10.1136/annrheumdis-2016-eular.3192
van der Heijde D, Deodhar A, FitzGerald O, Fleischmann R, Gladman D, Gottlieb AB, Hoepken B, Bauer L, Irvin-Sellers O, Khraishi M (2018) 4-year results from the RAPID-PsA phase 3 randomised placebo-controlled trial of certolizumab pegol in psoriatic arthritis. RMD Open 4(1):e000582
Mease PJ, Collier DH, Saunders KC, Li G, Kremer JM, Greenberg JD (2015) Comparative effectiveness of biologic monotherapy versus combination therapy for patients with psoriatic arthritis: results from the Corrona registry. RMD Open 1(1):e000181. https://doi.org/10.1136/rmdopen-2015-000181
Menter A, Gottlieb A, Feldman SR, Van Voorhees AS, Leonardi CL, Gordon KB, Lebwohl M, Koo JY, Elmets CA, Korman NJ (2008) Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol 58(5):826–850
Kurizky PS, Ferreira CC, Nogueira LSC, da Mota LMH (2015) Treatment of psoriasis and psoriatic arthritis during pregnancy and breastfeeding. An Bras Dermatol 90(3):367–375. https://doi.org/10.1590/abd1806-4841.20153113
Flint J, Panchal S, Hurrell A, van de Venne M, Gayed M, Schreiber K, Arthanari S, Cunningham J, Flanders L, Moore L (2016) BSR and BHPR guideline on prescribing drugs in pregnancy and breastfeeding—part I: standard and biologic disease modifying anti-rheumatic drugs and corticosteroids. Rheumatology 55(9):1693–1697
Papoutsaki M, Chimenti M-S, Costanzo A, Talamonti M, Zangrilli A, Giunta A, Bianchi L, Chimenti S (2007) Adalimumab for severe psoriasis and psoriatic arthritis: an open-label study in 30 patients previously treated with other biologics. J Am Acad Dermatol 57(2):269–275. https://doi.org/10.1016/j.jaad.2006.12.003
Mastroianni A, Minutilli E, Mussi A, Bordignon V, Trento E, D'Agosto G, Cordiali-Fei P, Berardesca E (2005) Cytokine profiles during infliximab monotherapy in psoriatic arthritis. Br J Dermatol 153(3):531–536. https://doi.org/10.1111/j.1365-2133.2005.06648.x
Chimenti MS, Teoli M, Saraceno R, Dattola A, Ventura A, Chiricozzi A, Chiaramonte C, Perricone R, Chimenti S (2013) Golimumab in patients affected by moderate to severe psoriatic arthritis: an open-label study in thirty-two patients previously treated with other biologics. Dermatology 227(4):305–310. https://doi.org/10.1159/000354263
Combe B, Behrens F, McHugh N, Brock F, Kerkmann U, Kola B, Gallo G (2016) Comparison of etanercept monotherapy and combination therapy with methotrexate in psoriatic arthritis: results from 2 clinical trials. J Rheumatol 43(6):1063–1067. https://doi.org/10.3899/jrheum.151290
Mease PJ, Goffe BS, Metz J, VanderStoep A, Finck B, Burge DJ (2000) Etanercept in the treatment of psoriatic arthritis and psoriasis: a randomised trial. Lancet 356(9227):385–390
Mease PJ, Kivitz AJ, Burch FX, Siegel EL, Cohen SB, Ory P, Salonen D, Rubenstein J, Sharp JT, Tsuji W (2004) Etanercept treatment of psoriatic arthritis: safety, efficacy, and effect on disease progression. Arthritis Rheumatol 50(7):2264–2272
Ritchlin C, Rahman P, Kavanaugh A, McInnes IB, Puig L, Li S, Wang Y, Shen Y-K, Doyle MK, Mendelsohn AM (2014) Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the phase 3, multicentre, double-blind, placebo-controlled, randomised PSUMMIT 2 trial. Ann Rheum Dis 73(6):990–999
Walsh J, Gottlieb AB, Hoepken B, Nurminen T, Mease PJ (2017) The efficacy of certolizumab pegol over 4 years in psoriatic arthritis patients with and without concomitant use of DMARDs. J Am Acad Dermatol 76(6):AB253 (Poster 5507)
Mariette X, Förger F, Abraham B, Flynn AD, Moltó A, Flipo R-M, van Tubergen A, Shaughnessy L, Simpson J, Teil M, Helmer E, Wang M, Chakravarty EF (2018) Lack of placental transfer of certolizumab pegol during pregnancy: results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann Rheum Dis 77:228–233
Clowse ME, Förger F, Hwang C, Thorp J, Dolhain RJ, van Tubergen A, Shaughnessy L, Simpson J, Teil M, Toublanc N, Wang M, Hale TW (2017) Minimal to no transfer of certolizumab pegol into breast milk: results from CRADLE, a prospective, postmarketing, multicentre, pharmacokinetic study. Ann Rheum Dis 76(11):1890–1896. https://doi.org/10.1136/annrheumdis-2017-211384
Acknowledgements
The authors thank the patients, the investigators, and their teams who took part in this study. The authors also acknowledge Mylene Serna, PhD, from UCB Pharma, Smyrna, GA, USA for publication coordination and Sarah Jayne Clements, PhD, from Costello Medical Consulting, Cambridge, UK, for medical writing and editorial assistance in preparing this manuscript for publication, based on the authors’ input and direction.
Author information
Authors and Affiliations
Contributions
Substantial contributions to study conception and design: JAW, ABG, BH, PJM; substantial contributions to the acquisition of data: JAW, ABG, BH, PJM; substantial contributions to analysis and interpretation of the data: JAW, ABG, BH, TN, PJM; drafting the article or revising it critically for important intellectual content: JAW, ABG, BH, TN, PJM; and final approval of the article to be published: JAW, ABG, BH, TN, PJM.
Corresponding author
Ethics declarations
Conflict of interest
JAW: Consultancies: Abbott, Celgene, UCB Pharma, and Novartis; ABG: Consultancies, speaking fees and honoraria: AbbVie, Janssen, Eli Lilly, Novartis, UCB Pharma, Allergan, Sienna, BMC, Sun Pharma; BH: Employee of UCB Pharma; TN: Employee of UCB Pharma; PJM: Research grants: AbbVie, Amgen, Bristol-Myers Squibb, Janssen, Eli Lilly, Novartis, Pfizer, Sun Pharma, UCB Pharma; Consultancies: AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Janssen, Eli Lilly, Novartis, Pfizer, Sun Pharma, UCB Pharma; Speakers’ bureau: AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Genentech, Janssen, Novartis, Pfizer, UCB Pharma.
Study sponsor information
UCB sponsored the study and the development of the manuscript and reviewed the text to ensure that from a UCB perspective, the data presented in the publication are scientifically, technically, and medically supportable, that they do not contain any information that has the potential to damage the intellectual property of UCB, and that the publication complies with applicable laws, regulations, guidelines, and good industry practice. The authors approved the final version to be published after critically revising the manuscript for important intellectual content.
Additional information
The original version of this article was revised: The above article originally published with an error present in Discussion section, “cavailable in the published literature, ...” should instead have read “Given the paucity of data currently available in the published literature, ...”
Electronic supplementary material
ESM 1
(DOCX 150 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Walsh, J.A., Gottlieb, A.B., Hoepken, B. et al. Efficacy of certolizumab pegol with and without concomitant use of disease-modifying anti-rheumatic drugs over 4 years in psoriatic arthritis patients: results from the RAPID-PsA randomized controlled trial. Clin Rheumatol 37, 3285–3296 (2018). https://doi.org/10.1007/s10067-018-4227-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-018-4227-7