
Abstract
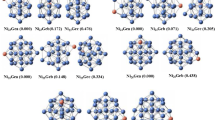
The stable geometries and atomization energies for the clusters Ni n (n = 2–5) are predicted with all-electron density functional theory (DFT), using the BMK hybrid functional and a Gaussian basis set. Possible isomers and several spin states of these nickel clusters are considered systematically. The ground spin state and the lowest energy isomers are identified for each cluster size. The results are compared to available experimental and other theoretical data. The molecular orbitals of the largest cluster are plotted for all spin states. The relative stabilities of these states are interpreted in terms of superatom orbitals and no-pair bonding.

The stable geometries and atomization energies for the clusters Ni n (n = 2–5) are predicted with all-electron density functional theory (DFT), using the BMK hybrid functional and a Gaussian basis set. Possible isomers and several spin states of these nickel clusters are systematically considered. The ground spin state and the lowest energy isomers are identified for each cluster size. The results are compared to available experimental and other theoretical data. The molecular orbitals of the largest cluster are plotted for all spin states. The relative stabilities of these states are interpreted in terms of superatom orbitals and no-pair bonding

Similar content being viewed by others

References
Ovsitser O, Kondratenko EV (2009) Similarity and differences in the oxidative dehydrogenation of C2–C4 alkanes over nano-sized VOx species using N2O and O2. Catal Today 142:138–142. doi:10.1016/j.cattod.2008.09.012
Uddin J, Morales CM, Maynard JH, Landis CR (2006) Computational studies of metal–ligand bond enthalpies across the transition metal series. Organometallics 25:5566–5581. doi:10.1021/om0603058
Simoes JAM, Beauchamp JL (1990) Transition metal hydrogen and metal carbon bond strengths: the keys to catalysis. Chem Rev 90:629–688
Zubarev DY, Boldyrev AI (2008) Developing paradigms of chemical bonding: adaptive natural density partitioning. Phys Chem Chem Phys 10:5207–5217. doi:10.1039/b804083d
Zubarev DY, Boldyrev AI (2009) Deciphering chemical bonding in golden cages. J Phys Chem A 113:866–868. doi:10.1021/jp808103t
Jensen KP, Roos BO, Ryde U (2007) Performance of density functionals for first row transition metal systems. J Chem Phys 126:014103–014116
Furche F, Perdew JP (2006) The performance of semilocal and hybrid density functionals in 3d transition-metal chemistry. J Chem Phys 124:044103–044129
Harrison JG (1983) Density functional calculations for atoms in the 1st transition Series. J Chem Phys 79:2265–2269
Arvizu GL, Calaminici P (2007) Assessment of density functional theory optimized basis sets for gradient corrected functionals to transition metal systems: the case of small Ni n (n ≤ 5) clusters. J Chem Phys 126:194102–194111. doi:19410210.1063/1.2735311
Kant A (1964) Dissociation energies of diatomic molecules of the transition elements. I. Nickel. J Chem Phys 41:1872–1876
Knickelbein MB, Yang S, Riley SJ (1990) Near-threshold photoionization of nickel clusters: ionization potentials for Ni3 to Ni90. J Chem Phys 93:94–104
Luo CL (2000) The structure of small nickel clusters: Ni2–Ni19. Model Simul Mater Sci Eng 8:95–101
Michelini MC, Diez RP, Jubert AH (2004) Density functional study of the ionization potentials and electron affinities of small Ni n clusters with n = 2–6 and 8. Comput Mater Sci 31:292–298. doi:10.1016/j.commatsci.2004.03.018
Moskovits M, Hulse JE (1977) Ultraviolet-visible spectra of diatomic, triatomic, and higher nickel clusters. J Chem Phys 66:3988–3994
Nygren MA, Siegbahn PEM, Wahlgren U, Akeby H (1992) Theoretical ionization energies and geometries for Ni N (4 ≤ N ≤9). J Phys Chem 96:3633–3640
Onal I, Sayar A, Uzun A, Ozkar S (2009) A density functional study of Ni2 and Ni13 nanoclusters. J Comput Theor Nanos 6:867–872. doi:10.1166/jctn.2009.1119
Parks EK, Zhu L, Ho J, Riley SJ (1994) The structure of small nickel clusters Ni3–Ni15. J Chem Phys 100:7206–7222
Pinegar JC, Langenberg JD, Arrington CA, Spain EM, Morse MD (1995) Ni2 revisited: reassignment of the ground electronic state. J Chem Phys 102:666–674
Reuse FA, Khanna SN (1995) Geometry, electronic-structure, and magnetism of small Ni N (N = 2–6, 8, 13) clusters. Chem Phys Lett 234:77–81
Reuse FA, Khanna SN (1999) Photoabsorption spectrum of small Ni n (n = 2–6, 13) clusters. Eur Phys J D 6:77–81
Pouamerigo R, Merchan M, Nebotgil I, Malmqvist PA, Roos BO (1994) The chemical-bonds in CuH, Cu2, NiH, and Ni2 studied with multiconfigurational 2nd-order perturbation theory. J Chem Phys 101:4893–4902
Grigoryan VG, Springborg M (2004) Structural and energetic properties of nickel clusters: 2 ≤ N ≤ 150. Phys Rev B 70:205415–205429. doi:10.1103/PhysRevB.70.205415
St Petkov P, Vayssilov GN, Kruger S, Rosch N (2006) Structure, stability, electronic and magnetic properties of Ni4 clusters containing impurity atoms. Phys Chem Chem Phys 8:1282–1291. doi:10.1039/b518175e
Xie Z, Ma QM, Liu Y, Li YC (2005) First-principles study of the stability and Jahn–Teller distortion of nickel clusters. Phys Lett A 342:459–467. doi:10.1016/j.physleta.2005.05.067
Boese AD, Martin JML (2004) Development of density functionals for thermochemical kinetics. J Chem Phys 121:3405–3416
Frisch MJ et al. (1994–2003, 2004) Gaussian 03, revision D.01. Gaussian Inc., Wallingford
Wachters AJ (1970) Gaussian basis set for molecular wavefunctions containing third-row atoms. J Chem Phys 52:1033–1038
Hay PJ (1977) Gaussian basis sets for molecular calculations—representation of 3d orbitals in transition-metal atoms. J Chem Phys 66:4377–4384
Harris J (1985) Simplified method for calculating the energy of weakly interacting fragments. Phys Rev B 31:1770–1779
Goel S, Masunov AE (2008) Potential energy curves and electronic structure of 3d transition metal hydrides and their cations. J Chem Phys 129:214302–14
Goel S, Masunov AE (2008) First-principles study of transition metal diatomics as the first step in multiscale simulations of carbon nanotube growth process. In: 4th Int Conf on Multiscale Material Modeling, Tallahassee, FL, USA, 27–31 Oct 2008, pp 110–113
Rabuck AD, Scuseria GE (1999) Improving self-consistent field convergence by varying occupation numbers. J Chem Phys 110:695–700
Wang HM, Haouari H, Craig R, Lombardi JR, Lindsay DM (1996) Raman spectra of mass-selected nickel dimers in argon matrices. J Chem Phys 104:3420–3422
Ham FS (2000) The Jahn–Teller effect: a retrospective view. J Lumin 85:193–197
Jiang DE, Whetten RL, Luo WD, Dai S (2009) The smallest thiolated gold superatom complexes. J Phys Chem C 113:17291–17295. doi:10.1021/jp9035937
Monari A, Pitarch-Ruiz J, Bendazzoli GL, Evangelisti S, Sanchez-Marin J (2010) High-spin states in tetrahedral X4 clusters (X = H, Li, Na, K). Int J Quantum Chem 110:874–884. doi:10.1002/qua.21987
Olson JK, Boldyrev AI (2009) Ab initio search for global minimum structures of the novel B3H y (y = 4–7) neutral and anionic clusters. Inorg Chem 48:10060–10067. doi:10.1021/ic900905h
Acknowledgments
This work was supported in part by a University of Central Florida (UCF) start-up grant. SG gratefully acknowledges an I2lab fellowship. The computer time was generously provided by the Department of Energy’s (DOE) National Energy Research Scientific Computing Center (NERSC), the Institute for Simulation and Training’s supercomputing facility (STOKES), and the UCF I2lab.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Goel, S., Masunov, A.E. Density functional theory study of small nickel clusters. J Mol Model 18, 783–790 (2012). https://doi.org/10.1007/s00894-011-1100-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-011-1100-x