
Abstract
Purpose
Nicorandil is a hybrid between nitrates and KATP channel opener activators. The aim of this study was to evaluate the nicorandil’s effects on ischemia–reperfusion (IR) lung injury and examine the mechanism of its effects.
Methods
Isolated rat lungs were divided into 6 groups. In the sham group, the lungs were perfused and ventilated for 150 min. In the IR group, after perfusion and ventilation for 30 min, they were interrupted (ischemia) for 60 min, and then resumed for 60 min. In the nicorandil (N) + IR group, nicorandil 6 mg was added before ischemia (nicorandil concentration was 75 µg ml−1). In the glibenclamide + N + IR group, the L-NAME (Nω-Nitro-l-arginine methyl ester) + N + IR group and ODQ (1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one) + N + IR group, glibenclamide 3 µM, L-NAME 100 µM, and ODQ 30 µM were added 5 min before nicorandil administration, respectively. We measured the coefficient of filtration (Kfc) of the lungs, total pulmonary vascular resistance, and the wet-to-dry lung weight ratio (WW/DW ratio).
Results
Kfc was significantly increased after 60 min reperfusion compared with baseline in the IR group, but no change in the sham group. An increase in Kfc was inhibited in the N + IR group compared with the IR group (0.92 ± 0.28 vs. 2.82 ± 0.68 ml min−1 mmHg−1 100 g−1; P < 0.01). Also, nicorandil attenuated WW/DW ratio was compared with IR group (8.3 ± 0.41 vs. 10.9 ± 2.5; P < 0.05). Nicorandil’s inhibitory effect was blocked by glibenclamide and ODQ (P < 0.01), but not by L-NAME.
Conclusions
Nicorandil attenuated IR injury in isolated rat lungs. This protective effect appears to involve its activation as KATP channel opener as well as that of the sGC-cGMP pathway.
Similar content being viewed by others


Introduction
Ischemia–reperfusion (IR) injury in patients undergoing lung transplant surgery remains a problematic and unresolved issue. IR injury is known to cause primary graft dysfunction, also known as primary graft failure, and to be associated with increased short- and long-term mortality after lung transplantation [1, 2]. However, the underlying mechanisms, especially how and where endothelial cells are impaired during and after IR lung injury, remain unclear.
Nicorandil is a hybrid compound with nitrate and KATP channel opener properties [3] that has shown protective effects against coronary events in patients with stable angina [4]. Although the protective effects of nicorandil on IR injury may be because of its KATP channel opener properties [5], its effects on microvascular permeability both during and after IR lung injury have yet to be evaluated. In addition, nicorandil’s nitrate properties have yet to be investigated in patients with IR lung injury.
Therefore, the objective of the present study was to investigate whether nicorandil reduces the risk of IR injury in an isolated buffer perfused rat lung model, and whether nicorandil’s beneficial effects are a result of its KATP channel opener properties and nitric oxide production, or are attributable to activation of the soluble guanylyl cyclase (sGC)/cyclic guanosine monophosphate (cGMP) pathway.
Materials and Methods
The present experimental protocol was approved by the Animal Experiment Committee of the Akita University Graduate School of Medicine in Akita, Japan, and was performed in accordance with the relevant aspects of the ARRVE guidelines. First, male Sprague–Dawley rats ranging in weight from 300–350 g were anesthetized intraperitoneally with sodium pentobarbital (30 mg/kg). Following local anesthesia with 1.0% lidocaine, tracheostomy and sternotomy were performed, and the lungs were mechanically ventilated using a ventilator (Ugo Basile Muromachi Kikai Co., Ltd., Tokyo, Japan) with room air at a rate of 50 breaths/min, a tidal volume of 2.5 ml, and a positive end-expiratory pressure of 2 cmH2O. Following a median sternotomy, heparin (200 U) was injected into the right ventricle and allowed to circulate for approximately 3 min. Next, the pulmonary artery (PA) was cannulated through the right ventricle, and the snare around the PA was tightened to isolate the lung inflow. Similarly, the left atrium (LA) was cannulated though the left ventricle and tightened. Next, the lungs were ventilated with the same gas mixture of 21% O2, 5% CO2, and balanced nitrogen, and then perfused with a bicarbonate-buffered physiological salt solution containing 119 mM NaCl, 4.7 mM KCl, 1.17 mM MgSO4, 22.6 mM NaHCO3, 1.18 mM KH2PO4, and 3.2 mM CaCl2. We then added 100 mg dextrose, 20 mU insulin, and 5 g bovine serum albumin (Sigma-Aldrich, Co., St. Louis, MO, USA) to each 100 ml of this stock solution. The perfusate pH was adjusted to 7.35–7.45 through the addition of sodium bicarbonate. Next, to monitor weight changes, the heart and lungs were removed en bloc and placed on an electronic balance (GX-400; A and D, Tokyo, Japan) in a humidified chamber with a set temperature of 37 ºC (Fig. 1). We then discarded the first 50 ml of perfusate, which contained large amounts of residual blood cells and plasma, and used 80 ml of perfusate for recirculation.

Schematic presentation of the experimental setup. Arterial and venous reservoirs are elevated simultaneously by 6 cmH2O during measurement of the coefficient of filtration (Kfc). The pump continuously circles the perfusate from the venous reservoir to the arterial reservoir to maintain a constant driving pressure. AD analog-to-digital, LA left atrium, PA pulmonary artery, Paw airway pressure, Ppa pulmonary arterial pressure, Ppv pulmonary venous pressure
We monitored and recorded pulmonary arterial pressure (Ppa) and pulmonary venous pressure (Ppv) continuously using calibrated pressure transducers (Baxter, IL, USA) and a multichannel recorder (RM 6000; Nihon Kohden, Tokyo, Japan), respectively. Next, a flow probe (FF-030T; Nihon Kohden) connected to an electromagnetic flowmeter (MFV2100; Nihon Kohden) was placed in the perfusion circuit for continuous monitoring of blood flow (Q). Ppa was adjusted by maintaining the flow at 0.04 ml/g body weight/min through the adjustment of the arterial reservoir level at the beginning of the experiment. Consequently, the driving pressure was kept constant throughout the experiment. The zero level for pressures was set at the position of the right atrium. Total pulmonary vascular resistance (PVR) (Rt) was calculated using the following formula:
Measurement of Microvascular Permeability
The change in lung weight induced by the elevation of venous pressure was used to determine the index of microvascular permeability to water (Kfc). The measurement of Kfc in isolated lungs has been described in detail [6, 7]. During ventilation and lung perfusion, the venous and arterial reservoirs were rapidly elevated by 6 cmH2O for 7 min, and the gain in lung weight, which consisted of two phases—rapid weight gain owing to changes in blood volume, and a slower, more prolonged phase of weight gain owing to transcapillary filtration—was recorded. Next, the logarithm of the rate of lung weight gain (∆W/∆t) was plotted as a function of time. The ∆W/∆t was then analyzed during a 3- to 6-min interval of elevated reservoirs. The initial rate of fluid filtration [(∆W/∆t)t = 0] was calculated by extrapolating ∆W/∆t at time 0. Finally, we calculated Kfc by dividing ∆W/∆t at time 0 [(∆W/∆t)t = 0] by changes in pulmonary capillary pressure (Ppc), and normalized using the baseline lung wet weight (WW) and expressed in ml min−1 mmHg−1 100 g−1 lung tissue.
To determine Ppc, the arterial and venous lines were simultaneously occluded for more than 5 s, and pulmonary arterial and venous pressures were converged to a specific level. Change in Ppc was calculated as the difference between that measured before versus at 7 min after elevation of the reservoir level. Although ventilation was discontinued during Ppc measurement, a constant flow of mixed gas (2 l/min) was administered at 2 cmH2O of airway pressure. At the end of the experiment, baseline lung WW was estimated by subtracting the weight of extrapulmonary tissues from the total weight of the lungs and extrapulmonary tissues, which was measured before perfusion.
Wet Weight to Dry Weight (WW/DW) Ratio
The lungs were used to estimate the tissue wet weight to dry weight (WW/DW) ratio. After recording the WW of the lung tissue, the lungs were placed in a drying oven at 60 ºC for 2 weeks and reweighed [6]. The lung WW/DW ratio was calculated using the following formula: (WW – DW)/DW [6].
Experimental Protocol
After the start of perfusion, the isolated perfused rat lungs were observed for 30 min to establish an isogravimetric state (Fig. 2). The lung preparations were then classified into six experimental groups. In the sham group (n = 6), the lungs were continuously perfused and ventilated for 150 min without IR, whereas in the IR group (n = 6), ventilation and perfusion were interrupted (ischemia) after equilibration and the lungs were maintained in the humidified chamber for 60 min, while the airway was kept open. The lungs were reperfused and reventilated for 60 min after ischemia. Meanwhile, in the nicorandil (N) + IR group (n = 6), nicorandil (6 mg) was added to the perfusate (nicorandil concentration in the perfusate was 75 µg/ml) followed by ischemia for 60 min; the preparation was then perfused for 60 min. In the glibenclamide (Gli) + N + IR group (n = 6), glibenclamide (3 µM) was added to the perfusate 5 min before nicorandil administration, followed by ischemia for 60 min; the preparation was then perfused for 60 min. In the Nω-Nitro-l-arginine methyl ester (L-NAME) + N + IR (n = 6) and 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) + N + IR (n = 6) groups, a similar protocol was used, except that glibenclamide was replaced with L-NAME (100 µM), an inhibitor of endogenous NO synthesis, and ODQ (30 µM), a sGC inhibitor.

Experimental protocol. M1, baseline measurement microvascular permeability to water (Kfc); M2, Kfc measurement 30 min after reperfusion; M3, Kfc measurement 60 min after reperfusion. Ischemia indicates no ventilation without perfusion, while reperfusion restoration of ventilation and perfusion
Statistical Analysis
Values are expressed as means ± standard deviation. We did not calculate a priori statistical power. The sample size was chosen based on related studies with a similar design [6, 8]. Within-group differences were analyzed using one-way repeated measures analysis of variance (ANOVA) (Statview 5.0; Abacus Concepts, Berkeley, CA, USA). Multiple samples of the same time interval were analyzed using one-way ANOVA, followed by a Bonferroni–Dunn test for post hoc comparisons. P values less than 0.05 were considered statistically significant.
Results
We used 11, 12, 11, 10, 10, and 12 rats in the sham, IR, N + IR, Gli + N + IR, L-NAME + N + IR, and ODQ + IR groups to obtain 6 samples in each group resulting from the failure of surgical technique or PA/LA tube positioning.
Perfusate Gas Analysis
No significant differences were observed in pH, PCO2, PO2 or base excess of perfusate just before ischemia between any groups (Table 1).
Changes in Total Pulmonary Vascular Resistance (PVR)
Changes in PVR tended to increase 60 min after IR in all groups compared with baseline; however, none of these differences was significant (Table 2). In addition, no significant differences were found between any of the groups at baseline or 60 min after IR.
Changes in Microvascular Permeability
Baseline Kfc values were similar in all groups. In the IR group, the Kfc after 60 min of reperfusion was significantly increased compared with baseline (0.81 ± 0.25 vs. 2.82 ± 0.68 ml min−1 mmHg−1 100 g−1, P < 0.01), whereas no changes were seen in Kfc in the sham group (0.87 ± 0.25 vs. 1.35 ± 0.37 ml min−1 mmHg−1 100 g−1, no significant difference). Compared with the IR group, administration of nicorandil significantly attenuated the increase of IR-induced Kfc after 60 min of reperfusion (0.92 ± 0.28 vs. 2.82 ± 0.68 ml min−1 mmHg−1 100 g−1, P < 0.01). Nicorandil’s inhibitory effect on the increase of IR-induced Kfc was blocked by glibenclamide (0.85 ± 0.26 vs. 2.81 ± 0.75 ml min−1 mmHg−1 100 g−1; P < 0.01) and ODQ (0.76 ± 0.77 vs. 2.43 ± 0.89 ml min−1 mmHg−1 100 g−1; P < 0.01), but not by L-NAME (1.02 ± 0.28 vs. 1.07 ± 0.32 ml min−1 mmHg−1 100 g−1; no significant difference) (Fig. 3).

Changes in the coefficient of filtration (Kfc). Data are mean ± SD (n = 6 per group). Open squares = IR group; closed squares = N + IR group; open triangles = Glb + N + IR group; closed triangles = ODQ + N + IR group; open circles = L-NAME + N + IR group; closed circles = sham group. *P < 0.01 versus baseline. #P < 0.01 versus IR group at the end of reperfusion
Changes in the Lung WW/DW Ratio
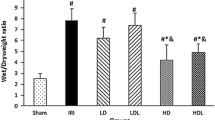
In the IR group, the lung WW/DW ratio increased after IR. By contrast, the lung WW/DW ratio remained unchanged after IR in the N + IR and L-NAME + N + IR groups, similar to the sham group. However, compared with the N + IR group, significant increases in the lung WW/DW ratio were observed in the Gli + N + IR and ODQ + N + IR groups (Fig. 4).

Changes in lung wet-to-dry weight ratio. Data are mean ± SD (n = 6 per group). *P < 0.01 versus sham group. #P < 0.01 versus IR group
Discussion
The major findings of the present study are as follows: (1) nicorandil administration before ischemia ameliorated increases in pulmonary microvascular permeability after IR in isolated rat lung preparations; (2) the protective effects of nicorandil against IR lung injury was blocked by glibenclamide as a KATP channel blocker and ODQ as a sGC inhibitor. These findings indicate that the salutary effects of nicorandil involve at least two mechanisms, the effect of the KATP channel opener and that of the activation of the sGC-cGMP pathway.
No significant differences were found in PVR between groups at baseline or at 30 and 60 min after IR. According to the experimental findings by Liu et al. [6], isoflurane–sevoflurane administration before ischemia attenuated IR injury without a significant PVR change in isolated rat lungs which was similar to the results in the present study. Meanwhile, functional vascular impairment from exposure to IR in the rat lung is likely to be restricted to the endothelial cell layer by the inhibition of the pulmonary vasodilator response to endothelium-dependent pulmonary vasodilators such as histamine and acetylcholine [9]. However, IR has been shown not to affect the response to sodium nitroprusside, an endothelium-independent pulmonary vasodilator [9]. These findings suggest that functional pulmonary vascular injury stemming from exposure to IR seems is restricted to the endothelial cell layer.
In the present study, nicorandil attenuated the increase in microvascular permeability after IR in rat lungs, and that effect was blocked by glibenclamide. Because glibenclamide, a KATP channel blocker, did not affect the increase in microvascular permeability produced by IR in rat lungs [8] when administered without nicorandil, the activation of KATP channels by nicorandil is likely to have attenuated the increase in pulmonary microvascular permeability after IR. The activation of KATP channels in the lung by administration of cromakalim could help protect against and even reverse the endothelial damage associated with reperfusion injury after ischemia [8]. The protective effect of a KATP channel opener appears to be due to blockade of superoxide anion production by leukocytes or endothelial cells because the opening of the KATP channel inhibited oxygen-derived free radical production by neutrophils [10].
In addition, our results showed that administration of ODQ as a sGC inhibitor before ischemia blocked the salutary effect of nicorandil. Since ODQ has been demonstrated to exert no effect on Kfc with and without IR in the rat lung [11], nicorandil also seems to exert salutary effects on IR lung injury through increasing intracellular cGMP by activating sGC as another mechanism [3]. Based on these considerations, the beneficial effects of nicorandil could be due the activation of the sGC-cGMP pathway. Indeed, nicorandil, because of its chemical structure, is considered a nitrate generator and a NO donor. Since sGC is a NO receptor, several reports have shown that NO donors reduced IR injury through activating sGC [12, 13]. During IR injury, endogenous NO and cGMP levels in tissues or organs were rapidly reduced [14]. In fact, the administration of NO to non-heart beating rat lungs during warm IR was associated with reduced IR injury and increased cGMP levels [13]. IR-induced pulmonary microvascular leak, which requires the activation of sGC, was also attenuated by inhaled NO [11]. Therefore, in the present study, the administration of nicorandil may have increased cGMP levels by the activation of sGC.
Another study reported that nicorandil elevated cGMP levels, but not NO generation, which was confirmed by ozone chemiluminescence reactions using human or rat liver microsomes (P450-rich fractions) with the addition of NADPH [15]. However, elevated cGMP levels in examined tissues were not inhibited by the NO trapping agent, carboxy-2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide [16]. In the present study, it is possible that the administration of nicorandil increased cGMP due to the direct activation of sGC.
The administration of L-NAME before ischemia did not attenuate the protective effect of nicorandil. This result may indicate that enhanced endothelial-derived NO activity by nicorandil does not account for the increase in pulmonary permeability under IR lung injury. The higher antiplatelet aggregation activity of endothelial cells caused by nicorandil was reduced when endothelial cells were treated with L-NAME and exposed to a hypoxia-reoxygenation condition [17], suggesting that the anti-aggregation activity of endothelial NO was enhanced by nicorandil. Considered together with the current results, the enhancing effect of nicorandil in endothelial-derived NO activity on IR injury may be involved only when blood cell components exist.
The findings of the present study may be transferrable not only to lung transplantation but also to thoracic surgery with long-term cardiopulmonary bypass or lung injury with ischemia–reperfusion. However, blood flow in the bronchial arteries is often maintained during cardiopulmonary bypass unlike lung transplantation.
This study did have some limitations. First, we used bicarbonate-buffered physiologic salt solution instead of blood as a perfusate. Blood components, especially neutrophils, play an important rule on IR injury. The interaction between neutrophils and endothelial cells releases various proteases and causes cell injury. To exclude these effects and examine nicorandil’s effects on endothelial cells, we used bicarbonate-buffered physiologic salt solution. Second, we did not measure NO generation. Therefore, whether NO generation was involved in the mechanism of nicorandil’s protective effect in our present study remains unclear. However, using ozone chemiluminescence detection methods, nicorandil has been shown not to generate NO [15]. Hence, the administration of nicorandil is likely to increase cGMP because of the direct activation of sGC. Third, the dose of nicorandil used in the present study was higher than the clinical dosage. Yamashita et al. used about tenfold of the clinical nicorandil dose and demonstrated that nicorandil ameliorated the IR injury of a lung allograft in a canine lung transplantation model [5]. In addition, Furuya et al. [17]. demonstrated that 300 µM of nicorandil improved post-ischemic cardiac function in a rat heart–lung reperfusion model [16]. It is, therefore, possible that rats are less sensible than humans to KATP channels.
In summary, nicorandil attenuated IR injury in isolated rat lungs. This protective effect was blocked by glibenclamide, a KATP channel blocker, and ODQ, a sGC inhibitor, but not by L-NAME. These results suggest that the protective effects of nicorandil after IR lung injury can be explained by the activation of a KATP channel opener as well as that of the sGC-cGMP pathway.
References
Suzuki Y, Cantu E, Christie JD (2007) Impact of immediate primary lung allograft dysfunction on bronchiolitis obliterans syndrome. Am J Respir Cirt Care Med 175:507–513
Daud SA, Yusen RD, Meyers BF et al (2007) Impact of immediate primary lung allograft dysfunction on bronchiolitis obliterans syndrome. Am J Respir Cirt Care Med 175:507–513
Taira N (1989) Nicorandil as a hybrid between nitrates and potassium channel activators. Am J Cardiol 63:18J–24J
IONA Study Group (2002) Effect of nicorandil on coronary events in patients with stable angina: the Impact Of Nicorandil in Angina (IONA) randomised trial. Lancet 359:1269–1275
Yamashita M, Schmid RA, Fujino S, Cooper JD, Patterson GA (2008) Nicorandil, a potent adenosine triphosphate-sensitive potassium-channel opener, ameliorates lung allograft reperfusion injury. J Thorac Cardiovasc Surg 112:1307–1314
Liu R, Ishibe Y, Ueda M (2000) Isoflurane-sevoflurane adminstration before ischemia attenuates ischemia–reperfusion-induced injury in isolated rat lungs. Anesthesiology 92:833–840
Drake R, Gaar KA, Taylor AE (1978) Estimation of the filtration coefficient of pulmonary exchange vessels. Am J Physiol 234:H266–274
Khimenko PL, Moore TM, Taylor AE (1995) ATP-sensitive K+ channels are not involved in ischemia-reperfusion lung endothelial injury. J Appl Physiol 79:554–559
Kandilci HB, Gümüşel B, Demiryürek AT, Lippton H (2006) Preconditioning modulates pulmonary endothelial dysfunction following ischemia-reperfusion injury in the rat lung: role of potassium channels. Life Sci 79:2172–2178
Pieper GM, Gross GJ (1992) EMD 52692 (bimakalim), a new potassium channel opener, attenuates luminol-enhanced chemiluminescence and superoxide anion radical formation by zymosan-activated polymorphonuclear leukocytes. Immunopharmacology 23:191–197
Chetham PM, Sefton WD, Bridges JP, Stevens T, McMurtry IF (1997) Inhaled nitric oxide pretreatment but not posttreatment attenuates ischemia-reperfusion-induced pulmonary microvascular leak. Anesthesiology 86:895–902
Egan TM, Hoffmann SC, Sevala M, Sadoff JD, Schlidt SA (2006) Nitroglycerin reperfusion reduces ischemia-reperfusion injury in non-heart-beating donor lungs. J Heart Lung Transplant 25:110–119
Takashima S, Koukoulis G, Inokawa H, Sevala M, Egan TM (2006) Inhaled nitric oxide reduces ischemia–reperfusion injury in rat lungs from non-heart-beating donors. J Thorac Cardiovasc Surg 132:132–139
Pinsky DJ, Oz MC, Koga S et al (1994) Cardiac preservation is enhanced in a heterotopic rat transplant model by supplementing the nitric oxide pathway. J Clin Investig 93:2291–2297
Minamiyama Y, Takemura S, Hai S, Suehiro S, Okada S, Funae Y (2007) Nicorandil elevates tissue cGMP levels in a nitric-oxide-independent manner. J Pharmacol Sci 103:33–39
Tajima M, Ishizuka N, Saitoh K, Sakagami H (2008) Nicorandil enhances the effect of endothelial nitric oxide under hypoxia-reoxygenation: role of the KATP channel. Eur J Pharmacol 579:86–92
Furuya A, Kashimoto S, Kumazawa T (2002) Effects of nicorandil on myocardial function and metabolism in the post-ischaemic reperfused heart with or without inhalation anaesthetics. Acta Anaesthesiol Scand 46:24–29
Acknowledgements
We are grateful to Yoshitsugu Tobe (laboratory technician, Department of Anesthesia and Intensive Care Medicine, Akita University Graduate School of Medicine, Akita City, Akita, Japan) for their technical assistance in performing the animal experiments.
Funding
This work was supported by Grants in Aid for Scientific Research from Japan Society for the Promotion of Science (No. 19591775).
Author information
Authors and Affiliations
Contributions
KA: This author helped design and conduct the study, collect the data, perform the analysis, and write the manuscript. TH: This author helped design the study and write the manuscript. KE: This author helped design the study and revise the manuscript. YM: This author helped conduct the study and revise the manuscript. TN, TK: This author helped review the original study data and revise the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Abe, K., Horiguchi, T., Enzan, K. et al. Nicorandil, a KATP Channel Opener, Attenuates Ischemia–Reperfusion Injury in Isolated Rat Lungs. Lung 198, 315–321 (2020). https://doi.org/10.1007/s00408-020-00339-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00408-020-00339-0