
Abstract
Although colorectal cancer is the third leading cause of death in Morocco, there are no studies of the microbiome changes associated with the disease in the Moroccan population. The aim of our study was to compare the stool microbiome of Moroccan cancer patients with healthy individuals. We analyzed the microbiome composition of samples from 11 CRC patients and 12 healthy individuals by 16S rRNA amplicon sequencing. Principal coordinate analysis of samples revealed defined cancer versus healthy clusters. Our findings showed that cancer samples had higher proportions of Firmicutes (T = 50.5%; N = 28.4%; p = 0.04), specifically of Clostridia (T = 48.3%; N = 19.0%; p = 0.002), and Fusobacteria (T = 0.1%; N = 0.0%; p = 0.02), especially of Fusobacteriia (T = 0.1%; N = 0.0%; p = 0.02), while Bacteroidetes were enriched in healthy samples (T = 35.1%; N = 62.8%; p = 0.06), particularly the class Bacteroidia (T = 35.1%; N = 62.6%; p = 0.06). Porphyromonas, Clostridium, Ruminococcus, Selenomonas, and Fusobacterium were significantly overrepresented in diseased patients, similarly to other studies. Predicted functional information showed that bacterial motility proteins, flagellar assembly, and fatty acid biosynthesis metabolism were significantly overrepresented in cancer patients, while amino acid metabolism and glycan biosynthesis were overrepresented in controls. This suggests that involvement of these functional metagenomes is similar and relevant in the carcinogenesis process, independent of the origin of the samples. Results from this study allowed identification of bacterial taxa relevant to the Moroccan population and encourages larger studies to facilitate population-directed therapeutic approaches.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is one of the most common cancers worldwide, and is the third cause of cancer mortality in the world [1,2,3]. In Morocco, CRC is as prevalent, behind breast and cervical cancer for women, and lung and prostate cancer for men [4, 5]. The number of patients affected has increased over the last decade, with an increase in incidence from 6.0 per 100,000 to 10.4 per 100,000 from 2005 to 2008 in Rabat [5] and from 10.8 per 100,000 to 12.9 per 100,000 from 2004 to 2007 in Casablanca [4, 6].
CRC is a multifactorial disease with both environmental and genetic contributions to its pathogenesis. CRC can be classified by etiology as hereditary [7] (e.g. familial adenomatous polyposis due to an initiating mutation in the Adenomatous Polyposis Coli (APC) gene), inflammatory (associated with Crohn’s disease and ulcerative colitis), or sporadic (in more than 80% of cases) [8]. Risk factors for sporadic CRC include diet, age, alcohol consumption, smoking, physical activity and body mass index [9,10,11,12]. The incidence and mortality of CRC show geographical variation, with a high prevalence in Western countries, reflecting the importance of environmental factors [13,14,15]. Indeed, immigrants from low-incidence areas acquire similar CRC rates over time upon arrival in higher incidence areas, and eating habits likely contribute to this observation [16,17,18,19]. We have previously shown that geographic location and diet habits may impact the composition of the gut microbiome as reflected by significant differences in bacterial populations in tumor and tumor-adjacent tissues in individuals from Spain and the US [20]. Our study demonstrated an association between Eikenella and tumor tissues only in US individuals, while tumors from Spaniards were enriched for Fusobacterium, Bulleida, Gemella, Parvimonas, Campylobacter, and Streptococcus. In another study, a comparison of healthy African American and native African groups identified significant differences between the two populations due to higher dietary intakes of animal products by the African American population, with major butyrate-producing bacterial groups overrepresented in native African populations [16]. Finally, CRC rates in migrant groups from high-incidence southern European countries declined after more than 15 years of residence in Australia, approaching the rates of the host country [18].
Research studies indicate that composition and functionality of the gut microbiome play a major role in modulating CRC risk [12, 21,22,23,24,25,26,27,28,29,30,31]. Studies seeking to identify specific bacterial signatures associated with cancer incidence have not been successful [32,33,34] due to the complexity of the gut microbiome [35,36,37,38], and the diversity within and between individuals and populations [39, 40]. However, metagenomics studies have implicated certain bacterial species correlated with the presence of CRC [41,42,43,44,45,46,47,48]. Overrepresentation of species of Fusobacterium in CRC has been demonstrated in several studies in both stool and mucosal samples [20, 49, 50], raising the possibility that this species may play a causative role in carcinogenesis [51,52,53]. Fusobacterium is a rare inhabitant of the colon [54], but a well-known pathogen of the mouth, responsible for periodontitis and inflammation [55, 56]. A study by Rubinstein et al. showed that binding of Fusobacterium nucleatum to a specific receptor of the epithelial cells activated the proliferation of human colon cancer cells [57].
The Moroccan diet, rich in fruits, vegetables, and olive oil, is consistent with a Mediterranean diet. Despite dietary similarities, different regions display specific habits depending on cultural influences, religion, and lifestyles [58,59,60]. A study comparing dietary habits of Mediterranean populations from Spain, Morocco and Palestine reported high carbohydrate intake and low protein intake in Palestine compared to Spain and Morocco, while the Moroccan population had the highest consumption of fruits, vegetables, dairy products, and fish [61]. Considering the impact of diet on the composition of the gut microbiome [62,63,64], research studies from different geographic regions with different dietary habits are essential to advance the collective knowledge and allow tailored and effective CRC treatments. The aim of our study was to compare the composition of the gut microbiome of Moroccan CRC patients versus healthy individuals. We performed 16S rRNA amplicon sequencing of stool samples to determine composition, followed by predictive functional analysis of data. The findings of this study provide new insights on the gut microbiome composition and on specific bacterial communities related to CRC in an understudied population.
Materials and methods
Ethics statement
This study was approved by the University Hospital Center Hassan II of Fez, Morocco. A written informed consent was obtained from all patients and healthy individuals.
Samples metadata and dietary questionnaires
Fecal samples were obtained from 11 colorectal cancer patients and 12 healthy subjects. Inclusion selection of CRC patients for this study was based on the following criteria: no gastrointestinal disorders, no antibiotic use during the last 3 months and those who had been recently diagnosed and had not yet started treatment. Inclusion criteria for healthy individuals were: absence of gastrointestinal disorders and no antibiotic treatment during the last 3 months. CRC and healthy individuals were of similar ages, came from the same region, and had similar diets and lifestyle. Individuals from the Oriental region and from Casablanca are served by the same University Hospitals. Colorectal cancer patients and healthy subjects were given a food survey questionnaire and were requested to report their diet over the past 5 days before collecting their stool samples. The food survey reports all food consumed by an individual during the day. From this survey, we measured the frequency of consumption of fruits, vegetables and red meat (e.g. times per day, daily, weekly). Dietary data were divided into two groups: (1) high consumption of fruits and vegetables/low consumption of meat and (2) low consumption of fruits and vegetables/high consumption of meat. Daily consumption of fruits, vegetables, and meat was measured in grams for each subject (patients and healthy individuals). Individuals in group 1 consumed more than 250 g of fruits and vegetables and less than 50 g of meat per day. Conversely, individuals in group 2 consumed less than 250 g of fruits and vegetables and more than 50 g of meat per day.
In addition to the food survey, information related to age, sex, body mass index (BMI) and family history of CRC were collected from the subjects. BMI was measured for both groups [BMI is calculated from body mass (M) and height (H). BMI = M/(H × H), where M = body mass in kilograms and H = height in meters]. Dietary data were self-reported.
Sample storage and DNA isolation
All stool samples were collected in sterile Eppendorf tubes and then frozen at − 80 °C until DNA extraction. DNA isolation was carried out using the QIAmp DNA Stool kit supplied by Qiagen (Hilden, Germany). The subsequent steps were performed as recommended by the manufacturer’s protocol with minor modifications. Briefly, 200 mg of stool samples was added to a tube containing 1.4 ml buffer ASL. Samples were homogenized using a Tissue Lyser (Qiagen) for 1 min at 25 Hz. 15 ml of proteinase K and 200 ml AL buffer were added to samples, vortexed and incubated at 70 °C for 10 min according to the manufacturer’s instruction. Then, 200 ml of 100% ethanol was added to the mixture and they were transferred into a column. Following this, 500 ml of buffers AW1 and AW2 was added to the column separately and the flow-through was discarded in each step. Finally, 200 ml of buffer AE was added to the column to elute the DNA.
16S rRNA amplicon sequencing
Sequencing of 16S rRNA amplicons was done at the UNC Microbiome Core Facility. DNA was amplified using primers targeting the V1–V2 region of the bacterial 16S rRNA gene [65, 66] and overhang adapter sequences appended to the primer pair for compatibility with Illumina index and sequencing adapters. Master mixes contained 12.5 ng of total DNA, 2× KAPA HiFi HotStart ReadyMix (KAPA Biosystems, Wilmington, MA, USA). The thermal profile for the amplification of each sample had an initial denaturing step at 95 °C for 3 min, followed by a cycling of denaturing of 95 °C for 30 s, annealing at 55 °C for 30 s and a 30 s extension at 72 °C (25 cycles), a 5 min extension at 72 °C and a final hold at 4 °C. Each 16S rRNA amplicon was purified using AMPure XP reagent (Beckman Coulter, Indianapolis, IN, USA). Each sample was then amplified using a limited cycle PCR program, adding Illumina sequencing adapters and dual-index barcodes [index 1(i7) and index 2(i5)] (Illumina, San Diego, CA, USA) to the amplicon target. For the second round of amplification, the thermal profile consisted of an initial denaturing step at 95 °C for 3 min, followed by a denaturing cycle of 95 °C for 30 s, annealing at 55 °C for 30 s and a 30 s extension at 72 °C (8 cycles), and 5 min extension at 72 °C. The final libraries were again purified using AMPure XP reagent (Beckman Coulter), quantified and normalized prior to pooling. The DNA library pool was then denatured with NaOH, diluted with hybridization buffer and heat denatured before loading on the MiSeq reagent cartridge (Illumina) and on the MiSeq instrument (Illumina). Automated cluster generation and paired-end sequencing with dual reads were performed according to the respective manufacturer’s instructions.
Bioinformatics sequencing data analysis
The Quantitative Insights Into Microbial Ecology (QIIME v.1.8.0) software pipeline [67] was conducted for the bioinformatics analysis of our bacterial 16S sequencing data. The raw sequences were demultiplexed and filtered; all reads with a length above 200 bp and with a quality score above 25 were kept. The resulting reads were clustered into operational taxonomic units (OTU) at 97% similarity threshold using UCLUST [68] from QIIME. After OTU picking, chimeras and singletons were removed using Chimera Slayer [69, 70]. Then, the sequences were aligned in order to build a phylogenetic tree using Fast Tree 2.1.3 [71]. The species level assignment was determined from the QIIME output using a biom file from the OTU picking. Additionally, to measure alpha diversity using observed species (S) and phylogenetic diversity (PD) metrics a random selection of 9090 sequences from each sample was used. Beta diversity and principal coordinates analysis (PCoA) were also calculated within QIIME using weighted and unweighted Unifrac distances [72] between samples at a depth of 9090 sequences per sample to evaluate dissimilarities between the samples. PD corresponds to the sum of branches on the phylogenetic tree among taxa occurring in a sample [73] and S is the number of OTUs per sample.
Functional metagenome prediction
To predict the functional metagenome profiles from 16S rRNA amplicon sequencing input data, we used the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) [74] (version 1.0.0) algorithm. Closed Reference OTUs were picked using UCLUST [68] against the GreenGenes database in order to create the OTU table for input into PICRUSt. The OTUs table result was normalized by dividing each OTU by the predicted 16S rRNA gene abundance before predicting the functional metagenome based on the KEGG orthology groups (KOs). The resulting functional metagenome by PICRUSt was used in the HMP Unified Metabolic Analysis (HUMAnN) [75] pipeline (version 0.99) to determine the presence or absence and the abundance of microbial KEGG pathways in our 16S rRNA amplicon sequencing data.
Statistical analyses
T-Tests were performed to evaluate significant (p < 0.05) differences in phylogenetic diversity (PD) and species richness (S) indexes between healthy individuals and colorectal cancer patients. Analysis of Similarities (ANOSIM) and Permutational Multivariate Analysis of Variance (PERMANOVA) analyses were used to evaluate similarities between the two groups. The non-parametric Steel–Dwass method, which performs multiple comparisons while controlling the overall experiment-wise error rate, was applied to microbiome data. Significant differences (p < 0.05) in relative abundances of bacterial taxa and in metabolic pathways and enzymes between cohorts were computed using JMP Genomics (SAS, JMP Genomics 10.0). The Steel–Dwass All Pairs test corrects for multiple comparisons.
Results
Eleven stool samples were collected from patients who had been diagnosed with CRC between October 2013 and December 2013, but who had not yet received treatment. The samples were collected from patients at the University Hospital Hassan II of Fez, Morocco. Twelve stool samples from healthy individuals were collected from the Oriental region (Northeastern area of Morocco) and Casablanca. CRC patients and healthy subjects were asked to give a detailed food record for the 5 days before sample collection, and data were collected regarding geographic origin, age, weight, family history, and risk factors. Characteristics of patients and healthy individuals are summarized in (Table 1). No statistical differences were observed between the two groups in age, sex and BMI with (p = 0.5), (p = 0.9), (p = 0.1) respectively.
After 16S rRNA amplicon sequencing of DNA extracted from stool samples, a total of 1,633,421 sequences passed our quality filtering (length > 200 bp, quality scores > 25). The average quality score was 35.5 ± 4.1, the average number of reads per sample was 67,505 ± 28,344, and the average of sequences length distribution was 315.2 ± 19.3. Almost the entirety of sequences (98.8%) was assigned to a taxonomic group, while 1.2% of the reads were unassigned. A total of 5081 Operational Taxonomic Units (OTUs) were identified in the cohort after clustering sequences at a 97% similarity threshold.
The CRC microbiome had a higher diversity than the non-CRC microbiome
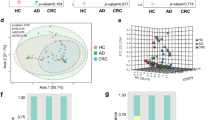
Rarefaction analyses at a sampling depth of 9090 reads/sample were conducted to determine phylogenetic diversity (PD) and species richness (S) indexes (Fig. 1a, b). In contrast to our previous study on biopsy samples [20], we observed a trend towards CRC samples having higher PD and S values than healthy samples (t test p < 0.1).

a Phylogenetic Diversity (PD) comparison between CRC and healthy individuals from the Moroccan population (*p < 0.1), b number of species identified in CRC and healthy individuals from the Moroccan population (*p > 0.1), c principal coordinates analysis—PCoA (unweighted UniFrac) of samples, d distribution of bacterial phyla in CRC versus healthy individuals, e distribution of bacterial phyla by individual (12 controls and 11 CRC)
Subsequent principal coordinates analysis (PCoA) with analysis of similarities (ANOSIM) and permutational multivariate analysis of variance (PERMANOVA) showed a low correlation between disease state in samples (ANOSIM, R = 0.2039, p = 0.008 and PERMANOVA, F = 1.8976, p = 0.005,) (Fig. 1c). No statistically significant differences were observed between control and CRC groups in the age category from 20 to 49 years old. However, a moderate but statistically significant effect was observed when we compared control and CRC groups in the second age category (ages 50–79) (ANOSIM, R = 0.3072, p = 0.036 and PERMANOVA, F = 1.7538, p = 0.036,). Comparison of samples according to sex, body mass index and diet showed no statistically significant differences between CRC and healthy groups.
Diet associated with the microbiome composition in healthy and CRC subjects
To assess the impact of diet on the gut microbiome composition, we Evaluated the phylogenetic diversity (PD) and species richness (S) indexes in healthy individuals by comparing samples from individuals following a diet rich in fruits and vegetables and low in red meat consumption with samples from individuals with a diet low in fruits and vegetables/high in red meat. Although we were not able to conduct any statistical analysis due to the low number of samples (high fruits and vegetables/low red meat group n = 6; low fruits and vegetables/high red meat group n = 6), we observed that high fruits and vegetables/low red meat subjects had high values of phylogenetic diversity and species richness (PD = 14.3 ± 4.6, S = 367.0 ± 164.7 vs PD = 12.6 ± 5.2, S = 299.0 ± 177.4). Veillonella, Shewanella, Lactococcus, and Bacteroides were statistically overrepresented in the high fruits and vegetables/low red meat group, while Pseudomonas was statistically overrepresented in the low fruits and vegetables/high red meat group (p ≤ 0.1) (Fig. 2). We also evaluated the phylogenetic diversity (PD) and species richness (S) indexes in CRC patients. We found that patients having a diet rich in fruits and vegetables/low in red meat had slightly higher values of PD and S than patients having a diet low in fruits and vegetables/high in red meat (PD = 20.4 ± 0.8, S = 518.7 ± 99.2 vs PD = 20.0 ± 1.5, S = 445.5 ± 28.9). However, no significant differences in bacterial communities were observed in CRC patients according to type of diet.

Relative abundances of significant bacterial genera in healthy individuals consuming a diet high in fruits and vegetables/low in red meat compared to healthy individuals following a diet low in fruits and vegetables/high in red meat (the boxplot scales are not the same)
Gut microbiome composition of CRC and control stools
Our analysis showed that sequences clustered into 13 phyla, 26 classes, 48 orders, 92 families, and 165 genera. The most represented phyla in both CRC and controls were Bacteroidetes, Firmicutes, and Proteobacteria (Fig. 1d, e). Other phyla detected at low relative abundance (< 1.0%) were Actinobacteria, Cyanobacteria, Elusimicrobia, Fusobacteria, Lentisphaerae, Synergistetes, TM7, Tenericutes, and Verrucomicrobia. In the CRC group, Fusobacteria (CRC = 0.1% vs control = 0.0%), Firmicutes (CRC = 50.5% vs control = 28.4%) and Proteobacteria (CRC = 9.5% vs control = 6.8%) were overrepresented (Steel Dwass all pairs, p < 0.05), while Bacteroidetes (CRC = 35.1% vs control = 62.6%) were more prevalent in controls (p = 0.06). The predominant genera in both cohorts were Bacteroides and Prevotella. However, Prevotella was overrepresented in the control group while Bacteroides showed a non-significant overrepresentation in CRC (p ≤ 0.1). CRC stools were markedly different from controls, showing an overrepresentation of 33 genera (Table 2). The most significantly overrepresented species in normal samples compared to CRC samples were Prevotella copri, Prevotella stercorea, and Faecalibacterium prausnitzii, while for the CRC samples we found that Collinsella aerofaciens (Actinobacteria), [Eubacterium] biforme (Firmicutes), Oxalobacter formigenes (Proteobacteria), Akkermansia municiphila (Verrucomicrobia) and Bacteroides fragilis (Bacteroidetes) were significantly overrepresented.
Predicted functional differences between the CRC and control cohorts
We used the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) [75] to identify differences in metagenome functional prediction based on Greengenes 16S rRNA database and KEGG Orthologs (KO). A total of 328 functional metagenomes were predicted in both CRC and control cohorts (Fig. 3, Supplementary Table 1). Cellular processes (bacterial chemotaxis, bacterial motility proteins, and flagellar assembly), environmental information processing (membrane transport and signal transduction), lipid (fatty acid biosynthesis and fatty acid metabolism) and carbohydrate metabolism (pentose phosphate pathway), and xenobiotics biodegradation and metabolism were overrepresented in the CRC cohort (Steel Dwass All Pairs, p < 0.05). In contrast, genetic information processing (chaperones and folding catalysts, RNA degradation, and protein processing in endoplasmic reticulum), organismal systems (carbohydrate digestion and absorption, protein digestion and absorption, and NOD-like receptor signaling pathway), amino acid metabolism (glycine, serine and threonine metabolism), energy metabolism (oxidative phosphorylation), glycan biosynthesis and metabolism (glycosyltransferases and lipopolysaccharide biosynthesis) and metabolism of other amino acids (glutathione metabolism) were significantly overrepresented in the control cohort. The significantly overrepresented enzymes between CRC and controls samples are listed in (Table 3).

Relative abundance of the predicted functional pathways in control versus CRC individuals
Discussion
CRC incidence in African populations is low compared to European and North American populations. This low incidence has been attributed to anthropomorphic or environmental factors [76, 77]. However, incidence has been increasing over the last decade due to the westernization of the diet in many African countries [4,5,6, 13, 14, 78, 79].
Studies to understand the impact of geographical and cultural differences affecting potential roles of the gut microbiome on CRC, especially in understudied populations, are essential. In this analysis, we compared 11 stool samples from CRC patients with 12 stool samples from healthy Moroccan subjects. We observed a trend towards a higher phylogenetic diversity (PD) and species richness (S) in CRC versus controls, but the differences were not significant. Other studies showed no significant changes in diversity and species richness between CRC and healthy subjects [80,81,82,83] and similar observations were made in tissue samples. A comparison of 90 matched pairs of colorectal carcinoma and tumor-adjacent (normal) tissues from cohorts from the US and Spain showed no significant differences between normal and tumor tissues for both cohorts [20]. In contrast, other studies have reported significant differences in diversity and species richness in both tissue [84,85,86] and stool samples [21].
The genus Bacteroides was overrepresented in CRC while Prevotella was increased significantly in controls. Specifically, Bacteroides fragilis was more common in CRC patients. Our findings agree with previous studies that showed that Bacteroides were enriched in CRC patients [20, 34, 75]. Prevotella has been repeatedly associated with diets rich in fiber, while diets rich in fat and animal protein are conducive to a Bacteroides-dominated gut microbiota [62, 63, 87]. More recently, the enterotoxigenic Bacteroides fragilis toxin (ETBF) has been shown to cause chronic inflammation that could promote CRC [34, 87,88,89,90]. One of the three isoforms of ETBF indirectly induces cleavage of E-cadherin, resulting in increased epithelial cell permeability and exposure to bacterial antigens, which contribute to chronic inflammation [91,92,93]. ETBF has been associated with inflammatory bowel disease and CRC. It is characterized by the activation of Stat3 in mucosal immune and epithelial cells, with a subsequent colonic mucosal Th17 response that induces robust colonic tumors [94]. Additionally, it has been shown that treatment with antibody-mediated IL-17, a key cytokine amplifying Th17, reduced ETBF and tumor formation [95].
Similarly, Fusobacterium was overrepresented in our CRC cohort. This genus has been recurrently associated with CRC [20, 48, 50,51,52,53, 57, 96,97,98]. Moreover, Fusobacterium was not detected in healthy control samples, as it is a relatively uncommon bacterium in the gut microbiome. F. nucleatum [51] has been associated with CRC through its ability to stimulate the proliferation of tumor cells through the FadA (fluffy autolytic dominant A) adhesion gene [99,100,101]. FadA expression has been associated with increased expression of oncogenic and inflammatory genes; it may have a major role in the transformation of epithelial cells and promotion of colon tumorigenesis. Additionally, FadA binds E-cadherin, activating the beta-catenin signaling pathway, promoting the gut inflammatory response [57]. Administration of F. nucleatum to Apc(Min/+) mice increased the number of colon tumors and recruited tumor-infiltrating myeloid cells, inducing a pro-inflammatory state similar to that observed in humans [52].
Our study confirmed the role of other bacterial biomarkers in CRC, including Porphyromonas, overrepresented in CRC samples in accordance with previous reports [21, 83, 84, 102]. An association between oral bacteria, periodontal disease, and cancer has been established, specifically between Porphyromonas gingivalis and F. nucleatum [99, 103,104,105,106]. Porphyromonas has been associated with oral periodontal disease. It can penetrate periodontal tissue and alters the composition of the oral microbiome [89, 107]. Porphyromonas gingivalis has been linked to orodigestive cancer [108], pancreatic cancer [109] and colorectal cancer [100]. Invasion of epithelial cells by P. gingivalis causes suppression of the apoptotic pathways JAK1, STAT3 and Akt [110, 111] and stimulates cell proliferation [112].
The role of other genera overrepresented in our CRC cohort is less clear. In our study, Clostridium, Butyricimonas, Peptostreptococcus, and Ruminococcus were significantly overrepresented in CRC samples. Conversely, Faecalibacterium prausnitzii, an anti-inflammatory commensal bacterium able to block NF-κB and IL-8 secretion [113, 114] was significantly overrepresented in healthy individuals. Finally, we observed a non-significant overrepresentation of the beneficial bacteria Lactobacillus and Bifidobacterium in healthy individuals.
Predictive functional analysis of sequencing data showed a higher significant relative abundance of genes responsible for cellular processes including bacterial chemotaxis, bacterial motility proteins, and flagellar assembly in CRC samples. Flagellin is the primary component of bacterial flagella. This protein has the potential to bind to Toll-like receptor 5 (TLR5) activating the transcription nuclear factor-κβ (NF-κβ) signaling-pathway with inflammatory and anti-apoptotic outcomes [115, 116]. Additionally, we found that acetaldehyde dehydrogenase and acetyl-CoA synthetase involved in glycolysis/gluconeogenesis were overrepresented in CRC samples. It is well known that acetaldehyde is highly toxic and is recognized as a carcinogenic molecule to humans [117]. Moreover, acetaldehyde is considered a CRC biomarker and plays a crucial role in cancer initiation and progression [118]. Finally, relative abundance of genes of the pentose phosphate pathway was significantly higher in CRC samples. This pathway plays a critical role in cancer cells by generating high levels of NADPH, which may be used in the synthesis of nucleic acids and is also required for both fatty acids synthesis and cell survival under stress conditions [119, 120].
This is the first study conducted on the CRC-associated gut microbiome in the Moroccan population. Studies have shown that populations from different geographic locations may have different healthy and disease-associated microbiota composition [20, 39, 121, 122], making this study of particular relevance. The low number of samples limited the power of this study; however, the study had the advantage of giving a first insight into the CRC gut microbiota composition of the Moroccan population. Likewise, the use of self-reported dietary information could have resulted in less accurate data. Although self-reported data are one the most used methods for data collection in health research, it could introduce biases and impact data reliability in analysis and potentially, the validity of the conclusions. Future, large-scale gut microbiome studies will confirm data from our pilot study in order to better understand the role of nutrition and other environmental factors on cancer etiology in the Moroccan population.
References
Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray, F. GLOBOCAN (2012) v1.0, Cancer Incidence and Mortality Worldwide, IARC CancerBase No. 11, International Agency for Research on Cancer, Lyon
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. Cancer J Clin 61(2):69–90. https://doi.org/10.3322/caac.20107
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136(5):E359–E386. https://doi.org/10.1002/ijc.29210
Bouchbika Z, Haddad H, Benchakroun N, Eddakaoui H, Kotbi S, Megrini A, Bourezgui H, Sahraoui S, Corbex M, Harif M, Benider A (2013) Cancer incidence in Morocco: report from Casablanca registry 2005-2007. Pan Afr Med J 16:31. https://doi.org/10.11604/pamj.2013.16.31.2791
Tazi MA, Er-Raki A, Benjaafar N (2013) Cancer incidence in Rabat, Morocco: 2006–2008. Ecancermedicalscience 7. https://doi.org/10.3332/ecancer.2013.338
Greater Casablanca cancer registry report 2007 (French) 2004. Available at: http://www.contrelecancer.ma/site_media/uploaded_files/Registre_des_Cancers_de_la_Re%C3%BCgion_du_grand_Casablanca_2004.pdf
Lynch H, De la Chapelle A (2003) Hereditary colorectal cancer. N Engl J Med 348(10):919–932
Hope ME, Hold GL, Kain R, El-Omar EM (2005) Sporadic colorectal cancer—role of the commensal microbiota. FEMS Microbiol Lett 244(1):1–7. https://doi.org/10.1016/j.femsle.2005.01.029
Bingham SA (2000) Diet and colorectal cancer prevention. Biochem Soc Trans 28(2):12–16
Yusof AS, Isa ZM, Shah SA (2012) Dietary patterns and risk of colorectal cancer: a systematic review of cohort studies (2000–2011). Asian Pac J Cancer Prev 13(9):4713–4717
Le Marchand L, Wilkens LR, Kolonel LN, Hankin JH, Lyu LC (1997) Associations of sedentary lifestyle, obesity, smoking, alcohol use, and diabetes with the risk of colorectal cancer. Cancer Res 57(21):4787–4794
Azcarate-Peril MA, Sikes M, Bruno-Barcena JM (2011) The intestinal microbiota, gastrointestinal environment and colorectal cancer: a putative role for probiotics in prevention of colorectal cancer? Am J Physiol Gastrointest Liver Physiol 301(3):G401–G424. https://doi.org/10.1152/ajpgi.00110.2011
Center MM, Jemal A, Smith RA, Ward E (2009) Worldwide variations in colorectal cancer. Cancer J Clin 59(6):366–378. https://doi.org/10.3322/caac.20038
Center MM, Jemal A, Ward E (2009) International trends in colorectal cancer incidence rates. Cancer Epidemiol Biomark Prev 18(6):1688–1694. https://doi.org/10.1158/1055-9965.EPI-09-0090
Vargas AJ, Thompson PA (2012) Diet and nutrient factors in colorectal cancer risk. Nutr Clin Pract 27(5):613–623. https://doi.org/10.1177/0884533612454885
Ou J, Carbonero F, Zoetendal EG, DeLany JP, Wang M, Newton K, Gaskins HR, O’Keefe SJ (2013) Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am J Clin Nutr 98(1):111–120. https://doi.org/10.3945/ajcn.112.056689
Yavari P, Hislop TG, Bajdik C, Sadjadi A, Nouraie M, Babai M, Malekzadeh R (2006) Comparison of cancer incidence in Iran and Iranian immigrants to British Columbia, Canada. Asian Pac J Cancer Prev 7(1):86–90
McMichael AJ, Giles GG (1988) Cancer in migrants to Australia: extending the descriptive epidemiological data. Cancer Res 48(3):751–756
Flood DM, Weiss NS, Cook LS, Emerson JC, Schwartz SM, Potter JD (2000) Colorectal cancer incidence in Asian migrants to the United States and their descendants. Cancer Causes Control 11(5):403–411
Allali I, Delgado S, Marron PI, Astudillo A, Yeh JJ, Ghazal H, Amzazi S, Keku T, Azcarate-Peril MA (2015) Gut microbiome compositional and functional differences between tumor and non-tumor adjacent tissues from cohorts from the US and Spain. Gut Microbes 6(3):161–172. https://doi.org/10.1080/19490976.2015.1039223
Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, Goedert JJ, Hayes RB, Yang L (2013) Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst 105(24):1907–1911. https://doi.org/10.1093/jnci/djt300
Zhu Q, Gao R, Wu W, Qin H (2013) The role of gut microbiota in the pathogenesis of colorectal cancer. Tumour Biol 34(3):1285–1300. https://doi.org/10.1007/s13277-013-0684-4
Rowland IR (2009) The role of the gastrointestinal microbiota in colorectal cancer. Curr Pharm Des 15(13):1524–1527
Sears CL, Garrett WS (2014) Microbes, microbiota, and colon cancer. Cell Host Microbe 15(3):317–328. https://doi.org/10.1016/j.chom.2014.02.007
Nistal E, Fernandez-Fernandez N, Vivas S, Olcoz JL (2015) Factors determining colorectal cancer: the role of the intestinal microbiota. Front Oncol 5:220. https://doi.org/10.3389/fonc.2015.00220
Dulal S, Keku TO (2014) Gut microbiome and colorectal adenomas. Cancer J 20(3):225–231. https://doi.org/10.1097/PPO.0000000000000050
Yang L, Pei Z (2006) Bacteria, inflammation, and colon cancer. World J Gastroenterol 12(42):6741–6746
Candela M, Guidotti M, Fabbri A, Brigidi P, Franceschi C, Fiorentini C (2011) Human intestinal microbiota: cross-talk with the host and its potential role in colorectal cancer. Crit Rev Microbiol 37(1):1–14. https://doi.org/10.3109/1040841X.2010.501760
Gagnière J, Raisch J, Veziant J, Barnich N, Bonnet R, Buc E, Bringer MA, Pezet D, Bonnet M (2016) Gut microbiota imbalance and colorectal cancer. World J Gastroenterol 22(2):501–518
Arnold JW, Roach J, Azcarate-Peril MA (2016) Emerging technologies for gut microbiome research. Cell Press 24(11):887–901
Verma M (2017) Mechanistic and technical challenges in studying the human microbiome and cancer epidemiology. Technol Cancer Res Treat 16(2):150–158. https://doi.org/10.1177/1533034616645219
Finegold SM, Flora DJ, Attebery HR, Sutter VL (1975) Fecal bacteriology of colonic polyp patients and control patients. Cancer Res 35(11 Pt.2):3407–3417
Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, Corthier G, Tran Van Nhieu J, Furet JP (2011) Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One 6(1):e16393. https://doi.org/10.1371/journal.pone.0016393
Scanlan PD, Shanahan F, Clune Y, Collins JK, O’Sullivan GC, O’Riordan M, Holmes E, Wang Y, Marchesi JR (2008) Culture-independent analysis of the gut microbiota in colorectal cancer and polyposis. Environ Microbiol 10(3):789–798. https://doi.org/10.1111/j.1462-2920.2007.01503.x
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin J, Sicheritz-Ponten T, Tims S, Torrents D, Ugarte E, Zoetendal EG, Wang J, Guarner F, Pedersen O, de Vos WM, Brunak S, Dore J, Meta HITC., Antolin M, Artiguenave F, Blottiere HM, Almeida M, Brechot C, Cara C, Chervaux C, Cultrone A, Delorme C, Denariaz G, Dervyn R, Foerstner KU, Friss C, van de Guchte M, Guedon E, Haimet F, Huber W, van Hylckama-Vlieg J, Jamet A, Juste C, Kaci G, Knol J, Lakhdari O, Layec S, Le Roux K, Maguin E, Merieux A, Melo Minardi R, M’Rini C, Muller J, Oozeer R, Parkhill J, Renault P, Rescigno M, Sanchez N, Sunagawa S, Torrejon A, Turner K, Vandemeulebrouck G, Varela E, Winogradsky Y, Zeller G, Weissenbach J, Ehrlich SD, Bork P (2011) Enterotypes of the human gut microbiome. Nature 473(7346):174–180. https://doi.org/10.1038/nature09944
Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE (2006) Metagenomic analysis of the human distal gut microbiome. Science 312(5778):1355–1359. https://doi.org/10.1126/science.1124234
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA (2005) Diversity of the human intestinal microbial flora. Science 308(5728):1635–1638. https://doi.org/10.1126/science.1110591
Bi Y, Qin N, Yang R (2015) Human microbiota: a neglected “organ” in precision medicine. Infect Dis Transl Med 1(2):63–72
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Meta HITC., Bork P, Ehrlich SD, Wang J (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464(7285):59–65. https://doi.org/10.1038/nature08821
Maccaferri S, Biagi E, Brigidi P (2011) Metagenomics: key to human gut microbiota. Dig Dis 29(6):525–530. https://doi.org/10.1159/000332966
Tjalsma H, Boleij A, Marchesi JR, Dutilh BE (2012) A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol 10(8):575–582. https://doi.org/10.1038/nrmicro2819
Marchesi JR, Dutilh BE, Hall N, Peters WH, Roelofs R, Boleij A, Tjalsma H (2011) Towards the human colorectal cancer microbiome. PLoS One 6(5):e20447. https://doi.org/10.1371/journal.pone.0020447
Boleij A, Schaeps RM, Tjalsma H (2009) Association between Streptococcus bovis and colon cancer. J Clin Microbiol 47(2):516. https://doi.org/10.1128/JCM.01755-08
Mager DL (2006) Bacteria and cancer: cause, coincidence or cure? A review. J Transl Med 4:14. https://doi.org/10.1186/1479-5876-4-14
Roberts S, Scott JR, Husmann LK, Zurawski CA (2006) Murine models of Streptococcus pyogenes infection. Curr Protoc Microbiol Chap 9:Unit 9:5. https://doi.org/10.1002/9780471729259.mc09d05s02 D .
Seder CW, Kramer M, Long G, Uzieblo MR, Shanley CJ, Bove P (2009) Clostridium septicum aortitis: report of two cases and review of the literature. J Vasc Surg 49(5):1304–1309. https://doi.org/10.1016/j.jvs.2008.11.058
Waisberg J, Matheus Cde O, Pimenta J (2002) Infectious endocarditis from Streptococcus bovis associated with colonic carcinoma: case report and literature review. Arq Gastroenterol 39(3):177–180
Geng J, Fan H, Tang X, Zhai H, Zhang Z (2013) Diversified pattern of the human colorectal cancer microbiome. Gut Pathog 5(1):2. https://doi.org/10.1186/1757-4749-5-2
Brim H, Yooseph S, Zoetendal EG, Lee E, Torralbo M, Laiyemo AO, Shokrani B, Nelson K, Ashktorab H (2013) Microbiome analysis of stool samples from African Americans with colon polyps. PLoS One 8(12):e81352. https://doi.org/10.1371/journal.pone.0081352
Tahara T, Yamamoto E, Suzuki H, Maruyama R, Chung W, Garriga J, Jelinek J, Yamano HO, Sugai T, An B, Shureiqi I, Toyota M, Kondo Y, Estecio MR, Issa JP (2014) Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res 74(5):1311–1318. https://doi.org/10.1158/0008-5472.CAN-13-1865
Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, Holt RA (2012) Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res 22(2):299–306. https://doi.org/10.1101/gr.126516.111
Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, El-Omar EM, Brenner D, Fuchs CS, Meyerson M, Garrett WS (2013) Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14(2):207–215. https://doi.org/10.1016/j.chom.2013.07.007
Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, Baselga J, Liu C, Shivdasani RA, Ogino S, Birren BW, Huttenhower C, Garrett WS, Meyerson M (2012) Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res 22(2):292–298. https://doi.org/10.1101/gr.126573.111
Segata N, Haake SK, Mannon P, Lemon KP, Waldron L, Gevers D, Huttenhower C, Izard J (2012) Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol 13(6):R42. https://doi.org/10.1186/gb-2012-13-6-r42
Sigusch BW, Engelbrecht M, Volpel A, Holletschke A, Pfister W, Schutze J (2010) Full-mouth antimicrobial photodynamic therapy in Fusobacterium nucleatum—infected periodontitis patients. J Periodontol 81(7):975–981. https://doi.org/10.1902/jop.2010.090246
Gmur R, Munson MA, Wade WG (2006) Genotypic and phenotypic characterization of fusobacteria from Chinese and European patients with inflammatory periodontal diseases. Syst Appl Microbiol 29(2):120–130. https://doi.org/10.1016/j.syapm.2005.07.011
Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW (2013) Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 14(2):195–206. https://doi.org/10.1016/j.chom.2013.07.012
Noah A, Truswell AS (2001) There are many Mediterranean diets. Asia Pac J Clin Nutr 10(1):2–9
Trichopoulou A, Vasilopoulou E (2000) Mediterranean diet and longevity. Br J Nutr 84(Suppl 2):S205–S209
Veronese N, Stubbs B, Noale M, Solmi M, Luchini C, Smith TO, Cooper C, Guglielmi G, Reginster JY, Rizzoli R, Maggi S (2016) Adherence to a Mediterranean diet is associated with lower prevalence of osteoarthritis: data from the osteoarthritis initiative. Clin Nutr. https://doi.org/10.1016/j.clnu.2016.09.035
Benhammou S, Heras-Gonzalez L, Ibanez-Peinado D, Barcelo C, Hamdan M, Rivas A, Mariscal-Arcas M, Olea-Serrano F, Monteagudo C (2016) Comparison of Mediterranean diet compliance between European and non-European populations in the Mediterranean basin. Appetite 107:521–526. https://doi.org/10.1016/j.appet.2016.08.117
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ (2014) Diet rapidly and reproducibly alters the human gut microbiome. Nature 505(7484):559–563. https://doi.org/10.1038/nature12820
Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, Sinha R, Gilroy E, Gupta K, Baldassano R, Nessel L, Li H, Bushman FD, Lewis JD (2011) Linking long-term dietary patterns with gut microbial enterotypes. Science 334(6052):105–108. https://doi.org/10.1126/science.1208344
Amato KR, Yeoman CJ, Cerda G, Schmitt CA, Cramer JD, Berg Miller ME, Gomez A, Turner TR, Wilson BA, Stumpf RM, Nelson KE, White BA, Knight R, Leigh SR (2015) Variable responses of human and non-human primate gut microbiomes to a Western diet. Microbiome 3:(53)
Edwards U, Rogall T, Blocker H, Emde M, Bottger EC (1989) Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucl Acids Res 17(19):7843–7853
Fierer N, Hamady M, Lauber CL, Knight R (2008) The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci USA 105(46):17994–17999. https://doi.org/10.1073/pnas.0807920105
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336. https://doi.org/10.1038/nmeth.f.303
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26(19):2460–2461. https://doi.org/10.1093/bioinformatics/btq461
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27(16):2194–2200. https://doi.org/10.1093/bioinformatics/btr381
Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, Methe B, DeSantis TZ, Human Microbiome C, Petrosino JF, Knight R, Birren BW (2011) Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21(3):494–504. https://doi.org/10.1101/gr.112730.110
Price MN, Dehal PS, Arkin AP (2010) FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5(3):e9490. https://doi.org/10.1371/journal.pone.0009490
Lozupone C, Hamady M, Knight R (2006) UniFrac—an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinf 7:371. https://doi.org/10.1186/1471-2105-7-371
Faith DP (1992) Conservation evaluation and phylogenetic diversity. Biol Convers 61:1–10
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31(9):814–821. https://doi.org/10.1038/nbt.2676
Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, White O, Kelley ST, Methe B, Schloss PD, Gevers D, Mitreva M, Huttenhower C (2012) Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 8(6):e1002358. https://doi.org/10.1371/journal.pcbi.1002358
Segal I, Edwards CA, Walker AR (2000) Continuing low colon cancer incidence in African populations. Am J Gastroenterol 95(4):859–860. https://doi.org/10.1111/j.1572-0241.2000.01922.x
Busolo DS, Woodgate RL (2015) Cancer prevention in Africa: a review of the literature. Glob Health Promot 22(2):31–39. https://doi.org/10.1177/1757975914537094
Irabor DO (2011) Colorectal carcinoma: why is there a lower incidence in Nigerians when compared to Caucasians? J Cancer Epidemiol 2011:675154. https://doi.org/10.1155/2011/675154
Siegel R, Naishadham D, Jemal A (2012) Cancer statistics, 2012. Cancer J Clin 62(1):10–29. https://doi.org/10.3322/caac.20138
Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI, Amiot A, Bohm J, Brunetti F, Habermann N, Hercog R, Koch M, Luciani A, Mende DR, Schneider MA, Schrotz-King P, Tournigand C, Tran Van Nhieu J, Yamada T, Zimmermann J, Benes V, Kloor M, Ulrich CM, von Knebel Doeberitz M, Sobhani I, Bork P (2014) Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol Syst Biol 10:766. https://doi.org/10.15252/msb.20145645
Hale VL, Chen J, Johnson S, Harrington SC, Yab TC, Smyrk TC, Nelson H, Boardman LA, Druliner BR, Levin TR, Rex DK, Ahnen DJ, Lance P, Ahlquist DA, Chia N (2017) Shifts in the fecal microbiota associated with Adenomatous polyps. Cancer Epidemiol Biomark Prev 26(1):85–94
Weir TL, Manter DK, Sheflin AM, Barnett BA, Heuberger AL, Ryan EP (2013) Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS One 8(8):e70803. https://doi.org/10.1371/journal.pone.0070803
Wu N, Yang X, Zhang R, Li J, Xiao X, Hu Y, Chen Y, Yang F, Lu N, Wang Z, Luan C, Liu Y, Wang B, Xiang C, Wang Y, Zhao F, Gao GF, Wang S, Li L, Zhang H, Zhu B (2013) Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb Ecol 66(2):462–470. https://doi.org/10.1007/s00248-013-0245-9
Chen W, Liu F, Ling Z, Tong X, Xiang C (2012) Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One 7(6):e39743. https://doi.org/10.1371/journal.pone.0039743
Sanapareddy N, Legge RM, Jovov B, McCoy A, Burcal L, Araujo-Perez F, Randall TA, Galanko J, Benson A, Sandler RS, Rawls JF, Abdo Z, Fodor AA, Keku TO (2012) Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. ISME J 6(10):1858–1868. https://doi.org/10.1038/ismej.2012.43
Burns MB, Lynch J, Starr TK, Knights D, Blekhman R (2015) Virulence genes are a signature of the microbiome in the colorectal tumor microenvironment. Genome Med 7(1):55. https://doi.org/10.1186/s13073-015-0177-8
Wu S, Morin PJ, Maouyo D, Sears CL (2003) Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology 124(2):392–400. https://doi.org/10.1053/gast.2003.50047
Keku TO, McCoy AN, Azcarate-Peril AM (2013) Fusobacterium spp. and colorectal cancer: cause or consequence? Trends Microbiol 21(10):506–508. https://doi.org/10.1016/j.tim.2013.08.004
Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA (2011) Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10(5):497–506. https://doi.org/10.1016/j.chom.2011.10.006
Belenguer A, Duncan SH, Calder AG, Holtrop G, Louis P, Lobley GE, Flint HJ (2006) Two routes of metabolic cross-feeding between Bifidobacterium adolescentis and butyrate-producing anaerobes from the human gut. Appl Environ Microbiol 72(5):3593–3599. https://doi.org/10.1128/AEM.72.5.3593-3599.2006
Sears CL (2009) Enterotoxigenic Bacteroides fragilis: a rogue among symbiotes. Clin Microbiol Rev 22(2):349–369. https://doi.org/10.1128/CMR.00053-08
Wu S, Lim KC, Huang J, Saidi RF, Sears CL (1998) Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proc Natl Acad Sci USA 95(25):14979–14984
Avila-Campos MJ, Liu C, Song Y, Rowlinson MC, Finegold SM (2007) Determination of bft gene subtypes in Bacteroides fragilis clinical isolates. J Clin Microbiol 45(4):1336–1338. https://doi.org/10.1128/JCM.02108-06
Wick EC, Rabizadeh S, Albesiano E, Wu X, Wu S, Chan J, Rhee KJ, Ortega G, Huso DL, Pardoll D, Housseau F, Sears CL (2014) Stat3 activation in murine colitis induced by enterotoxigenic Bacteroides fragilis. Inflamm Bowel Dis 20(5):821–834. https://doi.org/10.1097/MIB.0000000000000019
Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, Housseau F, Pardoll DM, Sears CL (2009) A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 15(9):1016–1022. https://doi.org/10.1038/nm.2015
Ray K (2011) Colorectal cancer: Fusobacterium nucleatum found in colon cancer tissue—could an infection cause colorectal cancer? Nat Rev Gastroenterol Hepatol 8(12):662. https://doi.org/10.1038/nrgastro.2011.208
McCoy AN, Araujo-Perez F, Azcarate-Peril A, Yeh JJ, Sandler RS, Keku TO (2013) Fusobacterium is associated with colorectal adenomas. PLoS One 8(1):e53653. https://doi.org/10.1371/journal.pone.0053653
Park CH, Han DS, Oh YH, Lee AR, Lee YR, Eun CS (2016) Role of Fusobacteria in the serrated pathway of colorectal carcinogenesis. Sci Rep 6:25271. https://doi.org/10.1038/srep25271
Binder Gallimidi A, Fischman S, Revach B, Bulvik R, Maliutina A, Rubinstein AM, Nussbaum G, Elkin M (2015) Periodontal pathogens Porphyromonas gingivalis and Fusobacterium nucleatum promote tumor progression in an oral-specific chemical carcinogenesis model. Oncotarget 6(26):22613–22623. https://doi.org/10.18632/oncotarget.4209
Flynn KJ, Baxter NT, Schloss PD (2016) Metabolic and community synergy of oral bacteria in colorectal cancer. mSphere. https://doi.org/10.1128/mSphere.00102-16
Louis P, Hold GL, Flint HJ (2014) The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 12(10):661–672. https://doi.org/10.1038/nrmicro3344
Vogtmann E, Hua X, Zeller G, Sunagawa S, Voigt AY, Hercog R, Goedert JJ, Shi J, Bork P, Sinha R (2016) Colorectal cancer and the human gut microbiome: reproducibility with whole-genome shotgun sequencing. PLoS One 11(5):e0155362. https://doi.org/10.1371/journal.pone.0155362
Fitzpatrick SG, Katz J (2010) The association between periodontal disease and cancer: a review of the literature. J Dentist 38(2):83–95. https://doi.org/10.1016/j.jdent.2009.10.007
Whitmore SE, Lamont RJ (2014) Oral bacteria and cancer. PLoS Pathogens 10(3):e1003933. https://doi.org/10.1371/journal.ppat.1003933
Atanasova KR, Yilmaz O (2014) Looking in the Porphyromonas gingivalis cabinet of curiosities: the microbium, the host and cancer association. Mol Oral Microbiol 29(2):55–66. https://doi.org/10.1111/omi.12047
Hajishengallis G (2015) Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol 15(1):30–44. https://doi.org/10.1038/nri3785
Harris JI, Russell RR, Curtis MA, Aduse-Opoku J, Taylor JJ (2002) Molecular mediators of Porphyromonas gingivalis-induced T-cell apoptosis. Oral Microbiol Immunol 17(4):224–230
Ahn J, Segers S, Hayes RB (2012) Periodontal disease, Porphyromonas gingivalis serum antibody levels and orodigestive cancer mortality. Carcinogenesis 33(5):1055–1058. https://doi.org/10.1093/carcin/bgs112
Michaud DS, Izard J, Wilhelm-Benartzi CS, You DH, Grote VA, Tjonneland A, Dahm CC, Overvad K, Jenab M, Fedirko V, Boutron-Ruault MC, Clavel-Chapelon F, Racine A, Kaaks R, Boeing H, Foerster J, Trichopoulou A, Lagiou P, Trichopoulos D, Sacerdote C, Sieri S, Palli D, Tumino R, Panico S, Siersema PD, Peeters PH, Lund E, Barricarte A, Huerta JM, Molina-Montes E, Dorronsoro M, Quiros JR, Duell EJ, Ye W, Sund M, Lindkvist B, Johansen D, Khaw KT, Wareham N, Travis RC, Vineis P, Bueno-de-Mesquita HB, Riboli E (2013) Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut 62(12):1764–1770. https://doi.org/10.1136/gutjnl-2012-303006
Yao L, Jermanus C, Barbetta B, Choi C, Verbeke P, Ojcius DM, Yilmaz O (2010) Porphyromonas gingivalis infection sequesters pro-apoptotic Bad through Akt in primary gingival epithelial cells. Mol Oral Microbiol 25(2):89–101. https://doi.org/10.1111/j.2041-1014.2010.00569.x
Yilmaz O, Sater AA, Yao L, Koutouzis T, Pettengill M, Ojcius DM (2010) ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cell Microbiol 12(2):188–198. https://doi.org/10.1111/j.1462-5822.2009.01390.x
Mao S, Park Y, Hasegawa Y, Tribble GD, James CE, Handfield M, Stavropoulos MF, Yilmaz O, Lamont RJ (2007) Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol 9(8):1997–2007. https://doi.org/10.1111/j.1462-5822.2007.00931.x
Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, Grangette C, Vasquez N, Pochart P, Trugnan G, Thomas G, Blottiere HM, Dore J, Marteau P, Seksik P, Langella P (2008) Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA 105(43):16731–16736. https://doi.org/10.1073/pnas.0804812105
Schippa S, Iebba V, Santangelo F, Gagliardi A, De Biase RV, Stamato A, Bertasi S, Lucarelli M, Conte MP, Quattrucci S (2013) Cystic fibrosis transmembrane conductance regulator (CFTR) allelic variants relate to shifts in faecal microbiota of cystic fibrosis patients. PLoS One 8(4):e61176. https://doi.org/10.1371/journal.pone.0061176
Zeng H, Wu H, Sloane V, Jones R, Yu Y, Lin P, Gewirtz AT, Neish AS (2006) Flagellin/TLR5 responses in epithelia reveal intertwined activation of inflammatory and apoptotic pathways. Am J Physiol Gastrointest Liver Physiol 290(1):G96–G108
Fröhlich EE, Mayerhofer R, Holzer P (2015) Reevaluating the hype: four bacterial metabolites under scrutiny. Eur J Microbiol Immunol 5(1):1–13
Yu HS, Oyama T, Isse T, Kitagawa K, Pham TT, Tanaka M, Kawamoto T (2010) Formation of acetaldehyde-derived DNA adducts due to alcohol exposure. Chem Biol Interact 188(3):367–375
Singh S, Arcaroli J, Thompson DC, Messersmith W, Vasiliou V (2015) Acetaldehyde and retinaldehyde-metabolizing enzymes in colon and pancreatic cancers. Adv Exp Med Biol 815:281–294
Patra KC, Hay N (2014) The pentose phosphate pathway and cancer. Trends Biochem Sci 39(8):347–354
Hagland HR, Søreide K (2015) Cellular metabolism in colorectal carcinogenesis: influence of lifestyle, gut microbiome and metabolic pathways. Cancer Lett 356:273–280
Suzuki TA, Worobey M (2014) Geographical variation of human gut microbial composition. Biol Lett 10(2):20131037. https://doi.org/10.1098/rsbl.2013.1037
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Caporaso JG, Lozupone CA, Lauber C, Clemente JC, Knights D, Knight R, Gordon JI (2012) Human gut microbiome viewed across age and geography. Nature 486(7402):222–227. https://doi.org/10.1038/nature11053
Acknowledgements
We are grateful to Prof. B. Benjelloun and Prof. A. Oussaden (Department of Surgery), Prof. M. Abkari and Prof. S. A. Ibrahimi (Department of Gastroenterology) and Dr. Imane Samri (Department of Molecular Genetics) from the University Hospital Hassan II of Fez for their support.
Funding
IA is a Fulbright scholar. This work was also supported by a Grant from the NIH for H3ABioNet/H3Africa to H.G (grant number U41HG006941).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No potential conflicts of interest were disclosed.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
. Predicted functional metagenome significantly over- or underrepresented in the colorectal cancer and control samples (DOCX 46 KB)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Allali, I., Boukhatem, N., Bouguenouch, L. et al. Gut microbiome of Moroccan colorectal cancer patients. Med Microbiol Immunol 207, 211–225 (2018). https://doi.org/10.1007/s00430-018-0542-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00430-018-0542-5