
Abstract
Gastrointestinal (GI) cancers are among the most common and lethal solid tumors worldwide. Unlike in malignancies such as lung, renal and skin cancers, the activity of immunotherapeutic agents in GI cancers has, on the whole, been much less remarkable and do not apply to the majority. Furthermore, while incremental progress has been made and approvals for use of immune checkpoint inhibitors (ICIs) in specific subsets of patients with GI cancers are coming through, in a population of ‘all-comers’, it is frequently unclear as to who may benefit most due to the relative lack of reliable predictive biomarkers. For most patients with newly diagnosed advanced or metastatic GI cancer, the mainstay of treatment still involves chemotherapy and/or a targeted agent however, beyond the second-line this paradigm confers minimal patient benefit. Thus, current research efforts are concentrating on broadening the applicability of ICIs in GI cancers by combining them with agents designed to beneficially remodel the tumor microenvironment (TME) for more effective anti-cancer immunity with intention of improving patient outcomes. This review will discuss the currently approved ICIs available for the treatment of GI cancers, the strategies underway focusing on combining ICIs with agents that target the TME and touch on recent progress toward identification of predictors of sensitivity to immune checkpoint blockade in GI cancers.
Similar content being viewed by others

Introduction
Gastrointestinal cancers are a huge global health problem. In 2018 almost 5 million new cases were diagnosed [1], the vast majority of which would have been at an advanced stage. While efforts to improve early diagnosis of these diseases are gaining momentum, gastrointestinal (GI) cancers are still the most deadly of all malignancies with colorectal, stomach and liver cancers representing the second, third and fourth leading causes of cancer-related deaths, respectively [1].
Over the past decade immunotherapy has revolutionized cancer treatment. Immune checkpoint inhibitors (ICIs) are now the cornerstone of managing malignancies such as lung, renal and skin cancers, among others. In some cases patients are experiencing marked radiological responses which may be sustained for years and side effects can be minimal compared with traditional chemotherapies. For an increasing number, this has meant a better quality and longer life. However, the activity of immunotherapeutic agents in GI cancers has, on the whole, been much less striking. Furthermore, in a population of ‘all-comers’, it is frequently unknown who may benefit due to the relative lack of reliable predictive biomarkers. For the majority of patients with newly diagnosed advanced/metastatic GI cancer, the mainstay of treatment still involves chemotherapy and/or a targeted agent. However, chemotherapy and/or targeted agents beyond the second-line have minimal efficacy in these diseases. More efficacious treatments are urgently needed.
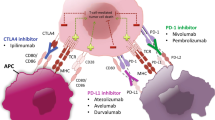
ICIs work by inhibiting the interaction between an immunosuppressive ligand or signal and their corresponding receptor or protein on immune or tumor cells, thus restoring the pre-existing host immune response against cancer [2]. Examples of ICIs include those that target the PD-1:PD-L1 interaction and reinvigorate cytotoxic T cell (CTL) function mainly in peripheral tissues e.g. pembrolizumab (anti-PD-1), nivolumab (anti-PD-1), atezolizumab (anti-PD-L1) or anti-CTLA-4 agents e.g. ipilimumab which act at an earlier stage of anti-cancer immunity by encouraging T cell activation in draining lymph nodes. The tumor microenvironment (TME) is a highly complex interplay of various different innate and adaptive immune cells such as NK cells, tumor-associated macrophages (TAMs) and T and B lymphocytes, stromal cells, such as cancer-associated fibroblasts (CAFs), endothelial cells, extracellular matrix (ECM) and secreted factors and it is of critical importance to supporting the success or failure of effective ICI therapy [3]. As such, where ICIs have not been effective as single agents, research has concentrated on combining ICIs with agents that target rational components of the TME to convert a ‘cold’ TME to an ‘immunogenic’ or ‘hot’ one, thus directing the immune response toward killing cancer cells and improving patient outcomes [4].
This article will discuss the currently approved ICIs available for the treatment of GI cancers, strategies underway focusing on combining ICIs with agents that target the TME and touch on recent progress toward predictive biomarker identification. Combinations of ICIs with cancer vaccines, cellular therapies or cytokine based therapies will not be covered.
Pre-requisites for anti-cancer immunity and the immune landscape of gastrointestinal cancers
The cancer-immunity cycle, a term first coined by Chen and Mellman in 2013, describes the seven main steps required for the immune system to carry out its final effector function of killing of cancer cells [5]. The majority of currently available ICIs work at this seventh step by blocking the PD-1/PD-L1 pathway, which usually acts as an ‘immunostat’ to prevent auto-immunity [5] but can also be up-regulated in patients with cancer [6], thereby removing the negative immune regulation on CTLs and reinstating the anti-cancer response. However if the other key events/steps in the cycle have not occurred beforehand, the anti-cancer response will not be fully potentiated by PD-1/PD-L1 blockade alone. Firstly, neoantigens are released by dying cancer cells. These are subsequently picked up by antigen presenting cells (APCs) which travel to lymph nodes to prime and activate T cells. This is a key step which requires the correct balance between co-stimulatory and inhibitory factors in the TME to promote stimulation of CTLs rather than pro-tumoral regulatory T cells (Tregs). Effector T cells then must travel to the site of the tumor, creep into the core of tumor, recognize the cancer cells on site and finally effectively kill them. In tumors which respond to single agent PD-1/PD-L1 blockade, only this final step in the sequence is flawed however, several tumor types, including most GI cancers, have other malfunctioning steps in the pathway which would also require correction.
In 2017, Chen and Mellman went on to describe three distinct immune phenotypes which could explain the mechanism of a tumour’s resistance to anti-cancer immunity [7]. So called “inflamed tumors” are most likely to respond to PD1/PD-L1 blockade as they are already permeated by plenty of different immune cells, including CTLs. The inflamed tumors might also harbor Tregs, myeloid-derived suppressor cells (MDSCs), B cells and cancer-associated fibroblasts (CAFs) which are generally inhibitory but PD-1/PD-L1 inhibitor administration is more likely to boost the CTL activity of this type of tumor than the other two “non-inflamed” phenotypes. In “immune-desert tumors”, CTLs are either a rarity or completely absent, possibly from a lack of appropriate T cell priming or activation in the lymph nodes, and in “immune-excluded tumors”, the T cells are present but they cannot get into the tumor due to inhibitory stromal or vascular factors. The Cancer Immunogram nicely plots the various factors in the immune milieu which may either be present or lacking in a patient’s tumor and thus outlines an agenda for successful individualized immunotherapy treatment [8].
Resistance to ICIs may be present from the outset or develop over a period of time on treatment due to mechanisms either intrinsic or extrinsic to the tumor cell [9, 10]. Overcoming de novo intrinsic resistance to ICIs is the main challenge in most GI cancers and may be due to the inability of the immune system to recognize cancer cells as foreign, a generally immunosuppressive TME or defective signaling pathways which would usually trigger an immune response. The first step in the Cancer Immunity Cycle requires the release of cancer neoantigens that can be recognized as non-self by the immune system [5]. It is now widely recognized that microsatellite unstable (MSI-H) or mismatch repair protein deficient (dMMR) tumors and those high tumor mutational burden (TMB) are characterized by high numbers of mutations or ‘neoantigens’ and predict sensitivity to ICIs [11,12,13]. However, dMMR/MSI-H GI tumors represent a very small proportion and the median somatic mutational load in these diseases is modest. Among GI cancers, oesophagogastric cancer is reported to have the highest TMB at 5 mutations/megabase, whereas pancreatic cancer has the lowest at 1 mutation/megabase [14]. Conversely, in melanoma, where ICIs exert the most efficacy, the median somatic mutational prevalence is 14 mutations/megabase [14]. Following neoantigen recoginition, antigen presenting cells e.g. dendritic cells (DCs) process the antigens and present them to T cells in lymph nodes which then become activated via major histocompatibility complex (MHC) class I or II co-stimulatory molecules. However, DCs in pancreatic cancer and cholangiocarcinoma (CCA) have been found to be limited in number or immature in advanced stages of disease [15,16,17], and mutations in the MHC I binding domain have been reported in colorectal cancer [18] thus potentially representing less effective antigen presentation and recognition in these tumors.
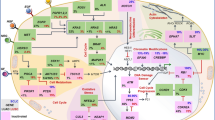
Detailed insight into TME components and their role in de novo resistance to checkpoint blockade in GI cancers has recently been reviewed by Batista et al. [19], and broadly outlined in Fig. 1. Within this article rational combinational strategies will be discussed in the context of reprogramming the TME for potentially effective ICI therapy.

Components of the TME which favor an immune suppressive milieu generally outweigh those which are associated with the T cell inflamed phenotype and response to immune checkpoint inhibitors in gastrointestinal cancers. CAF cancer-associated fibroblast, CCL C–C motif chemokine, CD cluster of differentiation, CSFR macrophage colony-stimulating factor, CXCL C–X–C motif ligand, DC dendritic cell, Fas apoptosis-mediating surface antigen FAS, GMCSF Granulocyte macrophage colony stimulating factor, HA hyaluronic acid, IDO indoleamine 2,3-dioxygenase, IFNγ interferon gamma, IL interleukin, MDSC myeloid-derived suppressor cells, NK natural killer, TAM tumor-associated macrophage, TGF-β transforming growth factor beta, Tregs T regulatory cells, VEGF vascular endothelial growth factor
The current role of checkpoint inhibitors in gastrointestinal cancers
Microsatellite unstable and mismatch repair deficient colorectal and other cancers
In 2017, the FDA approved both pembrolizumab and nivolumab, for use in the second-line treatment of MSI-H or dMMR metastatic colorectal cancer (mCRC) following the results of two pivotal trials. These studies demonstrated objective response rates (ORR) of 40% [20] and 32% [21] in patients with pre-treated MSI-H/dMMR mCRC who received 2 weekly pembrolizumab or nivolumab, respectively, with corresponding progression free survival (PFS) rates of 78% at almost 5 months and 50% at 12 months. In 2018, Overman et al. went onto show that giving nivolumab combined with ipilimumab every 3 weeks for 4 cycles followed by nivolumab alone to these patients, resulted in an ORR of 55% and 12 month PFS rate of 71% [22]. Accordingly the FDA granted accelerated approval to this regimen for patients with pre-treated MSI-H/dMMR mCRC in 2018. However only 3.5-5% of patients with mCRC display this MSI-H/dMMR phenotype [23, 24] and so checkpoint inhibitors currently play no role in the management of the majority of patients who have microsatellite stable (MSS) disease. Indeed the response rate to 2 weekly pembrolizumab in the MSS mCRC cohort from the aforementioned study was 0% [20].
In 2017 the FDA also granted the first tumor-agnostic approval to pembrolizumab for the treatment of any dMMR or MSI-H tumor following progression after standard treatment. The KEYNOTE-158 trial, which assessed the efficacy of 3 weekly pembrolizumab in 233 patients with 27 different types of advanced or metastatic non-colorectal MSI-H/dMMR tumors, subsequently reported an ORR of 33.4% in all patients which was durable [25]. This data reflect the findings from the earlier trials in patients with mCRC [20, 21]. However, once broken down into primary tumor type considerable variation in response rates are evident. For example, ORR was only 18.2% in patients with pancreatic cancer and 0% in patients with primary brain cancer [25] indicating that primary tumor location may influence sensitivity to ICIs even in the context of dMMR/MSI-H or that other immune suppressive factors in the TME may be in play or overall tumor burden too great [26].
Esophagogastric cancer
The role of ICIs in esophagogastric (OG) cancers is complex and rapidly evolving, with ongoing questions as to the most appropriate timing of immunotherapy. However, there now is established evidence for ICI use, particularly PD-1 inhibitors, in these tumors (Table 2).
KEYNOTE 181 was a phase III study in patients with advanced GOJ or esophageal cancer, showing superiority of pembrolizumab over investigator choice chemotherapy, producing prolonged median overall survival (mOS) of 9.3 vs. 6.7 months in 222 patients with PD-L1 positive tumors (CPS ≥ 10) [27]. Patients with metastatic esophageal squamous cell carcinoma (SCC) also demonstrated an improved mOS (8 vs. 7 months) however in the intention to treat (ITT) cohort there was no difference in mOS between the two arms. This study led to the FDA approval of pembrolizumab in metastatic or locally advanced esophageal SCC in second or later line treatment. The KEYNOTE 061 trial involved patients with metastatic gastric or GOJ adenocarcinoma in the second-line setting who were randomized to either pembrolizumab or taxane chemotherapy [28]. In patients with CPS ≥ 1 tumors, pembrolizumab was not superior to chemotherapy in terms of mOS. However pembrolizumab demonstrated benefit in those with CPS ≥ 10 in a post hoc analysis.
Two phase Ib studies (KEYNOTE 12 and KEYNOTE 28) have evaluated the role of pembrolizumab in PD-L1 expressing gastric cancer and esophageal cancer (SCC and adenocarcinoma), respectively [29, 30]. Both studies were in the second or later line and showed impressive ORR (22% in KEYNOTE 12 and 30% in KEYNOTE 28) (Table 1). Pembrolizumab is FDA approved for use in metastatic esophagogastric adenocarcinoma (OGA) with CPS > 1 in the third or later line setting. There is little evidence to support the use of pembrolizumab in the case of PD-L1 negative OGA in the later line setting.
Nivolumab has efficacy in advanced gastric and GOJ adenocarcinoma in the third line setting regardless of PD-L1 expression, as seen in the ATTRACTION-2 study [31]. In this study, 493 patients from Asian countries were randomly assigned to either nivolumab or placebo, following progression on 2 or more regimens. The ORR was 11% with a survival benefit (12 month survival 27% vs. 11%). Nivolumab is approved in Japan for use in the second-line treatment of advanced gastric cancer, regardless of PD-L1 expression (Table 2). The ATTRACTION-3 study has since also confirmed efficacy for nivolumab in advanced esophageal SCC however this indication is not yet approved [32]. This study assigned 419 patients to either nivolumab or taxane chemotherapy in the second line, regardless of PD-L1 expression. Nivolumab improved mOS by an addition of almost 3 months (10.9 vs. 8.4 months) with an improved toxicity profile. Again, there was no signal to suggest PD-L1 expression impacted efficacy.
Pancreatic cancer
Trials of single-agent immune checkpoint inhibitors have been particularly disappointing in pancreatic cancer. In a phase II study of ipilimumab monotherapy in 27 patients with advanced pancreatic ductal adenocarcinoma (PDAC), one patient had an objective response after initial pseudoprogression however there were no responses in the other 26 patients [33] Similarly, the anti-PD-L1 antibody BMS-936559 yielded 0% ORR in 14 patients with advanced PDAC in a multi-tumor type phase I study [34] Postulated reasons for such dramatic failures include low tumor mutational burden [35] and particularly dense desmoplastic stroma within the TME, impervious to adequate mobilization of immune cells [36].
Hepatocellular carcinoma
Following favorable results from the CheckMate 040 study, the FDA granted accelerated approval to nivolumab in September 2017 (Table 2). In this phase II study 246 patients with advanced HCC were treated with nivolumab in any line and at the 3 mg/kg dose which was taken forward to the dose expansion phase, the ORR was reported as 20% and the 9 month survival rate was 74%. PD-L1 positive status did not correlate with response in this study [37] The FDA subsequently granted approval for the use of pembrolizumab in second-line treatment of advanced HCC following the results of the phase II KEYNOTE 224 study where 104 patients who were intolerant of or had progressed on sorafenib were given 3 weekly pembrolizumab [38] In this study the response rate was 17% and the 12 month overall survival rate was 54% [38] Tremelimumab (anti-CTLA-4 antibody) has also proven efficacious in patients with advanced HCC due to chronic hepatitis C infection (Table 1). Interestingly, a reduction in viral load was also noted with response to tremelimumab therapy suggestive of improved T cell immunosurveillance with checkpoint inhibition [39].
Biliary tract cancer
Biliary tract cancers (BTCs) are rare malignancies so data from trials of checkpoint blockade in patients with BTC have come from basket trials with BTC cohorts. In KEYNOTE-028, pembrolizumab was given to patients with PD-L1 positive tumors including 24 patients with heavily pretreated PD-L1 positive BTC [40]. The ORR was 17% and responses were largely durable (over 40 weeks) [40]. In a larger study of 104 biomarker-unselected patients with BTCs (although 61 patients’ tumors did test positive for PD-L1), the response rate was only 5.8%. However responses were also durable and the 12 month OS rate was 37.8% [41] In KEYNOTE-028, dMMR/MSI-H status was not reported and in KEYNOTE-058, no tumor was MSI-H. In contrast, response rates of 41% have been reported in dMMR/MSI-high cholangiocarcinoma [25] Nivolumab has been proven to show equivalent efficacy in patients with previously treated BTCs (Table 1), retrospective PD-L1 analysis is ongoing for this study [42].
Anal cancer
Human papilloma virus (HPV) infection has been associated with higher numbers of tumor infiltrating lymphocytes and up-regulation of PD-1 checkpoints and is attributable to over 80% of case of squamous cell carcinoma of the anal canal (SCCA) [43]. In the first phase II study of a checkpoint inhibitor trialed in patients with SCCA, 37 patients with refractory disease were treated with nivolumab, 24% of whom had a radiological response including 2 complete responses and 7 partial responses [44] Median duration of response was 5.8 months and the longest response was recorded as 10.4 months but was still continuing at the data cut-off date [44]. Archival tissue was available from 15 patients and every one was positive for HPV infection, PD-L1 expression on tumor cells was also higher in responders compared to non-responders [44]. In a subsequent phase Ib study of pembrolizumab in 24 patients with PD-L1 positive advanced SCCA, 17% achieved a radiological response and all of these patients had received a prior treatment [45]. HPV status was not collected in this study. Based on the responses demonstrated in these two trials, the latest NCCN guidelines recommend either nivolumab or pembrolizumab in the second or later line treatment of SCCA [46].
Candidate predictors of sensitivity to immune checkpoint blockade in gastrointestinal cancers
As more and more approvals are coming through for use of ICIs in patients with GI cancers, the key research focus has been to try and improve and refine patient selection to expand the potential benefit of these therapies. In parallel to MSI-H/dMMR status discussed above, tumor TMB status, a reflection of the number of mutations within the tumor and thus the presumed neoantigen load capable of triggering an immune response [47], is an important candidate to highlight. Generally TMB is defined as number of non-synonymous somatic mutations (single nucleotide variants and small insertions/deletions) in coding regions and reported as mutations per megabase as standard [14, 48]. TMB can be measured by sequencing, if enough tissue is available, or by a panel-based approach which have demonstrated strong concordance [14, 49]. In a phase Ib/II study of toripalimab (anti-PD1) in chemo-refractory gastric cancer there was markedly superior OS in the TMB-high compared to the TMB-low group (14.6 vs. 4 months; HR 0.48) [50]. A cutoff of the top 20% of the TMB (12 mutations/Mb) was selected as defining a tumor as TMB-High (n = 12, 54 patients in total in the study) and patients with TMB < 12 mutations/Mb were defined as TMB-Low [50]. TMB-High cut-offs vary depending upon underlying histology and if there is a wide range of TMB values then the cut-off might potentially be set too high however the top 20% boundary has been shown to predict survival in a number of other tumor types [51]. In another study, however, the survival benefit of ICI therapy in patients with TMB-high esophagogastric cancer disappeared when those with MSI-high tumors were omitted from the group [52]. It does appear that, irrespective of histology, some patients with TMB-high tumors do not respond to ICIs and conversely there are some with TMB-low tumors who do [12, 35] and so, at present, TMB alone is not the panacea of predictive biomarkers. Of course one of the major challenges is intra-tumoral heterogeneity which means that the area of tissue which is eventually analyzed may show a very different mutational profile depending on from where in the primary or metastasis the biopsy sample has been taken [53]. Blood-based TMB (bTMB) assessment by liquid biopsy may overcome this particular hurdle and indeed a method of bTMB assessment has recently been validated in lung cancer [54].
PD-L1 expression is another important biomarker to mention, not least because a number of approved uses of ICIs in esophagogastric cancers mandate tumor PD-L1 positivity (Table 2). Historically methods of PD-L1 determination have varied widely however now most ICIs have their own companion-diagnostic for PD-L1 assessment which is generally measured by either combined positive score (CPS) [55, 56] or tumor proportion score (TPS) [57]. Nevertheless, in the ATTRACTION-02 study, the survival benefit from nivolumab in pre-treated patients with esophagastric cancer was observed in both PD-L1 positive and negative patients [58]. Furthermore, in KEYNOTE 012, where patients were subjected to repeated on-treatment biopsies, there was substantial disparity in PD-L1 expression between samples [29]. These findings suggest that PD-L1 status alone may not be the most useful predictor of successful ICI therapy in esophagogastric cancer.
Virus-associated cancers also demonstrate increased mutational loads [59, 60] and immune exhaustion [61] proposing a potential predictor of ICI efficacy. Epstein Barr Virus (EBV)-positive gastric cancers in particular have relatively high levels of CTLs and IFN-γ [62] and indeed, single agent pembrolizumab yielded an immense ORR of 100% in Korean patients with advanced EBV-positive gastric cancer [63] suggesting that EBV positivity may be an important predictive biomarker in gastric cancer. Correspondingly, the high response rates to nivolumab seen in patients with SCCA highlighted in the previous section [44] may well also have been linked to the high rates of associated HPV infection in this disease. Conversely however in the Checkmate 040 study, response rates to nivolumab did not significantly differ in patients with HCC caused by hepatitis B or C or not [37].
While the candidate predictive biomarkers so far discussed have been helpful in determining which patients with GI cancers may benefit most from ICI therapy, more precise biomarkers are clearly needed. Another potential contender on the horizon is the gut microbiome particularly in colorectal cancer where, for example, a certain species of bacterium has been shown to be inversely correlated to levels of T cell infiltrates in MSH-H CRC [64] and thus potentially an inferior outcome to ICI therapy. There may also be mileage in a composite biomarker approach given that, for example, in gastric cancer the combination of EBV and PD-L1-positivity but not MSI-H status was associated with response to checkpoint inhibition [64, 65]. Specific TME phenotypes defined by TME infiltration patterns of immune cells may also develop into a promising predictor of ICI response in gastric cancer [66].
Combining immune checkpoint inhibitors with agents that target the TME
Combination checkpoint blockade with chemotherapy
Traditional cytotoxic chemotherapy exerts its anti-neoplastic activity through direct cytotoxicity, impacting the cell cycle and leading to apoptosis. However, studies have shown that some chemotherapeutic agents also have immune modulating effects, through down-regulating immune inhibitory cells and stimulating the production of pro-inflammatory cytokines, leading to a ‘hot’ or T cell inflamed TME [67] (Table 3). Combining cytotoxic chemotherapy with ICIs have shown impressive outcomes in the treatment of other solid malignancies, including lung and head and neck cancers [68,69,70] however, thus far, studies of combined chemotherapy with ICIs in GI cancers have shown mixed outcomes.
The KEYNOTE 059 trial (n = 25) determined that pembrolizumab combined with platinum/fluropyrimidine in the first-line treatment of advanced gastric cancer had a manageable safety profile and, furthermore, showed impressive ORR (60%) and mOS of 13.8 months [71]. KEYNOTE 062 subsequently investigated the role of pembrolizumab with or without chemotherapy, versus chemotherapy alone in first-line, advanced OGA [72]. This study of 763 patients showed non-inferiority in mOS between pembrolizumab and chemotherapy and in an exploratory analysis of patients with PD-L1 CPS ≥ 10, there was an improved mOS with pembrolizumab (17 vs. 11 months), with a more tolerable toxicity profile. Importantly, however, combination pembrolizumab with chemotherapy was not superior to chemotherapy alone. The phase III KEYNOTE 590 study of pembrolizumab with chemotherapy as first-line therapy in advanced PD-L1 positive CPS ≥ 10 oesopheageal cancer is ongoing (NCT03189719) and will clarify whether the combination of chemotherapy plus ICI can improve OS in these patients over chemotherapy alone. Preliminary results also suggest that the combination of chemotherapy, pembrolizumab and HER2 directed therapy for HER2 amplified OGA may be efficacious. The ongoing phase II, single arm study evaluating trastuzumab, capectiabine and oxliaplatin in combination with pembrolizumab in the first-line setting of advanced HER2 amplified OGA which showed a high ORR (83%) with mPFS 11.4 months which showed a high ORR (83%) with mPFS 11.4 months [73] and is currently still recruiting (Table 4).
Combination ICI plus chemotherapy has also been studied in mCRC. In a phase II trial, 30 patients with mCRC, irrespective of MMR status, were treated with pembrolizumab in combination with mFOLFOX in the first-line setting, the ORR was 53% with 100% disease control rate at 8 weeks [74]. Conversely, however, the combination of trifluridine/tipiracil with nivolumab did not demonstrate clinically significant benefit in a heavily pre-treated population [75].
Combination checkpoint blockade with anti-VEGF therapy
Vascular endothelial growth factor (VEGF) regulates the tumoral vascular environment which enhances angiogenesis [76], and is frequently upregulated in tumors to promote growth and metastases. Blocking VEGF has anti-angiogenic effects and improves the delivery of anti-neoplastic therapies and T cell trafficking to the tumor (see Fig. 2 step 4 of cancer immunity cycle) [77]. Anti-VEGF therapies including bevacizumab in combination with chemotherapy have been shown to improve survival in mCRC [78]. There is also data to support the use of ramucirumab, a monoclonal antibody to VEGFR-2, in combination with paclitaxel in advanced OGA in the later line setting [79]. Sorafenib and lenvatinib, both with anti-VEGF activity, are also effective for treatment of advanced HCC [80].

Examples of partner agents which may work in synergy with immune checkpoint inhibitors in gastrointestinal cancers by promoting progression through the cancer immunity cycle [5] at these various steps. PD-1/PD-L1 blockade alone is not sufficient to activate the anti-cancer immune response in most ‘immunologically cold’ gastrointestinal cancers but combination strategies involving agents which favorably modulate the tumor microenvironment and immune milieu may render checkpoint inhibitor therapy more efficacious in these diseases. VEGF vascular endothelial growth factor
There is evidence that VEGF also has immune-modulating effects, through impacting DCs and inhibiting T cell activity (see Fig. 2—step 2 and 7 of Cancer Immunity Cycle) [81]. Combination immunotherapy with anti-VEGF therapies have been used in the treatment of advanced renal cell cancer (RCC) [82, 83] and non-small cell lung cancer [84] with improvements in survival in these tumor types. The combination of ramucirumab and nivolumab was investigated in a phase I/II study in the second-line treatment of advanced gastric adenocarcinoma. This combination produced an ORR 27% and mOS of 9 months [85]. Furthermore, in a phase II study of 133 patients with chemo-refractory mCRC randomized to either capecitabine, bevacizumab with either atezolizumab or placebo. The addition of atezolizumab produced a non-significant improvement in mPFS (primary end point), with low ORR (8.5% vs. 4.3%) [86].
The combination of atezolizumab and bevacizumab has also demonstrated efficacy in the first-line setting of advanced HCC. The phase III IMbrave 150 study showed that this combination improves mOS compared with sorafenib and was granted FDA approved for this indication [87]. Table 4 outlines some of the currently ongoing clinical trials of anti-angiogenic agents combined with ICIs in GI cancers.
Dual checkpoint blockade
Dual ICI therapy targeting CTLA-4 and PD-1 inhibition has established efficacy in malignancies such as melanoma [88] and RCC [89]. The proposed mechanism being that simultaneous inhibition of CTLA-4 and PD-1 provides an enhanced anti-tumor T cell response. However the role of dual checkpoint blockade in GI cancers is still being investigated. The CheckMate 142 trial has recently established a role for nivolumab and ipilimumab in dMMR mCRC. Of the 119 patients treated with this regimen, 58% had a response to treatment [90]. This combination is now FDA approved for use in dMMR/MSI-H mCRC in the second-line setting. The CheckMate 032 study investigated the role of nivolumab with ipilimumab in patients with advanced gastric, GOJ and esophageal cancers. The combination produced ORR of 8% (for nivolumab 3 mg/kg and ipilimumab 1 mg/kg) and 24% (nivolumab 1 mg/kg and ipilimumab 3 mg/kg) [58].
Relatlimab is a monoclonal antibody against the lymphocyte activation gene-3 (LAG-3) on tumor infiltrating lymphocytes (TILs). Blockade of this inhibitory signal can promote T cell mediated tumor cell death and activate TILs in CRC typically express high levels of LAG3, which make this a promising therapeutic target [91]. Furthermore, anti-EGFR antibody treatment appears to be capable of upregulating PD-1 and LAG3 checkpoints in RAS/RAF wild-type mCRC [92]. A phase II study to assess the efficacy of combination nivolumab and relatlimab in patients with RAS/RAF wild-type mCRC who develop acquired resistance to anti-EGFR treatment is currently recruiting. (Table 4).
Combination checkpoint blockade and epigenetic modifiers
Epigenetics is the modification of gene expression without alteration of DNA’s nucleic acid sequence [93]. These modifications include DNA methylation, alterations to histone proteins and remodeling of chromatin and they are often aberrant in disease processes such as cancer [93, 94]. Notably however, alterations in the epigenome rather than the core genetic material of cells have been shown to be reversible and hence a potential anti-cancer strategy [95].
Various epigenetic therapies have been approved for use in the treatment of hematological malignancies however their use as single agents in solid tumors, including CRC, has been historically somewhat limited for reasons such as short half-lives, high toxicity or low or no responses [96]. This has led to more recent research instead using them as ‘sensitizers’ in combination with other anti-cancer therapies such as chemotherapy and targeted agents which have purported some success particularly in CRC [96, 97]. Furthermore, there is increasing evidence that epigenetic agents can promote the anti-cancer immune response via multiple mechanisms including increased antigen release and antigen presentation [steps 1 and 2 of the Cancer Immunity Cycle (Fig. 2)], T cell trafficking and infiltration into the tumor (steps 4 and 5) as well as restoring effector T cell function (step 7), suggesting potential synergy with checkpoint inhibitors particularly in immunologically ‘cold’ tumors [98].
By far the most studied epigenetic modulators are inhibitors of histone deacetylases (HDACi) and DNA methyltransferases (DNMTi). Pre-clinical testing demonstrates that the DNMTi 5-azacitadine (5-AZA) upregulated 15 gene sets involved in the immune response such as antigen presentation, interferon signaling and chemokine and cytokine signaling in colorectal cancer cell lines [99]. Mouse models of colorectal tumors were completely eradicated when 5-AZA and entinostat (a class I HDACi) were combined with PD-1 and CTLA-4 antibodies by markedly reducing immune inhibitory MSDC populations thus allowing for expansion of CD8+ T cell cohorts [100]. Entinostat also improved responses to checkpoint blockade in pancreatic cancer mouse models via the same beneficial effects on the TME [101]. Furthermore, after treatment of colorectal cancer PDX models with decitabine (DNA methyltransferase inhibitor), antigen processing and presenting genes were significantly upregulated in addition to some cytokine and chemokine-related genes such as CXCL1, cell proliferation genes were conversely downregulated and there was influx of CD4+ and CD8+ T cells into the tumor [102]. Successively the combination of decitabine and PD-1 significantly inhibited tumor growth and improved survival than either drug alone [102]. The epigenetic modifier EZH2 has also been shown to suppress IFN-y induced PDL-1 expression in HCC cell lines, thus representing a possible therapeutic target for inhibition and subsequent combination with anti-PD-1/PDL-1 therapy [103]. Reduction of EZH2 expression in Tregs has also translated into improved efficacy of anti-CTLA-4 therapy [104].
Several clinical studies of checkpoint inhibitors combined with epigenetic modifiers, including IDO inhibitors which have also demonstrated similarly favorable immunomodulatory effects pre-clinically [105], are currently underway in GI cancers (Table 4). A recently reported pilot study of ‘priming’ with either 5-AZA or romidepsin (HDACi) or the combination of both, followed by administration of pembrolizumab to patients with MSS mCRC established that the combination of these agents with checkpoint blockade was safe and tolerable [106]. It will be of interest to see the corresponding translational results from the sequential biopsies which were taken pre- and post- prime as to whether the TME was indeed favorably sensitized for subsequent checkpoint inhibitor therapy, consistent with the pre-clinical data.
Combination checkpoint blockade and Wnt signaling modulators
Abberations in the Wnt/β-catenin signaling pathway occur frequently in cancer and while the constituents of the pathway are known, the mechanisms underpinning how the proteins interact are yet to be fully elucidated [107]. The role of Wnt signaling in development of colorectal cancer, most commonly via mutations of the APC gene, is particularly well-described however increasing evidence suggests that Wnt signaling also plays a key role in the pathogenesis of other GI cancers including PDAC, HCC, CCA and OG cancers [107,108,109,110], proposing a potential target for therapy.
Preliminary clinical studies of Wnt signaling modulators as monotherapies including Porcupine inhibitors, Frizzled receptor targeting agents, a Wnt5a-mimetic and agents targeting components further downstream in the pathway, have so far demonstrated promising safety profiles in patients with GI cancers where side effects including bone toxicity appear to be manageable [111,112,113,114,115,116], however signals of single agent efficacy have yet to be confirmed. In parallel, mounting evidence indicates a critical role for Wnt/β-catenin signaling in immunomodulation of the TME at multiple steps of the Cancer Immunity Cycle [117]. and thus the possibility of combining Wnt modulators with immune checkpoint inhibitors has emerged as an attractive therapeutic strategy (Fig. 2).
Firstly, activated Wnt/β-catenin signaling appears to accentuate tumor immune exclusion by suppressing DC recruitment into the TME via down-regulation of the chemokine CCL4 as well as impaired priming of effector T cells in melanoma mouse models, which were subsequently resistant to immune checkpoint blockade [118, 119]. Successively, activated Wnt/β-catenin signaling has also been shown to drive development of a non-T cell inflamed TME in other tumor types including GI cancers, most notably, esophageal, HCC and CRC [120, 121]. By inactivating Wnt/β-catenin signaling, presentation of cancer associated antigens and T cell priming appears to be restored [122]. In addition to impairing the first 3 steps of the Cancer Immunity Cycle, aberrant Wnt/β-catenin signaling also seems to deter T cells from entering the tumor and preferentially favors influx and survival of inhibitory Tregs while inactivating or stimulating apoptosis of effector T cells in the immune milieu [123, 124]. Metastatic lung and breast tumors also seem to be able to evade immune detection by secreting the Wnt inhibitor DKK1 in an autocrine manner [125] and elevated levels of DKK1 appear to be associated with worse prognosis in OG cancer [126]. DKK1 has since been shown to increase tumor growth and support an immunosuppressive environment by signaling to MDSCs, and treatment with a DKK1 neutralizing antibody, DKN, 01, was able to mitigate tumor growth by reduced levels of MDSCs and increased entry of effector T cells into the TME [127, 128]. These results suggest that DKN-01 may favorably reprogram the immune milieu for collaboration with checkpoint inhibitors.
In the clinic, the combination of DKN-01 and pembrolizumab has been shown to be safe and tolerable in patients with advanced MSS OG cancer with a hint of potential efficacy as a PR was observed in 1 patient and 5 patients had SD [129]. Interestingly, these 6 patients also had a reduction of MDSC levels in peripheral blood on treatment compared to baseline, consistent with pre-clinical observations [129]. Another clinical study of DKN-01 and atezolizumab will similarly assess the safety and efficacy of combination Wnt inhibition and checkpoint blockade and studies of combined Porcupine inhibitors and ICIs are also currently recruiting (Table 4).
Combination checkpoint blockade and stromal targeting agents
The tumor stroma is comprised of dense connective tissue including CAFs, stromal cells, osteoblasts, chondrocytes and ECM and it is a critical component of the TME [130]. Activated desmoplastic stroma is particularly unique to PDAC and is a huge barrier to effective delivery of anti-cancer therapy including immunotherapy [36, 130]. Thus, potential combination strategies involving stromal targeting agents and checkpoint inhibitors have recently garnered increased attention in this disease.
High levels of focal adhesion kinases (FAK) in PDAC appear to correlate with low levels of CTLs and a generally immunosuppressive TME and in mouse models FAK inhibition (FAKi) appeared to reduce tumor fibrosis and halt PDAC progression as well as decrease populations of immunosuppressive cells such as TAMs and Tregs [131]. FAKi also demonstrated increased T cell infiltration and promoted tumor shrinkage with addition of ICI suggesting a potentially efficacious synergy [131]. A clinical study of defactinib (FAKi) and pembrolizumab is currently underway in patients with solid tumors including PDAC (Table 4).
Agents targeting chemokine proteins and chemokine receptors which are involved in cell migration have also demonstrated potential as valuable ICI partners in PDAC. For example, the chemokine CXCL12 secreted by CAFs and the chemokine receptor CXCR4 found on T cells appear to drive immunosuppression in the TME by increasing populations of MDSCs [132, 133]. CXC12 inhibition combined with checkpoint blockade showed improved T cell infiltration and halted tumor growth in mouse models [133] and the phase II COMBAT trial of combination CXCR4 blockade with BL-8040 plus pembrolizumab recently reported a DCR of 34.5% in an ITT of 29 patients and mOS of 7.5 months as a second-line therapy in patients with advanced PDAC [134]. Supportively, the corresponding translational analyses indicated that BL-8040 reduced MDSC and Treg populations and increased tumor infiltration with CTLs [134]. These exciting results are likely to lead onto confirmatory phase III studies. Preliminary results from the safety run of mogamulizumab, a CCR4 antibody, in combination with nivolumab revealed confirmed tumor responses in patients with HCC and PDAC and similar immunomodulatory effects on the TME which also holds promise [135]. Additionally, CXCR2 and CCR2 or CSF1R inhibition have been shown to improve T cell infiltration, reprogram TAMs, upregulate PD-L1 and CTLA-4 checkpoints and increase sensitivity to ICIs in PDAC mouse models [136,137,138] which has led onto further assessment of these combinations in clinical studies (Table 4).
Alternative combination approaches include use of Bruton’s tyrosine kinase (BTK) inhibitors, which have been shown to deplete MDSCs in the TME, improve CTL activity and ameliorate the fibrous stroma of PDAC pre-clinically [139,140,141] with ICIs. However while the phase II study of the BTK inhibitor, alacabrutinib, combined with pembrolizumab showed reduction of MDSCs in peripheral blood from patients on study and the combination was tolerable, the response rates were limited [142]. Thus highlighting the complexity of the TME in PDAC. More promising strategies in the pipeline may include partnering CD40 agonists which favorably alter the immune component of the PDAC stroma [143]. with chemotherapy and PD-1 blockade (Table 4) and use of the bispecific antibody, M7824, which blocks both immune-inhibitory TGFβ and PD-L1 pathways and has demonstrated some efficacy signals in patients with PDAC and BTC [144, 145].
Conclusion
Over the past few years, data supporting clinical benefit for immune checkpoint inhibitors in the treatment of gastrointestinal cancers has gradually mounted and approvals for specific indications have come through. However, the challenge of expanding the benefit of immunotherapy to the majority of the population with non-T cell inflamed, ‘cold’ gastrointestinal tumors with intrinsic resistance to these therapies is still very much an unmet need. Research efforts into effective predictive biomarker identification, fraught with hurdles such as intratumoral heterogeneity, is still being explored and refined however much headway has already been made. Furthermore, rational combinatorial strategies of immune checkpoint inhibitors with agents that produce beneficial immunomodulatory effects and reprogram the TME to overcome intrinsic resistance to effective anti-cancer immunity, hold significant promise. The corresponding translational components of the clinical studies discussed herein may be the key to gaining a deeper understanding of underlying mechanisms of response and resistance to these strategies and slowly unlock more about the intricacies of the immune milieu in these malignancies with the hope of discovering more efficacious treatments for our patients.
References
Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A J Clin. 2018;00:1–31.
Granier C, De Guillebon E, Blanc C, et al. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open. 2017;2:e000213.
Ribeiro Franco PI, Rodrigues AP, de Menezes LB, et al. Tumor microenvironment components: allies of cancer progression. Pathol Res Pract. 2020;216:152729.
Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–14.
Chen DS. Mellman I. Oncology meets immunology: The cancer-immunity cycle. Immunity; 2013. p. 1–10.
Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother. 2007;56:739–45.
Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–30.
Blank CU, Haanen JB, Ribas A, et al. The “cancer immunogram”. Science (80−). 2016;352:658.
Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol. 2020;20:25–39.
Fares CM, Van Allen EM, Drake CG, et al. Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am Soc Clin Oncol Educ B. 2019;147:64.
Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in Melanoma. N Engl J Med. 2014;371(23):2189–99.
Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (80−). 2015;348(6230):124–8.
Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (80−). 2017;357:409–13.
Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34.
Fukunaga A, Miyamoto M, Cho Y, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 2004;28:e26.
Takagi S, Miyagawa S-I, Ichikawa E, et al. Dendritic cells, T-cell infiltration, and grp94 expression in cholangiocellular carcinoma. Hum Pathol. 2004;35:881–6.
Yanagimoto H, Takai S, Satoi S, et al. Impaired function of circulating dendritic cells in patients with pancreatic cancer. Clin Immunol. 2005;114:52–60.
Giannakis M, Mu XJ, Shukla SA, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016;15:857–65.
Batista S, Gregório AC, Hanada Otake A, et al. The gastrointestinal tumor microenvironment: an updated biological and clinical perspective. J Oncol. 2019;2019:6240505.
Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20.
Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91.
Overman MJ, Lonardi S, Wong KYM, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair–deficient/microsatellite instability–high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–9.
Koopman M, Kortman GAM, Mekenkamp L, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer. 2009;100:266–73.
Venderbosch S, Nagtegaal ID, Maughan TS, et al. Mismatch repair status and mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS Studies. Clin Cancer Res. 2014;20:5322–30.
Marabelle A, Le DT, Ascierto PA, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair–deficient cancer: results from the phase II KEYNOTE-158 Study. J Clin Oncol. 2019;38:1–10.
Huang AC, Postow MA, Orlowski RJ, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–5.
Kojima T, Muro K, Francois E, et al. Pembrolizumab versus chemotherapy as second-line therapy for advanced esophageal cancer: phase III KEYNOTE-181 study. J Clin Oncol. 2019;37:2.
Shitara K, Ozguroglu M, Bang Y-J, et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet (London, England). 2018;392:123–33.
Muro K, Chung HC, Shankaran V, et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol. 2016;17:717–26.
Doi T, Piha-Paul SA, Jalal SI, et al. Safety and antitumor activity of the anti-programmed death-1 antibody pembrolizumab in patients with advanced esophageal carcinoma. J Clin Oncol. 2018;36:61–7.
Kang Y-K, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (London, England). 2017;390:2461–71.
Kato K, Cho BC, Takahashi M, et al. Nivolumab versus chemotherapy in patients with advanced oesophageal squamous cell carcinoma refractory or intolerant to previous chemotherapy (ATTRACTION-3): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20:1506–17.
Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33:828.
Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65.
Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21.
Kleeff J, Beckhove P, Esposito I, et al. Pancreatic cancer microenvironment. Int J Cancer. 2007;121:699–705.
El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–502.
Zhu AX, Finn RS, Edeline J, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19:940–52.
Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59:81–8.
Bang YJ, Doi T, De Braud F, et al. 525 Safety and efficacy of pembrolizumab (MK-3475) in patients (pts) with advanced biliary tract cancer: Interim results of KEYNOTE-028. Eur J Cancer. 2015;51:S112.
Ueno M, Chung HC, Nagrial A, et al. 625PDPembrolizumab for advanced biliary adenocarcinoma: Results from the multicohort, phase II KEYNOTE-158 study. Ann Oncol. 2018;29:205.
Kim RD, Kim DW, Alese OB, et al. A phase II study of nivolumab in patients with advanced refractory biliary tract cancers (BTC). J Clin Oncol. 2019;37:4097.
Arbyn M, de Sanjosé S, Saraiya M, et al. EUROGIN 2011 roadmap on prevention and treatment of HPV-related disease. Int J Cancer. 2012;131:1969–82.
Morris VK, Salem ME, Nimeiri H, et al. Nivolumab for previously treated unresectable metastatic anal cancer (NCI9673): a multicentre, single-arm, phase 2 study. Lancet Oncol. 2017;18:446–53.
Ott PA, Piha-Paul SA, Munster P, et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with recurrent carcinoma of the anal canal. Ann Oncol Off J Eur Soc Med Oncol. 2017;28:1036–41.
Benson AB, Venook AP, Al-Hawary MM, et al. Anal carcinoma, Version 2.2018, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw J Natl Compr Canc Netw. 2018;16:852–71.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (80−). 2015;348:69–74.
Merino DM, McShane LM, Fabrizio D, et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project. J Immunother cancer. 2020;8:e000147.
Campesato LF, Barroso-Sousa R, Jimenez L, et al. Comprehensive cancer-gene panels can be used to estimate mutational load and predict clinical benefit to PD-1 blockade in clinical practice. Oncotarget. 2015;6:34221–7.
Wang F, Wei XL, Wang FH, et al. Safety, efficacy and tumor mutational burden as a biomarker of overall survival benefit in chemo-refractory gastric cancer treated with toripalimab, a PD-1 antibody in phase Ib/II clinical trial NCT02915432. Ann Oncol Off J Eur Soc Med Oncol. 2019;30:1479–86.
Samstein RM, Lee C-H, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–6.
Greally M, Chou JF, Chatila WK, et al. Clinical and molecular predictors of response to immune checkpoint inhibitors in patients with advanced esophagogastric cancer. Clin Cancer Res. 2019;25:60.
Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92.
Wang Z, Duan J, Cai S, et al. Assessment of blood tumor mutational burden as a potential biomarker for immunotherapy in patients with non-small cell lung cancer with use of a next-generation sequencing cancer gene panel. JAMA Oncol. 2019;5:696–702.
Fuchs CS, Dhoi T, Jang RWJ, et al. KEYNOTE-059 cohort 1: efficacy and safety of pembrolizumab (pembro) monotherapy in patients with previously treated advanced gastric cancer. J Clin Oncol. 2018;35:4003.
Kulangara K, Zhang N, Corigliano E, et al. Clinical utility of the combined positive score for programmed death ligand-1 expression and the approval of pembrolizumab for treatment of gastric cancer. Arch Pathol Lab Med. 2019;143:330–7.
Hutarew G. PD-L1 testing, fit for routine evaluation? From a pathologist’s point of view. Memo. 2016;9:201–6.
Janjigian YY, Bendell J, Calvo E, et al. CheckMate-032 study: efficacy and safety of nivolumab and nivolumab plus ipilimumab in patients with metastatic esophagogastric cancer. J Clin Oncol. 2018;36:2836–44.
Yin H, Qu J, Peng Q, et al. Molecular mechanisms of ebv-driven cell cycle progression and oncogenesis. Med Microbiol Immunol. 2019;208:573–83.
Arzumanyan A, Reis HMGPV, Feitelson MA. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer. 2013;13:123–35.
Dyck L, Mills KHG. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur J Immunol. 2017;47:765–79.
Lee J, Kim K-M. Biomarkers for gastric cancer: molecular classification revisited. Precis Future Med. 2017;1:59–68.
Kim ST, Cristescu R, Bass AJ, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med. 2018;24:1449–58.
Hamada T, Zhang X, Mima K, et al. Fusobacterium nucleatum in colorectal cancer relates to immune response differentially by tumor microsatellite instability status. Cancer Immunol Res. 2018;6:1327–36.
Network CGAR. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9.
Zeng D, Li M, Zhou R, et al. Tumor microenvironment characterization in gastric cancer identifies prognostic and immunotherapeutically relevant gene signatures. Cancer Immunol Res. 2019;7:737–50.
Zitvogel L, Galluzzi L, Smyth MJ, et al. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. 2013;39:74–88.
Gandhi L, Rodriguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378:2078–92.
Burtness B, Harrington KJ, Greil R, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet (London, England). 2019;394:1915–28.
Horn L, Mansfield AS, Szczęsna A, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. 2018;379:2220–9.
Bang Y-J, Muro K, Fuchs CS, et al. KEYNOTE-059 cohort 2: safety and efficacy of pembrolizumab (pembro) plus 5-fluorouracil (5-FU) and cisplatin for first-line (1L) treatment of advanced gastric cancer. J Clin Oncol. 2017;35:4012.
Tabernero J, Van Cutsem E, Bang Y-J, et al. Pembrolizumab with or without chemotherapy versus chemotherapy for advanced gastric or gastroesophageal junction (G/GEJ) adenocarcinoma: the phase III KEYNOTE-062 study. J Clin Oncol. 2019;37:4007.
Janjigian YY, Chou JF, Simmons M, et al. First-line pembrolizumab (P), trastuzumab (T), capecitabine (C) and oxaliplatin (O) in HER2-positive metastatic esophagogastric adenocarcinoma (mEGA). J Clin Oncol. 2019;37:62.
Shahda S, Noonan AM, Bekaii-Saab TS, et al. A phase II study of pembrolizumab in combination with mFOLFOX6 for patients with advanced colorectal cancer. J Clin Oncol. 2017;35:3541.
Patel MR, Falchook GS, Hamada K, et al. Results of a phase II study evaluating trifluridine/tipiracil plus nivolumab in patients with heavily pretreated microsatellite-stable (MSS) metastatic colorectal cancer (mCRC). J Clin Oncol. 2019;37:48.
Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;3:669–76.
Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7:987–9.
Hurwitz HI, Tebbutt NC, Kabbinavar F, et al. Efficacy and safety of bevacizumab in metastatic colorectal cancer: pooled analysis from seven randomized controlled trials. Oncologist. 2013;18:1004–12.
Wilke H, Muro K, Van Cutsem E, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 2014;15:1224–35.
Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391:1163–73.
Gabrilovich D, Ishida T, Oyama T, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–66.
Motzer RJ, Penkov K, Haanen J, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380:1103–15.
Rini BI, Plimack ER, Stus V, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380:1116–27.
Socinski MA, Jotte RM, Cappuzzo F, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378:2288–301.
Hara H, Shoji H, Takahari D, et al. Phase I/II study of ramucirumab plus nivolumab in patients in second-line treatment for advanced gastric adenocarcinoma (NivoRam study). J Clin Oncol. 2019;37:129.
Mettu NB, Niedzwiecki D, Boland PM, et al. BACCI: A phase II randomized, double-blind, placebo-controlled study of capecitabine bevacizumab plus atezolizumab versus capecitabine bevacizumab plus placebo in patients with refractory metastatic colorectal cancer. J Clin Oncol. 2018;36:TPS873.
Cheng A-L, Qin S, Ikeda M, et al. IMbrave150: Efficacy and safety results from a ph III study evaluating atezolizumab (atezo) + bevacizumab (bev) vs sorafenib (Sor) as first treatment (tx) for patients (pts) with unresectable hepatocellular carcinoma (HCC). Ann Oncol. 2019;30:ix186–7.
Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345–56.
Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378:1277–90.
Overman MJ, Lonardi S, Wong KYM, et al. Nivolumab (NIVO) + low-dose ipilimumab (IPI) in previously treated patients (pts) with microsatellite instability-high/mismatch repair-deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC): long-term follow-up. J Clin Oncol. 2019;37:635.
Chen J, Chen Z. The effect of immune microenvironment on the progression and prognosis of colorectal cancer. Med Oncol. 2014;31:82.
Woolston A, Khan K, Spain G, et al. Genomic and transcriptomic determinants of therapy resistance and immune landscape evolution during anti-EGFR treatment in colorectal cancer. Cancer Cell. 2019;36(35–50):e9.
Abdelfatah E, Kerner Z, Nanda N, et al. Epigenetic therapy in gastrointestinal cancer: the right combination. Therap Adv Gastroenterol. 2016;9:560–79.
West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–9.
Suvà ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567–70.
Baretti M, Azad NS. The role of epigenetic therapies in colorectal cancer. Curr Probl Cancer. 2018;42:530–47.
McGoverne I, Dunn J, Batham J, et al. Epitherapy and immune checkpoint blockade: using epigenetic reinvigoration of exhausted and dysfunctional T cells to reimburse immunotherapy response. BMC Immunol. 2020;21:22.
Chen X, Pan X, Zhang W, et al. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm Sin B. 2019;10:723.
Li H, Chiappinelli KB, Guzzetta AA, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5:587–98.
Kim K, Skora AD, Li Z, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci USA. 2014;111:11774–9.
Christmas BJ, Rafie CI, Hopkins AC, et al. Entinostat converts immune-resistant breast and pancreatic cancers into checkpoint-responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol Res. 2018;6:1561–77.
Yu G, Wu Y, Wang W, et al. Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by re-modulating the tumor microenvironment. Cell Mol Immunol. 2019;16:401–9.
Xiao G, Jin L-L, Liu C-Q, et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J Immunother Cancer. 2019;7:300.
Goswami S, Apostolou I, Zhang J, et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J Clin Invest. 2018. https://doi.org/10.1172/JCI99760.
Prendergast GC, Malachowski WP, DuHadaway JB, et al. Discovery of IDO1 Inhibitors: from Bench to Bedside. Cancer Res. 2017;77:6795–811.
Murphy AG, Walker R, Lutz ER, et al. Epigenetic priming prior to pembrolizumab in mismatch repair-proficient advanced colorectal cancer. J Clin Oncol. 2019;37:591.
Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461–73.
White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/β-catenin signaling in gastrointestinal cancers. Gastroenterology. 2012;142:219–32.
Clément G, Braunschweig R, Pasquier N, et al. Alterations of the Wnt signaling pathway during the neoplastic progression of Barrett’s esophagus. Oncogene. 2006;25:3084–92.
Flanagan DJ, Vincan E, Phesse TJ. Winding back Wnt signalling: potential therapeutic targets for treating gastric cancers. Br J Pharmacol. 2017;174:4666–83.
Janku F, Connolly R, LoRusso P, et al. Abstract C45: phase I study of WNT974, a first-in-class Porcupine inhibitor, in advanced solid tumors. Mol Cancer Ther. 2015;14:C45.
Ng M, Tan DSP, Subbiah V, et al. First-in-human phase 1 study of ETC-159 an oral PORCN inhibitor in patients with advanced solid tumors. J Clin Oncol. 2017;35:2584.
Jimeno A, Gordon M, Chugh R, et al. A first-in-human phase i study of the anticancer stem cell agent ipafricept (OMP-54F28), a decoy receptor for Wnt ligands, in patients with advanced solid tumors. Clin Cancer Res. 2017;23:7490–7.
Smith DC, Rosen LS, Chugh R, et al. First-in-human evaluation of the human monoclonal antibody vantictumab (OMP-18R5; anti-Frizzled) targeting the WNT pathway in a phase I study for patients with advanced solid tumors. J Clin Oncol. 2013;31:2540.
Andersson T, Axelsson L, Mohapatra P, et al. Abstract A116: targeting the Wnt-5a signaling pathway as a novel anti-metastatic therapy. Mol Cancer Ther. 2015;14:A116.
El-Khoueiry AB, Ning Y, Yang D, et al. A phase I first-in-human study of PRI-724 in patients (pts) with advanced solid tumors. J Clin Oncol. 2013;31:2501.
Wang B, Tian T, Kalland K-H, et al. Targeting Wnt/β-catenin signaling for cancer immunotherapy. Trends Pharmacol Sci. 2018;39:648–58.
Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumor immunity. Nature. 2015;523:231–5.
Spranger S, Dai D, Horton B, et al. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31(711–723):e4.
Luke JJ, Bao R, Sweis RF, et al. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res. 2019;25:3074–83.
Xue J, Yu X, Xue L, et al. Intrinsic β-catenin signaling suppresses CD8+ T-cell infiltration in colorectal cancer. Biomed Pharmacother. 2019;115:108921.
Hong Y, Manoharan I, Suryawanshi A, et al. Deletion of LRP5 and LRP6 in dendritic cells enhances antitumor immunity. Oncoimmunology. 2015;5:e1115941.
Ding Y, Shen S, Lino AC, et al. Beta-catenin stabilization extends regulatory T cell survival and induces anergy in nonregulatory T cells. Nat Med. 2008;14:162–9.
Wong C, Chen C, Wu Q, et al. A critical role for the regulated wnt-myc pathway in naive T cell survival. J Immunol. 2015;194:158–67.
Malladi S, MacAlinao DG, Jin X, et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell. 2016;165:45–60.
Kagey MH, He X. Rationale for targeting the Wnt signalling modulator Dickkopf-1 for oncology. Br J Pharmacol. 2017;3:4637–50.
D’Amico L, Mahajan S, Capietto A-H, et al. Dickkopf-related protein 1 (Dkk1) regulates the accumulation and function of myeloid derived suppressor cells in cancer. J Exp Med. 2016;213:827–40.
Haas M, Heath H, Wu Y, et al. Abstract 1710: dKN-01, a therapeutic DKK1 neutralizing antibody, has immune modulatory activity in nonclinical tumor models. Cancer Res. 2018;78:1710.
Klempner SJ, Bendell J, Meucci Villaflor V, et al. 660PSafety and efficacy of a DKK1 inhibitor (DKN-01) in combination with pembrolizumab (P) in patients (Pts) with advanced gastroesophageal (GE) malignancies. Ann Oncol. 2018;29:12.
Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumor stroma to improve cancer therapy. Nat Rev Clin Oncol. 2018;15:366–81.
Jiang H, Hegde S, Knolhoff BL, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22:851–60.
Seo E-H, Namgung JH, Oh C-S, et al. Association of chemokines and chemokine receptor expression with monocytic-myeloid-derived suppressor cells during tumor progression. Immune Netw. 2018;18:e23.
Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci. 2013;110:20212–7.
Bockorny B, Semenisty V, Macarulla T, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med. 2020;26:878.
Doi T, Muro K, Ishii H, et al. A phase I study of the anti-CC chemokine receptor 4 antibody, mogamulizumab, in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin Cancer Res. 2019;25:6614–22.
Steele CW, Karim SA, Leach JDG, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. 2016;29:832–45.
Mitchem JB, Brennan DJ, Knolhoff BL, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73:1128–41.
Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–69.
Stiff A, Trikha P, Wesolowski R, et al. Myeloid-derived suppressor cells express Bruton’s Tyrosine kinase and can be depleted in tumor-bearing hosts by ibrutinib treatment. Cancer Res. 2016;76:2125–36.
Gunderson AJ, Kaneda MM, Tsujikawa T, et al. Bruton tyrosine kinase–dependent immune cell cross-talk drives pancreas cancer. Cancer Discov. 2016;6:270–85.
Massó-Vallés D, Jauset T, Serrano E, Sodir NM, Pedersen K, et al. Ibrutinib exerts potent antifibrotic and antitumor activities in mouse models of pancreatic adenocarcinoma. Cancer Res. 2015;75:1675.
Overman M, Javle M, Davis RE, et al. Randomized phase II study of the Bruton tyrosine kinase inhibitor acalabrutinib, alone or with pembrolizumab in patients with advanced pancreatic cancer. J Immunother Cancer. 2020;8:000587.
Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (80−). 2011;331:1612–6.
Strauss J, Heery CR, Schlom J, et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFβ, in advanced solid tumors. Clin Cancer Res. 2018;24:1287–95.
Yoo C, Oh D-Y, Choi HJ, et al. AB053 P-2 M7824 (MSB0011359C), a bifunctional fusion protein targeting transforming growth factor β (TGF-β) and PD-L1, in Asian patients with pretreated biliary tract cancer (BTC): efficacy by BTC subtype. Hepatobiliary Surg Nutr. 2019;8:053.
Le DT, Kim TW, Van Cutsem E, et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability–high/mismatch repair–deficient metastatic colorectal cancer: KEYNOTE-164. J Clin Oncol. 2019;38:11–9.
Fuchs CS, Doi T, Jang RW, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 Trial. JAMA Oncol. 2018;4:e180013.
Chen L-T, Satoh T, Ryu M-H, et al. A phase 3 study of nivolumab in previously treated advanced gastric or gastroesophageal junction cancer (ATTRACTION-2): 2-year update data. Gastric Cancer. 2020;23(3):510–9.
Doi T, Piha-Paul SA, Jalal SI, et al. Updated results for the advanced esophageal carcinoma cohort of the phase Ib KEYNOTE-028 study of pembrolizumab (MK-3475). J Clin Oncol. 2016;34:7.
Vincent J, Mignot G, Chalmin F, et al. 5-fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell–dependent antitumor immunity. Cancer Res. 2010;70:3052–61.
Ghiringhelli F, Bruchard M, Apetoh L. Immune effects of 5-fluorouracil: ambivalence matters. Oncoimmunology. 2013;2:e23139.
Bruchard M, Mignot G, Derangère V, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. 2013;19:57–64.
Beyranvand Nejad E, van der Sluis TC, van Duikeren S, et al. Tumor eradication by cisplatin is sustained by CD80/86-mediated costimulation of CD8 T cells. Cancer Res. 2016;76:6017–29.
Rébé C, Demontoux L, Pilot T, et al. Platinum derivatives effects on anticancer immune response. Biomolecules. 2019;10:13.
Tesniere A, Schlemmer F, Boige V, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–91.
de Biasi AR, Villena-Vargas J, Adusumilli PS. Cisplatin-induced antitumor immunomodulation: a review of preclinical and clinical evidence. Clin Cancer Res. 2014;20:5384–91.
Casares N, Pequignot MO, Tesniere A, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–701.
Sistigu A, Yamazaki T, Vacchelli E, et al. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20:1301–9.
Luthra P, Aguirre S, Yen BC, et al. Topoisomerase II inhibitors induce DNA damage-dependent interferon responses circumventing ebola virus immune evasion. MBio. 2017;8:e00368.
Wan S, Pestka S, Jubin RG, et al. Chemotherapeutics and radiation stimulate MHC Class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS ONE. 2012;7:e32542.
Kalsbeek D, Golsteyn RM. G2/M-phase checkpoint adaptation and micronuclei formation as mechanisms that contribute to genomic instability in human cells. Int J Mol Sci. 2017;18:2344.
Shevchenko I, Karakhanova S, Soltek S, et al. Low-dose gemcitabine depletes regulatory T cells and improves survival in the orthotopic Panc02 model of pancreatic cancer. Int J Cancer. 2013;133:98–107.
Eriksson E, Wenthe J, Irenaeus S, et al. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J Transl Med. 2016;14:282.
Acknowledgements
This work is supported by the National Institute for Health Research (NIHR), Biomedical Research Center (BRC) at the Royal Marsden NHS Foundation Trust, and the Institute of Cancer Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure statement
Fiona Turkes and Justin Mencel have no conflict of interest to disclose.
Naureen Starling has the following conflict of interest disclosures: Research Funding: AstraZeneca, BMS, Pfizer; Travel & Accommodation: AstraZeneca, BMS, Eli Lilly, Merck, Roche; Honoraria: AstraZeneca, Eli Lilly, Merck, Servier; Advisory Board: Pfizer, AstraZeneca, Servier.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Turkes, F., Mencel, J. & Starling, N. Targeting the immune milieu in gastrointestinal cancers. J Gastroenterol 55, 909–926 (2020). https://doi.org/10.1007/s00535-020-01710-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-020-01710-x