
Abstract
Sr-substitution in the crystal structure of bredigite has been studied in detail. Samples of a hypothetical solid-solution series with nominal composition Ca7-xSrxMg[SiO4]4 (x = 0, 2, …,7) have been prepared from sinter reactions in the temperature range between 1275 and 1325 °C and characterized using powder and single-crystal X-ray diffraction. Synthesis runs between x = 1 and x = 4 resulted in compounds with increasing Sr contents, for which single-crystal diffraction studies revealed the following Sr/(Sr + Ca) atomic ratios: 0.133,0.268, 0.409 and 0.559. They are isostructural to the pure calcium end-member (x = 0) and adopt the orthorhombic space group Pnnm. Evolution of the unit-cell parameters and cell volumes of the solid-solution series are defined by linear or nearly linear trends when plotted against the Sr/(Sr + Ca) atomic ratio. Replacement of calcium with strontium atoms on the different sites shows clear preferences for specific positions. For the experiment with x = 5, formation of bredigite-related single-crystals with Sr/(Sr + Ca) = 0.675 was observed. These samples, however, exhibited a halved c lattice parameter when compared with the corresponding value in the Pnnm structure, pointing to a compositionally induced phase transition somewhere in region between Sr/(Sr + Ca) = 0.559 and 0.675. The crystal structure of this new phase with composition Ca2.32Sr4.82Mg0.86[SiO4]4 was successfully determined in space group Pbam. Basic crystallographic data are as follows: a = 18.869(2) Å, b = 6.9445(8) Å, c = 5.5426(6) Å, V = 726.28(14) Å3, Z = 2. Structure determination was accomplished using charge flipping. Subsequent least-squares refinements resulted in a residual of R(|F|) = 2.70% for 822 independent reflections and 87 parameters. The Pbam- and the Pnnm-structures are in a group-subgroup relationship and topologically related. Both are based on so-called pinwheel-like MSi6O24 clusters consisting of a central magnesium-dominated [MO6]-octahedron as well as six attached [SiO4]-tetrahedra. The clusters are linked into chain-like elements running along [001]. Linkage between the chains is provided by mixed Sr/Ca positions with 6 to 10 oxygen ligands. Differences between the two phases result from changes in Sr-Ca site occupancies in combination with displacements of the atoms and tilts of the tetrahedra. The distortion pattern has been studied using group-theoretical methods including mode analysis. Notably, for the samples with x = 6 and x = 7—the latter corresponding to the hypothetical pure strontium end-member composition—no bredigite-type phases could be identified, indicating that there is an upper limit for the Sr-uptake.
Similar content being viewed by others

Introduction
Bredigite (ideally Ca7Mg[SiO4]4) is an exotic phase, presently known from only a handful of localities listed in the mindat.org database (http://www.mindat.org, accessed May 3rd, 2021) including some famous areas for the discovery of new minerals such as the Hatrurim complex, Negev Desert, Israel (Sharygin et al. 2008; Kahlenberg et al. 2019) or the Ettringer Bellerberg volcano in the Eifel region, Germany (Blaß and Kruijen 2014). Despite of its rareness, bredigite occurs in quite a number of different petrological settings all of which are related to pyrometamorphism (Tilley and Vincent 1948; Bridge 1966; Grapes 2011; Žáček et al. 2015; Kahlenberg et al. 2019). However, there are also bredigite samples in nature that are of anthropogenic origin due to combustion processes in coal mine dumps, which triggered thermal alteration of pre-existing sedimentary rocks (Sharygin 2010).
On the other hand, synthetic equivalents to bredigite have been described from several products related to the field of applied mineralogy. For example, bredigite is a component in steel and stainless steel making wastes (Bodor et al. 2013). Recent investigations (Bodor et al. 2013; Chiang et al. 2013) studied the carbonation of different compounds from these slags with the purpose of CO2 sequestration. Interestingly, bredigite exhibited the highest CO2-uptake of all relevant crystalline phases. Furthermore, bredigite-containing slags have been investigated as potential hydraulic binders (Pontikes et al. 2013; Ferreira Neto et al. 2017).
Another field of application where bredigite comes into play are bone replacement and bone formation ceramics or coatings, since bredigite combines excellent biocompatibility with apatite mineralization ability and mechanical properties close to cortical bone (Wu et al. 2005; Razavi et al. 2013; Yi et al. 2014; Dezfuli et al. 2017; Huang et al. 2017).
Structural characterization of bredigite—in early phase equilibrium studies also referred to as “phase T” (Sharp et al. 1960; Gutt 1961)—has seen several trials and tribulations that have been summarized by Moseley and Glasser (1981). Since the early publication of Tilley and Vincent (1948) bredigite was believed to be isostructural with α’-Ca2SiO4 for more than 25 years. However, already Douglas (1952) pointed out that the powder diffraction patterns of both compounds showed pronounced differences and concluded that their identity cannot be established with absolute certainty. The final proof that phase T is a stoichiometric Mg-containing compound with idealized composition Ca7Mg[SiO4]4 and not isotypic with α’-Ca2SiO4 was delivered by Saalfeld (1974) and the crystal-structure analysis of Moore and Araki (1976). The Moore and Araki model was slightly modified by Kahlenberg et al. (2019). Though principally correct, the previous study had described the bredigite structure in an unnecessarily low acentric Pnn2 space-group symmetry.
So far, information on cation substitutions into the bredigite structure is scarce. According to Moseley and Glasser (1981, 1982), Mg2+ ions cannot be replaced by divalent cations such as Zn2+, Co2+ or Ni2+ between 1300 and 1350° C using solid-state reactions. By contrast, it is well known that larger amounts of manganese can be found in bredigites from metallurgical slags (Moore and Araki 1976). Quite surprisingly, however, the targeted incorporation of Mn2+ by the abovementioned synthesis route was not possible either. Replacements of larger alkaline earth cations (Sr, Ba) for Ca were seemingly more successful. When compared with the data for the pure calcium end-member, samples with nominal compositions Ca6.5Sr0.5 Mg[SiO4]4, Ca6SrMg[SiO4]4 and Ca6.5Ba0.5 Mg[SiO4]4 showed a significant increase in the values of the unit-cell volumes pointing to a successful substitution among the larger divalent cations. However, due to the limitations of structural studies based on X-ray powder diffraction data in the era before the Rietveld method became a standard tool for this purpose, Moseley and Glasser (1982) did not perform any more detailed crystallographic investigations on their polycrystalline samples.
The main goals of the present study were (i) to investigate the potential solid-solution series (Ca7-xSrx)Mg[SiO4]4 in more detail and (ii) to present structural data on the individual members of the series.
Experimental details
Starting materials were based on mixtures of CaCO3 (Merck, > 99.0%), SrCO3 (Merck, > 99.0%), SiO2 (Alfa Aesar, 99.5%), and MgO (Merck, 99%). Eight different samples of 2 g each were prepared for a hypothetical solid-solution series with nominal composition Ca7-xSrxMg[SiO4]4 (x = 0, 1, …,7). For easy reference, we will denote these mixtures Bred-0 to Bred-7. Before weighing on an analytical balance the educts were dried for 12 h at 300 °C in order to eliminate any physically adsorbed water. Since magnesia is known to be gradually hydrated to Mg(OH)2 after longer storage periods, the material taken from the bottle was pre-heated at 700 °C for 24 h. A planetary mill operated at 600 rpm was used for homogenization for 45 min. under ethanol. The resulting slurry was dried at 50 °C to remove the alcohol completely and stored in a desiccator. Subsequently, the educts were pressed into pellets (approximately 0.5 g, having a diameter of 12 mm and a thickness of about 3 mm). Two pellets for each composition were placed on platinum foils contained in alumina ceramic combustion boats, transferred to a resistance heated chamber furnace and heated from ambient temperature with a heating ramp of 50 °C/h to 1275 °C. The porous sinter-pellets were removed after 100 h and quenched in air. The samples were re-ground, re-pressed and fired for a second time at 1325 °C, respectively. After the second cycle the compact sinter tablets were crushed in an agate mortar and split into two parts.
One half was further pulverized and prepared for powder X-ray diffraction (PXRD). The patterns of the bulk material from the synthesis products were recorded with a Bruker D8-Discover diffractometer in Bragg–Brentano geometry. The device is equipped with a primary beam Ge(111)-monochromator, primary and secondary Soller slits and a LYNXEYE™ silicon strip detector. Data have been acquired in the range between 10° and 90° 2θ with a step size of 0.01° and a measurement time of 2.5 s per step. The Cu-X-ray tube was operated with 40 kV and 40 mA. The software DIFFRAC.EVA (Bruker-AXS 2020) in combination with the ICDD PDF-4 + database version 4.2103 was used for qualitative phase analysis of the powder diffraction patterns which revealed the multi-phase character of the products. Subsequent quantitative phase analysis (QPA) of the crystalline products was based on the Rietveld method performed with the program DIFFRAC.TOPAS (Bruker-AXS 2018). Peak profiles were modeled using the fundamental parameter approach. For simulation of the background Chebychev polynomials were selected.
From the remaining part of each sample small single-crystals up to 100 μm in diameter could be extracted which were further checked by polarization microscopy. Notably, many crystals were intensively twinned when observed between crossed-polarizers. Crystals of sufficient optical quality were fixed on the top of glass fibers with nail polish and further examined on a Rigaku Oxford Diffraction Gemini R Ultra diffractometer equipped with a four-circle kappa-goniometer and a Ruby CCD detector. Measurements were conducted with Mo-radiation. For the samples Bred-1 to Bred-5, crystals showing bredigite-related unit cells could be found. From each of these sets, the crystal with the best overall diffraction quality was finally selected for full data collection (see Table 1). The CrysAlisPRO software package (Rigaku 2015) was employed to process the data. After indexing, the diffraction patterns were integrated. Further data reduction included Lorentz and polarization as well as an absorption correction. Diffraction symmetry of all crystals conformed to the orthorhombic Laue group 2/m 2/m 2/m. For the crystals from samples Bred-1 to Bred-4 the values of the lattice parameters indicated the presence of bredigite-type phases isostructural to the pure calcium end-member. Consequently, the structure model of Kahlenberg et al. (2019) for a natural bredigite in space group Pnnm was used as a starting point for structure analysis. The crystals retrieved from sample Bred-5 showed a halved c lattice parameter when compared with the corresponding value in bredigite, pointing to a compositionally induced phase transition. The crystal structure of this compound was successfully determined in space group Pbam using charge flipping (program SUPERFLIP; Palatinus and Chapuis 2007).
Full-matrix least-squares refinements for all five samples were performed with the program SHELX-97 (Sheldrick 2008) embedded in the software suite WinGX (Farrugia 1999). X-ray scattering factors were taken from the International Tables for Crystalllography, Vol. C (Wilson 1992). The optimization calculations including fractional atomic coordinates, anisotropic displacement parameters and calcium-strontium as well as calcium-magnesium atom populations on the relevant sites converged to residual values for R(|F|) between 2.70 and 4.80%. During the refinements, no constraints concerning the bulk chemistry of the crystals were applied. However, full occupancy of the different cation positions was assumed. Notably, the resulting bulk compositions depart somewhat from ideal Ca7-xSrxMg[SiO4]4 and consistently exhibit less than one Mg-atom per formula unit (see Table 1). Final coordinates, site occupancies as well as equivalent isotropic displacement parameters are given in Tables 2 and 3, respectively. Anisotropic displacement parameters are listed in the supplementary Tables S1 and S2. Selected interatomic distances are summarized in Tables 4 and 5. Figures showing structural features were prepared using the program VESTA3 (Momma and Izumi 2011). The same program was also employed for the calculation of the effective coordination numbers (ECoN) as well as volumes and distortion indices of the MOx-polyhedra which can be found in Tables 4 and 5.
Results
X-ray powder diffraction and phase analysis
Evaluation of the powder diffraction patterns confirmed the earlier observation of Moseley and Glasser (1982) that the kinetics of bredigite formation by solid-state reactions is sluggish. After two burning cycles, the bredigite yield for the pure calcium end-member composition (Bred-0) was quite low. Actually, the following phases were identified: Ca-bredigite (43 wt%), β-Ca2SiO4 (29 wt%), γ-Ca2SiO4 (18 wt%), merwinite (5 wt%), åkermanite (2 wt%), periclase (2 wt% and lime (1 wt%) (see Fig. 1a). The numbers in parentheses refer to weight percentages obtained from QPA based on the corresponding Rietveld refinements. Structure models for the individual phases were taken either from the literature or from the data reported in this paper. According to our experience the accuracy of the QPA results is estimated to be 0.5—2 wt%. At any rate the real errors are significantly higher than those from the Rietveld analysis software reflecting only the precision of the mathematical fit between measured and calculated step intensities. For the samples Bred-1 to Bred-4, the yields for the Sr-bredigite phases increased with increasing Sr-content reaching 98 wt% for Bred-4. Within these samples, Sr-åkermanite (Sr2MgSi2O7) was always observed as an impurity phase (see Fig. 1b). Notably, sample Bred-5 contained minor amounts of the Pnnm-phase (6 wt%) in addition to the Pbam-polymorph which accounted for 90 wt% of the sample. For Bred-6 and Bred-7 no bredigite formation could be observed – at least within the detection limit of PXRD which is about 1 wt%. The sample Bred-7, for instance, consisted of several Sr- and Sr-Mg-silicates including Sr3MgSi2O8 (39 wt%), Sr-åkermanite (5 wt%), α’-Sr2SiO4 (48 wt%) as well as β-Sr2SiO4 (8 wt%) (see Fig. 1c).

Rietveld plots of the powder X-ray diffraction patterns of selected samples synthesized from solid-state reactions: (a) Bred-0, (b) Bred-5 and (c) Bred-7. Observed step intensities are presented by red dots. Calculated step intensities (solid blue line) have been modeled based on a mixture of the crystalline phases listed in the insets. Tick marks for the Bragg peaks of each phase are given. The lower black line represents the difference curve between observed and calculated step intensities
Pnnm solid-solution series
As mentioned above, the crystals obtained from the samples Bred-1 to Bred-4 are isotypic with bredigite, whose atomic arrangement has been already described in detail by Moore and Araki (1976) as well as Kahlenberg et al. (2019). Therefore, we will refrain from an in-depth discussion of the crystal structure but rather focus on the most important aspects. Many of the structural features described in the following paragraphs will be also of relevance for the Pbam—modification which will be addressed later.
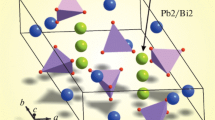
Bredigite belongs to the group of nesosilicates and is based on so-called pinwheel-like clusters, an expression that has been coined by Moore and Araki (1976). A single pinwheel consists of a central magnesium-dominated octahedron as well as six attached [SiO4]-tetrahedra (see Fig. 2a). Within the unit-cell four of these MSi6O24 polyhedral microensembles (PME) can be found, each of which resides on positions with site-symmetry 2/m. Using the classification of Ilyushin and Blatov (2002) based on the calculation of the coordination sequences up to k = 3, the PME can be denoted as {6,6,18}. The PME are located in about 7 Å wide layers which are parallel to (100). Within these slabs, chain-like elements along [001] can be identified (cf. Fig. 3a). A single chain consists of an alternating sequence of the pinwheels centered around M7 and M8, where a single octahedron shares two tetrahedra on each side with two adjacent octahedra. Linkage between proximate chains along [010] is provided by M1 and M2, whose coordination polyhedra can be described as strongly distorted trigonal antiprisms sharing common edges (for M2) or faces (for M1) with the [M7O6]- and [M8O6]-octahedra. Notably, for the antiprism around the M1 position four additional M–O bonds can be found up to 3.0 Å which should be actually considered for the coordination environment. Neighboring layers, in turn, are linked by the positions M3, M4, M5, and M6 having irregular coordination environments with 7 to 9 next oxygen neighbors (see Table 4). For the Sr-richest member of the Pnnm-type solid-solution series (Sr/(Ca + Sr) = 0.559) minor changes in the coordination environment have been observed. Actually, the following longer bonds have to be considered as well: M4: one bond to O3 with 2.983 Å and M5: two bonds to O3 with 2.949 Å each as well as one bond to O6 with 2.977 Å. Projections of the whole crystal structure for the representative with Sr/(Ca + Sr) = 0.133 are shown in Figs. 4a and 5a, respectively.

MSi6O24 polyhedral microensembles (PME) or pinwheel clusters in the (a) Pnnm and (b) Pbam structure-type. Blue spheres represent oxygen atoms. The six tetrahedra exhibit an UDUDUD conformation of up (U) and down (D) pointing apices

Chain-like elements of linked pinwheels in the (a) Pnnm and (b) Pbam structure-type. The chains are running along [001]

Projections of the whole structures along [010]: (a) Pnnm- and (b) Pbam-structure type. Sr- and Ca cations are given by pink and green spheres. Bi-colored spheres indicate mixed Sr-Ca positions. Occupancies refer to the values for samples Bred-1 and Bred-5, respectively

Projections of the whole structures along [001]: (a) Pnnm- and (b) Pbam-structure type. Sr- and Ca cations are given by pink and green spheres. Bi-colored spheres indicate mixed Sr-Ca positions. Occupancies refer to the values for the samples Bred-1 and Bred-5, respectively
The substitution of a larger cation like Sr2+ for the smaller Ca2+ increases, as expected, the size of the unit-cell volume. Actually, the volume gain between Bred-1 and Bred-4 corresponds to 67.7 Å3 or 4.97%. As can be seen from Fig. 6, the volume data do not exhibit a significant deviation from a linear behavior when plotted against the Sr/(Sr + Ca) atomic ratio. The volume changes are also reflected in an increase of the individual lattice parameters a, b and c (see Fig. 7a to c), which are enlarged by about 1.89, 1.90 and 1.11%, respectively. This indicates that the a and b directions (showing almost identical change rates) are more affected by the strontium incorporation when compared with c. Including the literature data for a natural bredigite having a composition close to the pure calcium end-member (Kahlenberg et al. 2019), the evolution of the unit-cell volume and the individual lattice parameters as a function of the Sr/(Sr + Ca) atomic ratio X can be described by the following linear expressions
where R2 is the coefficient of determination.

Evolution of the unit-cell volumes of the samples from the runs Bred-1 to Bred-4 (squares) and Bred-5 (filled circle) as a function of the Sr/(Sr + Ca) atomic ratio. The data point for Sr/(Sr + Ca) = 0 has been taken from Kahlenberg et al. (2019)

Evolution of the lattice parameters of the samples from the runs Bred-1 to Bred-4 (squares) and Bred-5 (filled circle) as a function of the Sr/(Sr + Ca) atomic ratio. The data point for Sr/(Sr + Ca) = 0 has been taken from Kahlenberg et al. (2019)
The octahedrally coordinated M7 and M8 sites in the centers of the pinwheel-like moieties are Mg-dominated. However, site-population refinements indicate the presence of small but nevertheless significant amounts of Ca2+-cations on these positions as well. Concerning the uptake of calcium atoms both sites exhibit an unlike behavior. While the Ca-Mg distributions on the M8-position do not allow to discern a clear trend as a function of the bulk composition, the M7 site incorporates increasing amounts of calcium atoms with increasing overall Sr/(Sr + Ca) atomic ratio of the crystals.
Strontium cation substitution in the solid-solution series is limited to the positions M1 to M6. However, these six crystallographically independent sites show different preferences for the incorporation of Sr2+ (see Fig. 8). The primary sink for Sr is M1. Even for the lowest Sr/(Sr + Ca) atomic ratio of X = 0.133 more than 90% of the calcium cations have already been replaced. The corresponding values for the remaining sites are drastically lower for this Ca-richest composition. From X = 0.409 onwards, M1 can be eventually considered a pure strontium position. The Sr-occupancies for the sites M2, M4, M5, and M6 start from comparatively low levels but increase almost linearly and finally reach values between 62 and 90%. By contrast, the M3-position exhibits only minor Sr-concentrations which vary between 0% (for X = 0.133) to 16% (for X = 0.559).

Variation of the Sr-concentrations for the six M sites outside the pinwheel-like building units as a function of the Sr/(Sr + Ca) atomic ratio
The Si–O bond distances as well as O-Si–O angles of the three crystallographically independent [SiO4]-tetrahedra are in the normal range for silicate compounds (Liebau 1985). When comparing the <Si–O>-distances for the different members of the solid- solution series, no clear trends could be observed, i.e. the influence of the Sr/(Sr + Ca) atomic ratio on the geometry of the tetrahedra is insignificant. On the other hand, there are clear trends concerning the tilting angles of the [SiO4]-moieties. For example, the orientation of the tetrahedron around Si1 residing on a general position can be specified by two angles Ψ and Φ. Ψ can be defined as the angle between the base plane given by the oxygen atoms O1, O2, O3 and the [010]-direction, while Φ represents the angle between the edge O1-O2 and [001] (see Figs. 3a and 5a, respectively). Ψ as well as Φ exhibit a continuous decrease with increasing Sr/(Sr + Ca) atomic ratio. Actually, the values are reduced from 8.82 to 4.99° (for Ψ) and 8.81 to 4.94° (for Φ), i.e. there is progress towards a less-tilted orientation of the [Si1O4]-groups and we will come back to this point in the Discussion section. The tetrahedra centered by Si2 and Si3 are located on mirror planes perpendicular to [001]. Therefore, changes in orientation are more restricted and can be conveniently described with only one rotational parameter each: Δ and Λ. They represent the angles between the b-axis and the O6-O7 edge (for the [Si2O4]-tetrahedron) as well as the O9-O10 edge (for the [Si3O4]-unit), respectively (see Fig. 5a). Whereas Δ decreases from 46.48 to 40.87°, Λ increases from 9.09 to 14.54°.
For the Sr-free octahedra around M7 and M8, there is a gap between the <M7-O>—and the <M8-O>-values which could be principally rationalized by the higher Ca-concentrations on M7, whose values increase from Bred-1 to Bred4. The average bond distances as well as the polyhedral volumes of the M7O6-octahedra, however, remain fairly constant within the series (see Table 4). For the M8 site, on the other hand, no clear trend concerning the Ca-Mg-incorporation could be detected, but nevertheless the <M8-O> distances and the volumes of the octahedra increase with increasing Sr-concentration of the mixed crystals. These findings point to the presence of an additional induced effect on the dimensions of the octahedra belonging to the pinwheels resulting from the Sr-incorporation of the remaining six M sites. The analysis of the evolution of the corresponding <M–O> values for these six positions shows an increase with rising overall Sr/(Sr + Ca) atomic ratios (see Table 4). Not surprisingly, the M1-position representing the “largest void” in the structure showing the largest polyhedral volume and the highest coordination number of 10 is especially prone to incorporation of strontium atoms. This finding confirms an earlier hypothesis presented by Moseley and Glasser (1982) who suggested a preferred uptake of larger alkaline-earth cations such as Sr or Ba on this site. As already mentioned, for the Sr-richest member of the solid-solution series an increase of the coordination number for two of the M sites from 9 to 10 (for M4) and from 7 to 10 (for M5) has been observed. A more detailed analysis of the evolution of the coordination environments for those cations that do not exhibit any changes in their coordination spheres can be achieved by the calculation of their effective coordination number (ECoN) as defined by Hoppe (1979) (see Table 4). The most pronounced changes have been observed for M1 (increase by about 20%), while the values for M3 remain almost constant. The low ECoN-values for the M6 site ranging from 4.14 to 4.58 indicate that the formal nine-fold coordination is more adequately described as a 4 + 5 coordination. Actually, this position also exhibits the most significant bond-lengths distortion index (see Table 4).
Pbam structure
The crystal structure of the Pbam polymorph is based on the same principal building blocks that have been identified in the Pnnm structure: chains of MSi6O24-clusters running along [001] (see Figs. 2b and 3b). However, for the Pbam case, adjacent pinwheels along the chains are equivalent by translation symmetry. Actually, the local symmetry of the pinwheels in both structure types are identical: 2/m. An important difference stems from the fact the bridging tetrahedra about Si1 in Pbam are located on mirror planes as well, whereas the corresponding groups in the Pnnm-structure are twisted and tilted away from the special positions which results in a loss of these mirror planes (see Fig. 2a and b). A more detailed analysis of the relative distortions between both modifications based on concepts of group-theory will be presented in the next section.
Concerning the geometry of the [SiO4]-tetrahedra it can be noted that the range of the individual Si–O distances in the Pbam modification is larger, but that distortion parameters such as quadratic elongations (λ) or bond angle variances (σ2) as defined by Robinson et al. (1971) are not that much influenced. While the values for Si1 and Si2 range from 1.001 to 1.003 (for λ) and 4.796 and 14.685 (for σ 2) the corresponding data for the three symmetrically independent tetrahedra in sample Bred-4 scatter between 1.001—1.005 and 4.634—22.395, respectively.
The most dramatic change with respect to Sr/Ca-occupancies can be observed for the M2 site corresponding to the position M3 in the Pnnm-structure. Actually, the Sr-content increases from 16% (in sample Bred-4) to about 65% in the Pbam-phase. Furthermore, the number of next oxygen neighbors shows an increase for this position from eight to nine, as do the average bond distances (rise from 2.529 to 2.630 Å, see Table 5).
Finally, it has to be emphasized that Moore and Araki (1976) in their landmark paper on the genealogy of alkaline-earth orthosilicates already sketched a structural model of a bredigite aristotype having a Pbam arrangement (or Pmcb according to their preferred setting) which conforms closely with our experimentally determined crystal structure for Ca2.32Sr4.82Mg0.86[SiO4]4. We are pleased that we were able to confirm the realization of this hypothetical structure whose derivation 45 years ago was based on purely geometrical considerations.
Discussion
Diffraction experiments point to a klassengleiche phase transition of index 2 somewhere in the compositional range between Sr/(Sr + Ca) = 0.559 and 0.675. The space groups Pbam (high symmetry) and Pnnm (low symmetry) are in a group-subgroup relationship and the crystal structures have similar topological features. In summary, one can say that the atomic coordinates of the structure types are related by the following transformation (P,p): a,b,2c (0,0,0).
A more detailed understanding of the phase transformation can be achieved by symmetry-mode analysis. In the course of this procedure implemented in the program AMPLIMODES (Orobengoa et al. 2009) the structural distortions present in the low-symmetry structure are decomposed into contributions from different modes, whose symmetries are given by the irreducible representations (irreps) of the space group of the parent or high-symmetry phase.
Due to the maximal group-subgroup relationship, the onset of one primary mode could already explain the observed symmetry break directly. It corresponds to the irrep \({Z}_{2}^{+}\) of space group Pbam associated with the point Z (0,0,½) of the first Brillouin zone. Furthermore, the trivial fully symmetrical \({\Gamma }_{1}^{+}\) distortion is also allowed that retains the symmetry of the Pbam-structure.
Using the crystallographic data for the boundary compositions with Sr/(Sr + Ca) = 0.559 (Pnnm) and 0.675 (Pbam), a reference structure was calculated in a first step representing the parent phase, but expressed in the low-symmetry space group (see supplementary Table S3). Notably, several Wyckoff orbits have split. Due to the splitting, the number of independent atoms in the asymmetric unit increases from 13 to 21. The resulting displacement field obtained from the comparison of the reference structure with the low-symmetry Pnnm-phase is given in Table 6. The displacement vectors u for the atoms in the asymmetric unit of the reference structure listed in this table completely define the displacive distortions relating both structures. It is obvious, that the oxygen atoms are distinctly more affected by the phase transformation. Their absolute shifts vary between 0.162 and 0.509 Å. The silicon atoms and most of the M sites show considerably smaller displacements. An exception, however, is the M2 position which is shifted by about 0.188 Å. Actually, this site shows largest increase in Sr-occupancy due to the phase transition. The average displacement of all corresponding atom pairs in both structures has a value of 0.159 Å.
In the next step of mode analysis, the absolute amplitudes for the two individual components of the global distortions were calculated. The relevant values are 2.104 Å (for \({Z}_{2}^{+}\)) and 0.234 Å (for \({\Gamma }_{1}^{+}\)), i.e. the \({Z}_{2}^{+}\)-mode is about ten times larger, indicating that the onset of this mode triggers the transition. However, for a complete understanding the contributions of the trivial non-symmetry breaking \({\Gamma }_{1}^{+}\)-mode have to be considered as well.
Finally, for each involved irrep the corresponding polarization vector was obtained. Supplementary Table S4 shows a crystallographic description of these normalized vectors. For the asymmetric unit of the reference structure the set of correlated atomic displacements (in relative units) is given for both irreps. The actual distortion of a specific mode can be easily obtained by multiplying the components of the polarization vector with the amplitudes mentioned above. For \({Z}_{2}^{+}\), for example, columns 5 to 7 have to multiplied with the corresponding amplitude of 2.104 Å. From inspection of Table S4 it is clear that neither \({Z}_{2}^{+}\) nor \({\Gamma }_{1}^{+}\) induce any displacement of the M1- or the M5-position. For the M2 site, for example, the displacement vectors related to \({Z}_{2}^{+}\) are strictly parallel or anti-parallel to [001]. In order to obtain a concise graphical overview of the distortion fields, individual displacements of the most affected atoms (M2, O1-O6) calculated for the dominant \({Z}_{2}^{+}\) representation only have been visualized using the program VESTA3 (see Fig. 9).

Displacement field induced by the irrep \({Z}_{2}^{+}\) on the most affected atoms (M2 site, oxygen anions) of the reference structure. Labeling has been performed in accordance with Table 6. For sake of better visibility, the magnitudes of the individual shift vectors (red) have been scaled with a common factor
The aforementioned orientation angles Ψ and Φ of the [SiO4]-tetrahedra could be used as order parameters for the characterization of the phase transformation: due to symmetry constraints their values are strictly zero in the Pbam-phase, while they can deviate from 0° in the Pnnm-phase. The evolution of Ψ as a function of the Sr/(Sr + Ca) atomic ratio, for example, is given in Fig. 10. In order to increase the number of data, we included the corresponding value for a natural bredigite sample with a composition close to the calcium end-member (Kahlenberg et al. 2019). A non-linear decrease of Ψ can be recognized. In principle, the present Pbam – Pnnm phase transformation could be continuous within the framework of Landau as well as renormalization theory (Stokes and Hatch 1988). However, the limited number of data points in the vicinity of the transition do not allow a final conclusion if the transition is really combined with a continuous evolution of the order parameter. Under the assumptions (i) that the transformation is of second order and that the critical composition Xc corresponds to the midpoint between the two boundary compositions (this is Xc = 0.617), the order parameter Ψ has been fitted using the relation Ψ ~ (Xc-X)β. The derived critical exponent β has a value of 0.24(2). The resulting curve is shown in Fig. 10 and should be regarded as a guide to the eye.

Evolution of the tilting angle Ψ of the [Si1O4]-tetrahedron as a function of the composition of the solid-solution series. The curve is a guide to the eye
Conclusions
Structural information on the Ca-Sr substitution in natural silicates is limited. This is related to the fact that strontium is generally present in minerals only as a minor or trace element. While there are some data in the literature concerning the Sr-content of natural feldspars (Lagache and Dujon 1987 and references cited therein) only a small number of other silicate minerals have been structurally characterized, where larger amounts of calcium have been replaced with strontium atoms. Examples include slawsonite (the Sr-analog of paracelsian, Griffen et al. 1977), piemontite (Catti et al. 1988) or melilite (Bindi et al. 2001). More systematic investigations on Ca-Sr substitutions among silicates are restricted to a few synthetic samples and involve α-(Ca1-xSrx)SiO3 (Ohsato et al. 2017), Ca2-xSrxSiO4 (Catti et al. 1984; Chen et al. 2015), Ca-Sr-diopside (Benna et al. 1987) or Ca-Sr-zoisite/clinozoisite (Dörsam et al. 2007). Similar to the present case, for the Ca2-xSrxSiO4 solid-solution series the existence of a compositional-dependent phase transition from the so-called β- to the α’-modification has been reported (Chen et al. 2015).
The present study focused on a better understanding of the fundamental structural impacts resulting from the Sr-Ca substitution into the bredigite structure type. However, more comprehensive information on this specific solid-solution series could be also of importance for the improvement of bredigite’s physical and biological properties in bioactive ceramics. Enhanced osteoporotic bone regeneration, for example, has been described for other alkaline earth silicates such as α- and β-CaSiO3 after doping with strontium (No et al. 2017) and the same effect may be observable for Sr-substituted bredigite as well.
The existence of Ba-substituted bredigite-type mixed-crystals has been already suggested by Moore and Araki (1976). Actually, Moseley and Glasser (1982) claimed successful synthesis experiments on Ca6.5Ba0.5[SiO4]4 and Ca6Ba[SiO4]4, respectively. While the powder pattern of the first compound could be satisfactorily indexed with a standard bredigite-type cell, the match between the observed diffractogram and the peak positions suggested by the orthorhombic primitive metric for the second composition was markedly less convincing. Therefore, Moseley and Glasser eventually proposed an orthorhombic B-centered cell with a doubled b lattice parameter for Ca6Ba[SiO4]4 (when referred to our preferred setting of the unit cell). Full details and an indexed powder pattern, though announced by the authors, have never been published. Attempts aiming on the growth of single-crystals at the compositions of the Ba-Ca solid-solution series combined with structural investigations could prove a fruitful investigation shedding more light on bredigite’s structural response to the replacement of calcium with barium.
Finally, we would like to mention that Sr3Mg[SiO4]2—one of the phases that occurred in the Bred-7 sample—is also based on pinwheel-like clusters and structurally related to merwinite (Yonesaki 2015). For this compound, cation substitutions among the larger alkaline-earth ions also triggered compositionally dependent phase transitions. Along the BaxSr3-xMg[SiO4]2 solid-solution, for example, three different but closely related structures could be distinguished. Similar to the present case, the observed symmetry changes have been attributed to tilts of the silicate tetrahedra caused by size mismatches of the alkaline-earth ions in the interstitial positions between the pinwheel clusters (Yonesaki 2015).
References
Baur WH (1974) The geometry of polyhedral distortions. Predictive relationships for the phosphate group. Acta Crystallogr B 30:1195–1215
Benna P, Chiari G, Bruno E (1987) Structural modifications in clinopyroxene solid solutions: the Ca-Mg and Ca-Sr substitutions in the diopside structure. Mineral Petrol 36:71–84
Bindi L, Bonazzi P, Fitton JG (2001) Crystal chemistry of strontian soda melilite from nephelinite. Eur J Mineral 13:121–125
Blaß G, Kruijen F (2014) Die Neuigkeiten aus der Vulkaneifel (2/14). Mineralien-Welt 25:62–75
Bodor M, Santos RM, Kristova L, Elsen J, Vlad M, van Gerven T (2013) Susceptibility of mineral phases of steel slags towards carbonation: mineralogical, morphological and chemical assessment. Eur J Mineral 25:533–549
Bridge TE (1966) Bredigite, larnite and γ-dicalcium silicates from the Marble Canyon. Am Mineral 51:1766–1774
Bruker AXS (2018) DIFFRAC.TOPAS, version 6.0.0.9: General profile and structure analysis software for powder diffraction data. Bruker-AXS GmbH, Karlsruhe, Germany
Bruker-AXS (2020) DIFFRAC.EVA, version 5.2.0.5. Bruker-AXS GmbH, Karlsruhe, Germany
Catti M, Gazzoni G, Ivaldi G (1984) Order-disorder in the α’-(Ca, Sr)2SiO4 solid solution: a structural and statistical-thermodynamic analysis. Acta Crystallogr B 40:537–544
Catti M, Ferraris G, Ivaldi G (1988) Thermal behavior of the crystal structure of strontian piemontite. Am Mineral 73:1370–1376
Chen M, Xia Z, Molokeev M, Liu Q (2015) Structural phase transformation and luminescent properties of Ca2-xSrxSiO4:Ce3+ orthosilicate phosphors. Inorg Chem 54:11369–11376
Chiang YW, Santos RM, Monballiu A, Ghyselbrecht K, Martens JA, Mattos MLT, van Gerven T, Meesschaert B (2013) Effects of bioleaching on the chemical, mineralogical and morphological properties of natural and waste-derived alkaline materials. Miner Eng 48:116–125
Dezfuli SN, Huan Z, Mol A, Leeflang S, Chang J, Zhou J (2017) Advanced bredigite-containing magnesium-matrix composites for biodegradable bone implant applications. Mat Sci Eng C 79:647–660
Dörsam G, Liebscher A, Wunder B, Franz G, Gottschalk M (2007) Crystal chemistry of synthetic Ca2Al3Si3O12OH-Sr2Al3Si3O12OH solid-solution series of zoisite and clinozoisite. Am Mineral 92:1133–1147
Douglas AMB (1952) X-ray investigation of bredigite. Mineral Mag 29:875–884
Farrugia LJ (1999) WinGX suite for small-molecule single-crystal crystallography. J Appl Crystallogr 32:837–838
Ferreira Neto JB, Fredericci C, Faria JOG, Chotoli FF, Ribeiro TR, Malynowskyj A, Silva ALN, Quarcioni VA, Lotto AA (2017) Modification of basic oxygen slag for cement manufacturing. J Sustain Metall 3:720–728
Grapes R (2011) Pyrometamorphism, 2nd edn. Springer, Heidelberg
Griffen DT, Ribbe PH, Gibbs GV (1977) The structure of slawsonite a strontium analog of paracelsian. Am Mineral 62:31–35
Gutt W (1961) A new calcium magnesiosilicate. Nature 190:339–340
Hoppe R (1979) Effective coordination numbers (ECoN) and mean fictive ionic radii (MEFIR). Z Kristallogr 150:23–52
Huang M, Zhang M, Yao D, Chen X, Pu X, Liao X, Huang Z, Yin G (2017) Dissolution behavior of CaO-MgO-SiO2-based bioceramic powders in simulated physiological environments. Ceram Int 13:9583–9592
Ilyushin GD, Blatov VA (2002) Crystal chemistry of zirconosilicates and their analogs: topological classification of MT frameworks and suprapolyhedral invariants. Acta Crystallogr B 58:198–218
Kahlenberg V, Galuskina I, Krüger B, Pauluhn A, Galuskin E (2019) Structural investigations on bredigite from the Hartrurim formation. Mineral Petrol 113:261–272
Lagache M, Dujon SC (1987) Distribution of strontium between plagioclase and 1 molar aqueous chloride solutions at 600°C, 1,5 kbar and 750 °C, 2 kbar. Bull Minéral 110:551–561
Liebau F (1985) Structural chemistry of silicates. Springer Verlag, Berlin
Momma K, Izumi F (2011) VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. J Appl Crystallogr 44:1272–1276
Moore PB, Araki T (1976) The crystal structure of bredigite and the genealogy of some alkaline earth orthosilicates. Am Mineral 61:74–87
Moseley D, Glasser FP (1981) Identity, composition and stability of bredigite phase T. Cement Concrete Res 11:559–565
Moseley D, Glasser FP (1982) Properties and composition of bredigite-structures phases. J Mater Sci 17:2736–2740
No YJ, Li JJ, Zeirat H (2017) Doped calcium silicate ceramics: a new class of candidates for synthetic bone substitutes. Materials 10:153
Ohsato H, Suzuki I, Kagomiya I (2017) Crystal structure and microwave dielectric properties of α-(Ca1-xSrx)SiO3 (x=1 and 0.8) ring silicates for millimeter-wave applications. Mat Res Bull 96:115–120
Orobengoa D, Capillas C, Aroyo MI, Perez-Mato JM (2009) Amplimodes: symmetry-mode analysis on the Bilbao Crystallographic Server. J Appl Crystallogr 42:820–833
Palatinus L, Chapuis G (2007) SUPERFLIP - A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J Appl Crystallogr 40:786–790
Pontikes Y, Kriskova L, Cizer Ö, Jones PT, Blanpain B (2013) On a new hydraulic binder from stainless steel converter slag. Adv Cem Res 25:21–31
Razavi M, Fathi M, Savabi O, Razavi SM, Beni BH, Vashaee D, Tayebi L (2013) Surface modification of magnesium alloy implants by nanostructured bredigite coating. Mater Lett 113:174–178
Rigaku (2015) CrysAlisPRO Software System, version 1.171.38.43. Rigaku Oxford Diffraction
Robinson K, Gibbs GV, Ribbe PH (1971) Quadratic elongation: a quantitative measure of distortion in coordination polyhedra. Science 172:567–570
Saalfeld H (1974) Kristallographische Untersuchungen über die Verbindung Ca7MgSi4O16 (Phase T) im System Ca2SiO4-Ca3Mg(SiO4)2. Ber Dt Keram Ges 51:295–298
Sharp JD, Johnson W, Andrews KW (1960) Visual method of primary slag control for the basic electric-arc furnace. J Iron Steel Inst 195:83–94
Sharygin VV, Sokol EV, Vapnik Y (2008) Minerals of the pseudobinary perovskite-brownmillerite series from combustion metamorphic larnite rocks of the Hartrurim Formation (Israel). Russ Geol Geophys 49:709–726
Sharygin VV (2010) Mineralogy of Ca-rich metacarbonate rocks from burned dumps of the Donetsk coal basin. In: Latest Developments in CoalFire Research. Bridging the Science, Economics, and Politics of a Global Disaster. Proc. ICCFR2–Second International Conference on Coal Fire Research. Berlin, Germany, pp 162–170
Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr A 64:112–122
Stokes HT, Hatch DM (1988) Isotropy groups of the 230 crystallographic space groups. World Scientific, Singapore
Tilley CE, Vincent HCG (1948) The occurrence of an orthorhombic high-temperature form of Ca2SiO4 (bregidite) in the Scawt Hill contact-zone and as a constituent of slags. Mineral Mag 28:255–271
Wilson AJC (1992) International Tables for Crystallography Volume C Mathematical Physical and Chemical Tables, 1st edn. Kluwer Academic Publishers, Dordrecht
Wu C, Chang J, Wang J, Ni S, Zhai W (2005) Preparation and characteristics of a calcium magnesium silicate (bredigite) bioactive ceramic. Biomaterials 26:2925–2931
Yi D, Wu C, Ma B, Ji H, Zheng X, Chang J (2014) Bioactice bredigite coating with improved bonding strength, rapid apatite mineralization and excellent cytocompatibility. J Biomater Appl 28:1343–1353
Yonesaki Y (2015) (Ba, Sr)3MgSi2O8 structure change caused by Ba/Sr replacement. Powder Diffr 30:40–51
Žáček V, Skála R, Dvořák Z (2015) Combustion metamorphism in the Most Basin. In: Stracher G, Prakash A, Sokol EV (eds) Coal and peat fires: a global perspective, vol 3, 1st edn. Case studies. Elsevier, Amsterdam, pp 162–202
Acknowledgements
The authors thank Herta Silvia Effenberger and Irina Galuskina for their recommendations and comments which improved the manuscript.
Funding
Open access funding provided by University of Innsbruck and Medical University of Innsbruck.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial handling: H. Poellmann
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kahlenberg, V., Prosser, L., Salzmann, M.F. et al. On the incorporation of strontium into the crystal structure of bredigite: structural effects and phase transition. Miner Petrol 116, 151–167 (2022). https://doi.org/10.1007/s00710-021-00771-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00710-021-00771-x