
Abstract
Purpose
To raise awareness of Stargardt disease (STGD1) patients without fundus abnormalities.
Methods
Medical records were evaluated for age at onset, initial symptoms and diagnosis, reason for delay of diagnosis, age at STGD1 diagnosis, best-corrected visual acuity (BCVA), ophthalmoscopy, fundus photography, fundus autofluorescence (FAF), fluorescein angiography (FA), spectral-domain optical coherence tomography (SD-OCT), full-field electroretinography (ffERG), color vision test, and the presence of ABCA4 variants.
Results
In 11.1% of our STGD1 cohort of 280 patients, no fundus abnormalities were observed at first ophthalmic consultation. The median age at onset was 8 years (range, 1–18). There was a median delay in diagnosis of 3 years (range, 0–19) in 27 out of 31 patients, which resulted in a median age at diagnosis of 12 years (range, 7–26). Patients were misdiagnosed with amblyopia, myopia, optic disk pathology, mental health problems, tension headache, neuritis bulbaris, and uveitis. Subtle abnormalities, such as lipofuscin accumulation, were seen on FAF at an earlier disease stage than in ophthalmoscopy. On SD-OCT, this included a thickened external limiting membrane. Color vision tests showed red-green insufficiency in 79% of patients. Reduced ERG amplitudes were only present in 26% (N = 8) and a dark choroid sign in 65% of the patients. Visual acuity considerably fluctuated in the first 5 years after onset. The majority of the patients (65%) carried a least one variant with a severe effect on ABCA4 function.
Conclusions
Childhood-onset STGD1 patients were diagnosed with a delay of median 3 years. The presence of accurate competence, equipment, and the possibility for genetic screening is required; therefore, we recommend to refer children with visual complaints without initial fundus abnormalities to a specialized ophthalmologic center. In particular, to diagnose patients at an early stage of disease is of increased importance with the advent of new therapeutic possibilities.
Similar content being viewed by others

Introduction
Stargardt disease (STGD1) is arguably the most common retinal dystrophy and affects 1:10000 people worldwide [1]. This autosomal recessive disease is caused by variants in the ABCA4 gene that encodes for a retinal-specific ATP-binding cassette transporter protein. Dysfunction of the ABCA4 protein leads to toxic accumulation of byproducts from the visual cycle in the photoreceptor cell and retina pigment epithelium (RPE), which eventually leads to irreversible damage of the outer retinal layers [2, 3].
Up to 5962 variants in the ABCA4 gene have been identified; the specific combinations of variants in conjunction with largely unknown modifying factors in each patient result in a highly heterogenic phenotype [4]. Patients with STGD1 present with progressive vision loss, which typically occurs in young adulthood, but early and late forms have been well recognized [5,6,7]. In general, the fundus picture is characterized by the presence of irregular yellow-white fundus flecks in the posterior pole. During the course of the disease, macular atrophy develops, sometimes with a “beaten bronze” aspect; in other patients, a bull’s eye pattern can be observed. Lipofuscin accumulates in the outer retinal layers, which results in a “dark choroid” on the fluorescein angiogram in approximately 80% of the patients [8,9,10,11]. In early forms with a disease onset ≤ 10 years of age, atrophy of the macula is a prominent and early feature; the yellow flecks may be absent or hardly notable [5, 12, 13]. The flecks are much more common in the classic form of STGD1 with an age of onset in the early teens, sometimes extending beyond the vascular arcades resulting in the fundus flavimaculatus phenotype [14, 15]. Late-onset forms of the disease are characterized by atrophy of the retinal pigment atrophy, subtle flecks, and foveal sparing [6, 16, 17].
The diagnosis of STGD1 can be challenging in early disease especially, as no apparent changes may be present on ophthalmoscopy despite the loss of visual function [5]. This lack of clinical signs in combination with the limited capabilities for expression in young children may delay the correct diagnosis. Not only is early identification of these patients essential for the emotional aspect of a timely diagnosis, it is also important in the light of emerging therapeutic options for STGD1 disease, such as gene augmentation (trial number NCT01367444, NCT01736592), stem cell therapy (trial number NCT01469832), and small molecule drugs (trial number NCT02402660).
In this study, we describe—in detail—the clinical and molecular genetic findings in a group of STGD1 patients, which presents without initial fundus abnormalities in ophthalmoscopy. We hope that a heightened awareness avoids misdiagnosis, such as functional visual loss in these children and, in worst case scenarios, years of inappropriate treatment.
Methods
Patients
The database with STGD1 patients of the Department of Ophthalmology, Radboud University Medical Center, (Nijmegen, the Netherlands) contains 280 patients of all ages and disease-onset, of whom one or more ABCA4 variants could be identified. We included 31 patients who did not show obvious fundus abnormalities at the first presentation. This study was approved by the Institutional Ethics Committee and was performed in accordance with the Declaration of Helsinki.
Clinical evaluation
We collected the clinical data from the medical records. These included age at onset, initial symptoms, initial diagnosis and examinations or therapy, age at STGD1 diagnosis, delay of diagnosis and reason for this delay, number of referrals before diagnosis, and general medical history. Age at onset was defined as the first manifestation of the disease, and these symptoms could have been noticed by not only the patient, but also their family members and/or the school physician. If visual complaints of the patient were initially diagnosed due to stress or need for attention, we used the term “mental health issues”.
The standard ophthalmic examination included best-corrected visual acuity (BCVA) using Early Treatment Diabetic Retinopathy Study (ETDRS) or Snellen charts, slit-lamp biomicroscopy, and detailed fundus examination. BCVA was transformed into the logarithm of the minimum angle of resolution (logMAR) for statistical analysis. For fundus photography, we used the Topcon TRC50IX (Topcon Corporation, Tokyo, Japan). Fluorescein angiography (FA) and cross-sectional images using spectral-domain optical coherence tomography (SD-OCT) centered at the macula were obtained with the Spectralis (HRA + OCT, Heidelberg Engineering, Heidelberg, Germany). Short-wave fundus autofluorescence imaging (FAF) (λ = 488 nm, emission 500–700 nm) was performed using a confocal scanning laser ophthalmoscope (Spectralis HRA + OCT or HRA2, Heidelberg Engineering, Heidelberg, Germany). The field of view was set at 30° × 30° or 55° × 55°, centered at the macula. For evaluation of color vision, we employed the Ishihara or Panel D-15 test. Full-field electroretinography (ERG) was performed using Dawson-Trick-Litzkow (DTL) electrodes and the RETI-port system (Roland Consults, Stasche & Finger GmbH, Brandenburg an der Havel, Germany). The recordings were performed in accordance with the guidelines of the International Society for Clinical Electrophysiology of Vision (ISCEV) [18]. We grouped ERG results as described by Lois et al. [19]: group 1—patients with normal ERG responses, group 2—patients with reduced photopic amplitudes (< 5% of normal range), and group 3—patients with reduced photopic and scotopic amplitudes (< 5% of normal range).
Genetic analyses
Genetic analysis of the ABCA4 gene was performed at the Department of Human Genetics at the Radboud University Medical Center using arrayed primer extension analysis (APEX, Asper Biotech, Tartu, Estonia). If the Asper microarray screening revealed only one ABCA4 variant, exon and intron-exon boundaries were sequenced in the ABCA4 gene to identify additional variants. All variants were confirmed with Sanger sequencing. The following variants were defined as severe: protein-truncating, canonical splice-site variants, and deletions spanning at least one exon.
Statistical analysis
We used SPSS version 22.0 (IBM Corp, Armonk, NY) for statistical data analysis, using descriptive statistics by median and range for continuous variables and percentages for categorical variables. We employed Kaplan-Meier estimators to analyze the interval between age at onset and age at which four different visual endpoints were reached. These four points were based on the classification of visual impairment of the World Health Organization: (near-)normal to mild visual impairment ≥ 0.2 logMAR (≤20/32 Snellen), moderate visual impairment ≥ 0.6 logMAR (≤ 20/80 Snellen), severe visual impairment ≥ 1.0 logMAR (≤ 20/200 Snellen), and blindness ≥ 1.4 logMAR (≤ 20/500 Snellen).
Results
Clinical characteristics
In 31 of 280 (11.1%) STGD1 patients, no obvious fundus abnormalities were observed at the first ophthalmic consultation. The group consisted of 15 males and 16 females with six siblings from three different families and 25 isolated cases. An overview of the clinical findings and the diagnostic process is given in Table 1. In one third of cases, symptoms of a decreased visual acuity were not noticed by the patient, but by the parents or a school physician. Age at onset occurred at a median age of 8 years (range, 1–18). In 87% of the patients, there was a delay in diagnosis: a median delay of 3 years (range, 0–19), which resulted in a median age at diagnosis of 12 years (range, 7–26). The main reason for delayed STGD1 diagnosis was misdiagnosis, in particular amblyopia treated with occlusion therapy (6 patients) and mental health issues (5 patients). The majority of patients (94%) visited more than two hospitals before the correct diagnosis was made. All patients were finally diagnosed with STGD1 in tertiary referral centers. The first fundus abnormalities were observed at a median time of 3 years (range, 0.5–16) after first symptoms. These included central retinal pigment epithelium (RPE) alterations (43%), bull’s eye maculopathy (33%), and/or parafoveal flecks (24%). Once these features had been observed, STGD1 was generally diagnosed relatively quickly in the majority of patients (median, 0.7 years, range, 0.1–3).
We could retrieve the BCVA at the first ophthalmic visit in 21 out of 31 patients; the median BCVA at that time was 20/32 Snellen (20/20–20/400). The median interval and 95% confidence interval (CI) between the age at onset and decline in BCVA to mild, moderate, and severe visual impairment and blindness was 1 year (95% CI, 0.0–2.25), 4 years (95% CI, 3.1–4.9), and 12 years (CI 95% 7.8–16.2). One patient reached blindness 34 years after the first symptoms of onset at age 9. In many patients, the visual acuity findings were quite variable early in the course of the disease as shown in Fig. 1.

Course of the best-corrected visual acuity (BCVA) in logMAR in ten patients. The visual acuity varies greatly during the first 5 years after first ophthalmic consultation. LogMAR 0 = 20/20 Snellen, LogMAR 0.5 = 20/63 Snellen, LogMAR 1.00 = 20/200 Snellen, LogMAR 1.50 ≈ 20/630 Snellen, LogMAR 2.00 = 20/2000 Snellen. Each patient (ID = identification) is shown in a different color
In 29 out of 31 patients, fundus flecks were eventually noticed at a median time of 3.5 years (range, 0.1–16.5) after the initial ophthalmic consultation. In 17 patients (59%), subtle parafoveal flecks could be seen; in 5 patients (17%), flecks were noticed within the vascular arcades; and in 7 patients (24%), flecks extended to the periphery. In 2 patients, no fundus flecks were reported at any time during the course of the disease (follow-up time, 1 and 10 years).
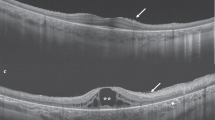
In 23 of 31 patients, the first SD-OCT was performed at 9 years (range, 0.1–24) after disease onset. No SD-OCT scans were performed in the remaining 8 patients. All OCT scans showed abnormalities by disorganized or absent RPE. A thickened external limiting membrane (ELM) was seen in 2 patients (0.5 and 1 year after disease onset). A dark choroid was observed in 15 of 23 (65%) patients in whom FA was performed. In 22 patients, the first FAF was performed 3.5 years (0.5–24) after onset. No atrophy was seen (median, 1 year after onset) in 5 patients, (peri)foveal atrophy in 9 (median, 4 years after onset), atrophy within the vascular arcades in 7 (median, 15 years after onset), and panretinal atrophy (15 years after onset) in 1. In 4 patients, abnormalities were seen on FAF, but were missed on ophthalmoscopy.
The first ERG was performed 2 years (0.1–27) after onset in 27 patients. Normal ERG recordings (group 1) were present in 20 patients (74%), 2 years (0.1–21) after disease onset. We found group 2 ERGs in 4 patients (15%) with 1 year (0.1–3.5) after onset, and group 3 recordings in 3 patients (11%) with 18 years (6–27) after onset. Follow-up for ERG recordings was available in 21 patients. In 4 of these patients progressed from group 1 to group 2 (median time, 8 (7–16) years), 2 patients progressed from group 2 to group 3 (within 2 and 3 years), and 1 patient progressed from group 1 to group 3 in 6 years. In 17 patients, color vision was tested. In 15 patients (79%), abnormalities were noticed, red-green defects in 14 patients, and a blue-yellow defect in 1 patient.
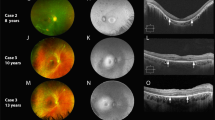
Various imaging modalities of patient 28 and patient 21 are depicted respectively in Figs. 2 and 3.

Three-and-a-half year follow-up in patient ID28. Age at onset at 6.5 years, 1 year later the fundus auto-fluorescence showed a perifoveal ring of hyper-fluorescence (A2), and on OCT, a discrete thickening of the external limiting membrane can be seen (A3, red arrow). Only 6 months later, a subtle hyper-fluorescent perifoveal lesion developed as shown in B2, red arrow. Over time, the hyper-fluorescent flecks become more visible on autofluorescence imaging. In addition, the flecks became noticeable on color fundus photographs. The thickened external limiting membrane remained present during the entire follow-up time (red arrows A3, C3, D3, E3, F3, H3)

Multimodal imaging in ID21 at age 25, 15 years after the first symptoms. The visual acuity is now 20/1000 Snellen OU. Color fundus photography (a) shows attenuated retinal vessels (especially the veins), para-arteriolar pigmentation, and diffuse chorioretinal atrophy. The fundus autofluorescence (b) shows widespread hypofluorescence, especially at the macula, indicative of RPE cell loss. The fluorescence angiogram (c) shows widespread granular hyperfluorescent lesions as result of RPE damage, and on the OCT (d), loss of the outer retinal layers, as well as the choriocapillaris, can readily be observed
Genetic characteristics
Overall, genetic analysis was not performed at first visit, but with a median delay of 5 years (range, 0–30) after the first visit. An overview of ABCA4 variants in our cohort is described in Table 2.
Variants in the ABCA4 gene were identified in 59 of 62 alleles (95%). Three variants were found in one patient, two ABCA4 variants in 27, and one variant in three. In total, 32 distinct variants were detected. The majority of the patients (65%) carried at least one variant with a severe effect on ABCA4 function (Table 2, in italics).
Discussion
A lack of obvious fundus abnormalities left a high number of children with STGD1 disease without the correct diagnosis. Instead, these patients underwent unnecessary investigations, such as psychic evaluations, brain MRIs or CTs, and lumbar punctures. Many of these children were treated for mental illness and/or amblyopia with pointless and possibly harmful treatments, including years of occlusion therapy.
In patients with adult-onset STGD1, initial ophthalmoscopic features typically include yellow-white flecks and central macular atrophy, and cases without fundus abnormalities have not been described [6]. In young children, the clinical presentation can be confusing for the general ophthalmologist. Fujinami et al. [7] noticed that one third of their child cohort (< 17 years) initially had a normal fundus appearance. Lambertus et al. [5] described a cohort of 41 STGD1 patients younger than 10 years, and 10 of these patients (24%) also did not have fundus abnormalities. These studies show that the absence of readily observable fundus abnormalities in young STGD1 patients is not an isolated finding, but a relative common part of the clinical spectrum.
Recently, Khan et al. reported the earliest features of ABCA4-associated retinopathy [20]. In their study, they included eight children (six prospectively) to describe their ophthalmologic features. At the first visit, four children were asymptomatic and four had complaints of visual decline. In six out of eight children, the central macula appeared normal or had an altered foveal reflex. This is similar to the findings in our study. In contrast, their study focused on the (most prospectively) appearance of abnormalities in ABCA4-related dystrophy even if they were asymptomatic. Our study is focused, retrospectively, on STGD1 patients initially without fundus abnormalities, despite having visual complaints. Besides the natural course of this phenotype, we describe the reasons of delay in diagnoses and to find similarities within this group of patients in order to provide recommendations for ophthalmologists in general. Therefore, this study is a contribution from another point of view of children with STGD1 which initially present without fundus abnormalities.
In our cohort, when fundus abnormalities did occur, these were often not the typical yellowish flecks but rather RPE alterations, often in a bull’s eye pattern. A hypothesis for the absence of typical fundus flecks may lie in the relative high pathogenicity of ABCA4 variants. As the majority of our cohort (65%) carried at least one severe variant, there may be little ABCA4 function left in these patients, leading to a very early manifestation of the disease. The buildup of toxic A2E in RPE cells develops rapidly, thereby causing early cell death without the opportunity for lipofuscin to accumulate and subsequent fleck formation.
Blocking of the choroidal vessels on FA resulting a dark or silent choroid is frequently used as a diagnostic marker for STGD1. The prevalence of this FA finding in STGD1 patients has been described in up to 86% of patients [21]. A dark choroid was present in 65% in our relatively small cohort. A correlation has been described between the presence of yellow-white fundus flecks and the appearance of dark choroid [9, 22], which might account for the relatively low percentage of patients with a dark choroid in this cohort. FAF imaging is a relatively new modality that may be used to identify early and subtle lipofuscin [23]. In addition, a thickened ELM on OCT may also serve as an early marker for STGD1 [24]. However, in the study of Lee et al., this distinct ELM thickening was described to occur in all (26/26) cases. In our cohort, only 2/23 cases with performed OCT (0.5 and 1 year after disease onset) were observed to have this feature. The delay of performing an OCT (mean, 9 years after disease onset), could be an explanation of the difference in the appearance of ELM thickening in both cohorts. Abnormal color vision was observed in 79% of our STGD1 patients, which corresponds with the previously reported percentages [25].
Fluctuation of visual acuity in 10/31 children in the first 5 years of ophthalmic consultation suggests that visual acuity is an ambiguous symptom at early stages of STGD1. This could be explained by lowered reading performance due to ring scotomas that allow for good visual acuity, but cause visual field impairment. [26]
Although STGD1 is the most common juvenile macular dystrophy, it remains a relatively rare disorder. Clinicians in a general ophthalmic practice may lack the experience to identify and interpret the subtle abnormalities in these young children. When we take into account the relative difficulty associated with the ophthalmic examination of (very) young children, the high number of misdiagnoses in the early-onset STGD1 patient group may not come as a surprise. Therefore, we want to make ophthalmologists aware of early findings in STGD1, especially appearing in children. First, visual acuity often fluctuates in these patients which should not automatically rule out the possibility of a photoreceptor disease. Second, a fundus photograph can be helpful in discerning very subtle fundus abnormalities and may be useful in follow-up. Third, non-invasive investigations such as OCT (thickening of the ELM), FAF (subtle lipofuscin accumulation), and color vision tests may help in the diagnostic process. FA is invasive, apart from the oral variant, but may also be less helpful in patients without fundus abnormalities in the light of the relatively low percentage of dark choroids in these patients.
In children with visual disturbances, retinal dystrophies should be considered and ruled out when possible, even in the absence of fundus abnormalities on ophthalmoscopy, because STGD1 may be a cause of low vision in normal fundus. Instead of wait and see, we would recommend referring these children to a tertiary ophthalmic center and performing OCT and FAF to define early findings of STGD1. In addition, early identification may prove important in the light of emerging therapeutic options.
References
Blacharski P (1988) Retinal dystrophies and degenerations. Newsome DA (ed): 135–159
Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA, Bernstein PS, Peiffer A, Zabriskie NA, Li Y, Hutchinson A, Dean M, Lupski JR, Leppert M (1997) Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science (New York, NY) 277: 1805–1807
Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, Rattner A, Smallwood P, Li Y, Anderson KL, Lewis RA, Nathans J, Leppert M, Dean M, Lupski JR (1997) A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 15:236–246. https://doi.org/10.1038/ng0397-236
Cornelis SS, Bax NM, Zernant J, Allikmets R, Fritsche LG, den Dunnen JT, Ajmal M, Hoyng CB, Cremers FP (2017) In silico functional meta-analysis of 5,962 ABCA4 variants in 3,928 retinal dystrophy cases. Hum Mutat 38:400–408. https://doi.org/10.1002/humu.23165
Lambertus S, van Huet RA, Bax NM, Hoefsloot LH, Cremers FP, Boon CJ, Klevering BJ, Hoyng CB (2015) Early-onset Stargardt disease: phenotypic and genotypic characteristics. Ophthalmology 122:335–344. https://doi.org/10.1016/j.ophtha.2014.08.032
Westeneng-van Haaften SC, Boon CJ, Cremers FP, Hoefsloot LH, den Hollander AI, Hoyng CB (2012) Clinical and genetic characteristics of late-onset Stargardt’s disease. Ophthalmology 119:1199–1210. https://doi.org/10.1016/j.ophtha.2012.01.005
Fujinami K, Zernant J, Chana RK, Wright GA, Tsunoda K, Ozawa Y, Tsubota K, Robson AG, Holder GE, Allikmets R, Michaelides M, Moore AT (2015) Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology 122:326–334. https://doi.org/10.1016/j.ophtha.2014.08.012
Klevering BJ, Deutman AF, Maugeri A, Cremers FP, Hoyng CB (2005) The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefes Arch Clin Exp Ophthalmol 243(2):90–100
Fishman GA, Stone EM, Grover S, Derlacki DJ, Haines HL, Hockey RR (1999) Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch Ophthalmol 117:504–510
Armstrong JD, Meyer D, Xu S, Elfervig JL (1998) Long-term follow-up of Stargardt's disease and fundus flavimaculatus. Ophthalmology 105:448–457; discussion 457-448. https://doi.org/10.1016/S0161-6420(98)93026-3
Querques G, Leveziel N, Benhamou N, Voigt M, Soubrane G, Souied EH (2006) Analysis of retinal flecks in fundus flavimaculatus using optical coherence tomography. Br J Ophthalmol 90:1157–1162. https://doi.org/10.1136/bjo.2006.094136
Klevering BJ, Blankenagel A, Maugeri A, Cremers FP, Hoyng CB, Rohrschneider K (2002) Phenotypic spectrum of autosomal recessive cone-rod dystrophies caused by mutations in the ABCA4 (ABCR) gene. Invest Ophthalmol Vis Sci 43:1980–1985
Michaelides M, Chen LL, Brantley MA Jr, Andorf JL, Isaak EM, Jenkins SA, Holder GE, Bird AC, Stone EM, Webster AR (2007) ABCA4 mutations and discordant ABCA4 alleles in patients and siblings with bull's-eye maculopathy. Br J Ophthalmol 91:1650–1655. https://doi.org/10.1136/bjo.2007.118356
Stargardt K (1909) Über familliäre, progressive Degeneration in der Maculagegend des Auges. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie:534–550
Franceschetti A (1965) A special form of tapetoretinal degeneration: fundus flavimaculatus. Transactions - American Academy of Ophthalmology and Otolaryngology American Academy of Ophthalmology and. Otolaryngology 69:1048–1053
van Huet RA, Bax NM, Westeneng-Van Haaften SC, Muhamad M, Zonneveld-Vrieling MN, Hoefsloot LH, Cremers FP, Boon CJ, Klevering BJ, Hoyng CB (2014) Foveal sparing in Stargardt disease. Invest Ophthalmol Vis Sci 55:7467–7478. https://doi.org/10.1167/iovs.13-13825
Fujinami K, Sergouniotis PI, Davidson AE, Wright G, Chana RK, Tsunoda K, Tsubota K, Egan CA, Robson AG, Moore AT, Holder GE, Michaelides M, Webster AR (2013) Clinical and molecular analysis of Stargardt disease with preserved foveal structure and function. Am J Ophthalmol 156(487–501):e481. https://doi.org/10.1016/j.ajo.2013.05.003
Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M, International Society for Clinical Electrophysiology of V (2009) ISCEV standard for full-field clinical electroretinography (2008 update). Documenta ophthalmologica Advances in ophthalmology 118:69–77. https://doi.org/10.1007/s10633-008-9155-4
Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC (2001) Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol 119:359–369
Khan KN, Kasilian M, Mahroo OAR, Tanna P, Kalitzeos A, Robson AG, Tsunoda K, Iwata T, Moore AT, Fujinami K, Michaelides M (2018) Early patterns of macular degeneration in ABCA4-associated retinopathy. Ophthalmology 125:735–746. https://doi.org/10.1016/j.ophtha.2017.11.020
Fishman GA, Farber M, Patel BS, Derlacki DJ (1987) Visual acuity loss in patients with Stargardt’s macular dystrophy. Ophthalmology 94:809–814
Gelisken O, De Laey JJ (1985) A clinical review of Stargardt’s disease and/or fundus flavimaculatus with follow-up. Int Ophthalmol 8:225–235
Chun R, Fishman GA, Collison FT, Stone EM, Zernant J, Allikmets R (2014) The value of retinal imaging with infrared scanning laser ophthalmoscopy in patients with Stargardt disease. Retina 34:1391–1399. https://doi.org/10.1097/IAE.0000000000000070
Lee W, Noupuu K, Oll M, Duncker T, Burke T, Zernant J, Bearelly S, Tsang SH, Sparrow JR, Allikmets R (2014) The external limiting membrane in early-onset Stargardt disease. Invest Ophthalmol Vis Sci 55:6139–6149. https://doi.org/10.1167/iovs.14-15126
Vandenbroucke T, Buyl R, De Zaeytijd J, Bauwens M, Uvijls A, De Baere E, Leroy BP (2015) Colour vision in Stargardt disease. Ophthalmic Res 54:181–194. https://doi.org/10.1159/000438906
Messias A, Reinhard J, Velasco e Cruz AA, Dietz K, MacKeben M, Trauzettel-Klosinski S (2007) Eccentric fixation in Stargardt’s disease assessed by Tubingen perimetry. Invest Ophthalmol Vis Sci 48:5815–5822. https://doi.org/10.1167/iovs.06-0367
Funding
This study was supported by the Stichting A.F. Deutman Oogheelkunde Researchfonds, Nijmegen, The Netherlands; the Nederlandse Oogonderzoek Stichting (NOS), Nijmegen, the Netherlands; the Stichting MaculaFonds, the Netherlands; and UitZicht projects (2013-25 and 2014-3) that were financed by Oogfonds, landelijke Stichting Blinden en Slechtzienden, and Maculafonds. The funding organizations had no role in the design or conduct of this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bax, N.M., Lambertus, S., Cremers, F.P.M. et al. The absence of fundus abnormalities in Stargardt disease. Graefes Arch Clin Exp Ophthalmol 257, 1147–1157 (2019). https://doi.org/10.1007/s00417-019-04280-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-019-04280-8