
Abstract
Purpose
An estimated 5–11% of patients with neurofibromatosis type 1 (NF1) harbour NF1 microdeletions encompassing the NF1 gene and its flanking regions. The purpose of this study was to evaluate the clinical phenotype in children and adolescents with NF1 microdeletions.
Methods
We retrospectively analysed 30 children and adolescents with NF1 microdeletions pertaining to externally visible neurofibromas. The internal tumour load was determined by volumetry of whole-body magnetic resonance imaging (MRI) in 20 children and adolescents with NF1 microdeletions. Furthermore, the prevalence of global developmental delay, autism spectrum disorder and attention deficit hyperactivity disorder (ADHD) were evaluated.
Results
Children and adolescents with NF1 microdeletions had significantly more often cutaneous, subcutaneous and externally visible plexiform neurofibromas than age-matched patients with intragenic NF1 mutations. Internal neurofibromas were detected in all 20 children and adolescents with NF1 microdeletions analysed by whole-body MRI. By contrast, only 17 (61%) of 28 age-matched NF1 patients without microdeletions had internal tumours. The total internal tumour load was significantly higher in NF1 microdeletion patients than in NF1 patients without microdeletions. Global developmental delay was observed in 28 (93%) of 30 children with NF1 microdeletions investigated. The mean full-scale intelligence quotient in our patient group was 77.7 which is significantly lower than that of patients with intragenic NF1 mutations. ADHD was diagnosed in 15 (88%) of 17 children and adolescents with NF1 microdeletion. Furthermore, 17 (71%) of the 24 patients investigated had T-scores ≥ 60 up to 75, indicative of mild to moderate autistic symptoms, which are consequently significantly more frequent in patients with NF1 microdeletions than in the general NF1 population. Also, the mean total T-score was significantly higher in patients with NF1 microdeletions than in the general NF1 population.
Conclusion
Our findings indicate that already at a very young age, NF1 microdeletions patients frequently exhibit a severe disease manifestation which requires specialized long-term clinical care.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neurofibromatosis type 1 (NF1) is a hereditary tumour predisposition syndrome with an incidence of 1 in 2000–3000 [1,2,3,4]. NF1 belongs to the group of phacomatoses which present with characteristic clinical symptoms of the skin, central and peripheral nervous system as well as tumours. The hallmark features of NF1 are café-au-lait spots, Lisch nodules, neurofibromas and an increased risk for malignant peripheral nerve sheath tumours as well as optic pathway gliomas. NF1 is caused in the majority of patients by mutations located within the NF1 gene on chromosome 17q11.2. However, 4.7–11% of all NF1 patients harbour large deletions encompassing the NF1 gene and its flanking regions at 17q11.2 [5,6,7,8]. These large NF1 deletions have also been termed ‘NF1 microdeletions’. Most frequent are type-1 NF1 microdeletions which encompass 1.4 Mb and include 14 protein-coding genes as well as four microRNA genes [9,10,11]. Type-1 deletions account for 70–80% of all large NF1 deletions and usually occur as germline deletions that are present in all cells of the affected patients [12, 13]. Less frequent than type-1 NF1 deletions are type-2, type-3 and atypical NF1 deletions that are distinguishable by their size and breakpoint location, by the number of genes located within the deletion region and the frequency of mosaicism with normal cells without the NF1 microdeletion [reviewed by 14].
Since the first identification of patients with NF1 microdeletions, it became clear that these patients often present with a severe clinical phenotype and exhibit features which are not common in patients with intragenic NF1 mutations including facial dysmorphic features, severe learning disabilities, high numbers of neurofibromas, overgrowth/tall-for-age stature, large hands and feet with excessive soft tissue, hyperflexibility of joints and congenital cardiac anomalies [8, 15,16,17,18,19,20,21]. The average intelligence in the group of patients with NF1 microdeletions was found to be lower than in patients with intragenic NF1 mutations [18, 22, 23].
Importantly, individuals with NF1 microdeletions have a lifetime MPNST risk in the range of 16–26% [18, 24], which is higher than the estimated lifetime MPNST risk in the general NF1 population of 8–13% according to Evans et al. [25, 26] or 15.8% according to Uusitalo et al. [27]. Further, MPNSTs occur significantly earlier in patients with NF1 microdeletions as compared with NF1 patients with intragenic mutations [24].
Several studies suggested that patients with NF1 microdeletions not only exhibit a high number of cutaneous neurofibromas but also an early (prepubertal) onset of cutaneous neurofibroma growth [9, 17, 28,29,30]. However, a comprehensive analysis of children has not yet been performed in order to ascertain whether an early (prepubertal) onset in growth of neurofibromas is significantly more prevalent in children with NF1 microdeletions as compared to children with intragenic NF1 mutations. Furthermore, the clinical phenotype in very young patients with NF1 microdeletions pertaining to cognitive abilities, developmental delay and the presence of autistic symptomatology has not been systematically investigated as yet.
In this study, we have addressed these issues and investigated the frequency of cutaneous, subcutaneous and plexiform neurofibromas as well as the internal tumour load in children and adolescents with NF1 microdeletions. In order to further characterize the clinical phenotype in this patient group, we also investigated cognition, the frequency of facial dysmorphic features, developmental delay, autistic symptoms, attention deficit, heart defects and skeletal anomalies. Our analyses indicate that the clinical phenotype associated with NF1 microdeletions is frequently very severe already at a young age of the patients who need long-term, specialized clinical care as well as intensive psychological and educational support.
Patients and methods
NF1 study population
In this study, we retrospectively characterized the clinical phenotype of 30 children and adolescents with NF1 microdeletions. The type and the extent of the NF1 deletions were analysed in our previous studies or in the course of the current study by either MLPA using the SALSA P122 C1 MLPA assay (MRC Holland, Amsterdam, Netherlands) or microarray analysis of blood leucocytes (Supp. Table S1) [31,32,33,34,35,36,37].
Eighteen of the 30 patients with NF microdeletions analysed were males, and 12 patients were females. One patient was a familial case, and the others were sporadic harbouring de novo NF1 microdeletions. The median age of the 30 patients was 4 years, and the mean age was 5.9 years (range: 1–15 years) at the time of investigation for the presence of externally visible neurofibromas. Eleven of these 30 patients were investigated for the presence of externally visible neurofibromas also in later years of their life (Supp. Table S2).
Six of the 30 patients with NF1 microdeletions analysed here have been clinically analysed by us already in a previous study which included a total of 29 patients with NF1 microdeletions [18]. In addition to the six children, this latter study included also 23 NF1 microdeletion patients who were mostly adults (mean age: 26.3 years).
Control group of patients with intragenic NF1 mutations
We also investigated 30 age-matched children and adolescents with intragenic NF1 mutations identified and clinically characterized at the Department of Neurology, University Medical Center Hamburg-Eppendorf. The median age of these patients was 5.0 years, and the mean age was 5.7 years (range: 1–15 years). This cohort of patients was analysed with regard to the presence or absence of cutaneous, subcutaneous and externally visible plexiform neurofibromas and used as control for the comparison with the number of patients with NF1 microdeletions exhibiting these tumours.
Externally visible neurofibromas
In total, 30 children and adolescents with NF1 microdeletions were evaluated for the presence of cutaneous, subcutaneous and externally visible plexiform neurofibromas. Cutaneous and subcutaneous neurofibromas were differentiated by the fact that cutaneous tumours move with the skin, whereas the skin can be moved over the top of subcutaneous tumours.
Internal tumour load
The load of internal neurofibromas of 20 children and adolescents with NF1 microdeletions was analysed by whole-body MRI performed at 1.5 T (Siemens Magnetom, Siemens Healthineers, Erlangen, Germany) between December 2004 and February 2017. The total volume of internal tumours in millilitres was determined by volumetric analysis of T2-weighted sequences in axial orientation (T2w HASTE TIRM: TR 1200 ms; TE 85 ms, FA 148°; Matrix 384 × 384; FOV 450 × 450mm; slice thickness 8 mm; intersection gap 10 mm) using MedX software (v3.42) as described previously [38]. The internal tumour load observed in the 20 NF1 microdeletion patients investigated in this study was compared with the internal tumour load of 28 age-matched NF1 patients without large deletions analysed by us in a previous study [39].
Brain tumours
In total, 29 NF1 microdeletion patients were analysed by head MRI in order to determine the presence of optic pathway gliomas (OP gliomas) or tumours located anatomically distinct from the optic pathway (non-optic pathway gliomas, non-OP gliomas).
Developmental delay and cognition
Global developmental delay in motor and language skills was assessed in 30 children with NF1 microdeletions by means the modified Munich Functional Developmental Diagnostics (Münchener Funktionelle Entwicklungsdiagnostik, MFED) for children at the age of 1–3 years [40, 41].
Cognitive ability in 24 patients with NF1 microdeletions was assessed at the age of 6–18 years by means of the German language versions of Wechsler Intelligence Scales (Hamburg-Wechsler Intelligence Scales HAWIK-R, HAWIK-III and HAWIK-IV) [42, 43]. Based on these tests, mean full-scale intelligence quotient (FSIQ) values was determined.
ASD and ADHD
Autism spectrum disorder (ASD) symptoms were assessed in 24 patients with NF1 microdeletions at the age of 4–18 years by means of the Social Responsiveness Scale (SRS) [44]. This questionnaire includes 65 items rated on a 4-point Likert scale (from 1 = never true to 4 = almost always true). It provides information concerning social awareness and cognition, reciprocal social communication, social motivation and autistic behaviours. The results of this questionnaire are summed into raw and T-scores based on population norms [45]. Total T-scores of 76 and higher indicate severe problems and are associated with a clinical diagnosis of ASD. Total T-scores of between 60 and 75 are considered as being in the mild to moderate range, indicating deficiencies in reciprocal social interaction, which may be clinically significant. T-scores below 59 are considered as being in the normal range.
Attention deficit hyperactivity disorder (ADHD) was assessed in 17 children and adolescents with NF1 microdeletions by means of test methods described in our previous study [18].
Scoliosis
The presence of scoliosis was determined in 30 children with NF1 microdeletions by X-ray analysis. A Cobb angle higher than 10 degree was considered as indicative of scoliosis.
Statistical analysis
The number of patients with NF1 microdeletions with or without externally visible or internal neurofibromas was compared with the number of patients from the general NF1 population or patients with intragenic NF1 mutations of the control cohort using the Fisher’s exact test (https://www.langsrud.com/stat/Fishertest.htm). The median total volume of internal tumours in millilitres was compared between deletion and non-deletion patients using a non-parametric Mann-Whitney U test (https://www.socscistatistics.com/tests/mannwhitney/default2.aspx.) Mean values were compared using an unpaired t test (https://www.graphpad.com/quickcalcs/ttest2/). A difference with p < 0.05 was considered as significant. All statistical tests performed were two-sided.
Results and discussion
In this study, we characterized the clinical phenotype of 30 children and adolescents with NF1 microdeletions. The type and the extent of the NF1 deletions were analysed in our previous studies or in the course of the current study as summarized in Supp. Table S1. Of these 30 patients, 27 harboured type-1 NF1 deletions spanning 1.4 Mb, whereas three patients had larger atypical NF1 deletions encompassing 2 Mb, 3 Mb and 4.7 Mb, respectively (Supp. Table S1).
Cutaneous and subcutaneous neurofibromas
Cutaneous neurofibromas (cnf) and subcutaneous neurofibromas (scnf) are age-related clinical features of NF1. In the general NF1 population, these benign tumours are often not present in very young children but may appear around the age of 8 years, and their numbers increase with age [46]. Puberty and pregnancy often represent periods of neurofibroma growth [reviewed by 47, 48]. In children with NF1 younger than 10 years of age, only 3% present with cnf [49] or 11% according to Huson et al. [50]. By contrast, many children with NF1 microdeletions seem to exhibit cnf and also scnf already at an early age [5, 9, 17, 29, 51, 52]. However, these studies included only a small number of young children, and the frequency of neurofibromas in children with NF1 microdeletions has not been systematically analysed in a larger group of these patients. In the study presented here, we determined the number of cnf and scnf in 30 children and adolescents with NF1 microdeletions (Supp. Table S2). We also assessed the absence or presence of these tumours in a control cohort of age-matched NF1 patients with intragenic NF1 mutations analysed at the University Medical Center Hamburg-Eppendorf (Supp. Table S3). The comparison of both patient groups indicated that children and adolescents with NF1 microdeletions exhibited significantly more often cnf and scnf than age-matched patients with intragenic NF1 mutations (Table 1). Importantly, none of the patients investigated by us had several hundreds of cnf or scnf at an early age. Such high numbers of neurofibromas are typically seen in a subgroup of adult patients with NF1. Nevertheless, seven of the 30 children and adolescents with NF1 microdeletions investigated by us had 50 or more cnf or scnf at the age of 18 years or younger (Supp. Table S2). In 13 children with NF1 microdeletions and neurofibromas present at an early age, the number of tumours was assessed also at later years of their life. A considerable increase in the number of tumours with advancing age was observed in 8 (61%) of these 13 patients (Supp. Table S2). This finding implies that if neurofibromas are present at an early age in patients with NF1 microdeletions, a further increase in the number of neurofibromas during childhood and adolescence is very likely.
We also compared the number of children and adolescents with NF1 microdeletions exhibiting cnf or scnf with those observed in the cohort of NF1 patients reported by Duong et al. [49]. The patients of this cohort were not selected pertaining to NF1 mutation type and thus represent the general NF1 population. The analysis of this control cohort demonstrated that only 3% of the children younger 10 years of age had cnf, whereas 10% exhibited scnf [49]. By contrast, 54% of the NF1 microdeletion patients younger than 10 years of age had cnf, and 41% exhibited scnf (Table 2).
In children and adolescents with NF1 microdeletions aged ≥ 10–19 years, 90% had cnf, and 67% exhibited scnf. By contrast, only 23% of the NF1 patients aged ≥ 10–19 years reported by Duong et al. [49] had cnf, and 26% of the patients exhibited scnf (Table 3). Taken together, children and adolescents with NF1 microdeletions had significantly more often cnf and scnf than patients of the general NF1 population (Tables 2 and 3). This difference remained significant if the three patients with atypical NF1 deletions larger than 1.4 Mb were not included in the comparison and only patients with type-1 NF1 microdeletions were analysed (Supp. Tables S4 and S5).
Also Huson et al. [50] investigated the proportion of children and adolescents with NF1 who exhibit cutaneous neurofibromas (cnf). The patients of this cohort were not selected with regard to NF1 mutation type and thus represent the general NF1 population. A comparison between the patients of this cohort and the children and adolescents with NF1 microdeletions analysed here indicated that children and adolescents with NF1 microdeletions had significantly more often cnf than age-matched patients of the general NF1 population (Supp. Tables S6 and S7).
It seems likely to assume that the hemizygous loss of a gene or genes located within the 1.4-Mb microdeletion region promotes early neurofibroma growth in NF1 microdeletion patients. The MIR193A gene is a good candidate for such a gene as it encodes for the microRNA miR-193a which represses c-kit expression [53, 54]. The tumour microenvironment, in particular c-kit expressing mast cells, has been shown to be important for neurofibroma growth in various mouse models [reviewed by 55]. Cutaneous neurofibromas are benign tumours that do not transform towards malignancy, and local inflammation mediated by activated mast cells plays a key role in the promotion of neurofibroma cell growth [reviewed by 55, 56, 57]. Mouse models indicated that the elimination of mast cells from the tumour microenvironment abrogated neurofibroma growth and that Nf1 heterozygous mast cells require c-kit for tumour formation and maintenance [58]. Hence, the reduced miR-193a expression in patients with NF1 microdeletions due to the hemizygous loss of the MIR193A gene may lead to enhanced c-kit expression and activation of mast cells which in turn contribute to early neurofibroma growth.
Externally visible plexiform neurofibromas
Previously performed studies reported the frequency of externally visible plexiform neurofibromas in patients with NF1 of all age groups to be 32% [50], 15% [59] and 16.8% [60]. Plexiform neurofibromas (pnf) are considered to be tumours of early embryonic origin, but not all pnf are readily detected in very young children if the tumours are of small size, even although the growth rate of pnf is highest in young children [60,61,62,63]. In the study presented here, we investigated the presence or absence of externally visible pnf in children and adolescents with NF1 microdeletions as well as age-matched patients with intragenic NF1 mutations. Pnf were observed in 16 (53%) of the 30 NF1 microdeletion patients. By contrast, only 7 (23%) of the 30 age-matched patients with intragenic NF1 mutations exhibited pnf (p = 0.032; two-tailed Fisher’s exact test; Table 1, Supp. Table S3). If only children in the age group of 1–3 years were analysed, 8 (67%) of 12 children with NF1 microdeletions had pnf. In contrast to this, only one child (9%) of the 11 children with intragenic NF1 mutations at the age of 1–3 years had a plexiform neurofibroma (Supp. Table S8). This difference in frequency of pnf among very young children at the age of 1–3 years was significant (p = 0.0094; two-tailed Fisher’s exact test) and suggests that pnf tend to be larger and therefore earlier to identify in children with NF1 microdeletions as compared with those with intragenic NF1 mutations.
In our previous study, pnf were observed in 22 (76%) of 29 NF1 microdeletion patients analysed [18]. These 29 patients were of different age (range: 4–43 years), and six of the young children analysed here were included in this previous study. If only the 19 adult NF1 microdeletion patients from our previous study are considered (mean age 29.5 years, SD 9.6), pnf were present in 15 (79%) of them. This proportion is much higher than the 15–32% of patients from the general NF1 population who exhibit pnf [50, 59, 60]. Hence, our present study including children and adolescents with NF1 microdeletions and our previous study on adult patients clearly indicate a very high frequency of pnf in patients with NF1 microdeletions.
Internal neurofibromas and MPNST
Whole-body MRI of NF1 patients indicates the burden of internal neurofibromas, most of which are not apparent on physical examination. At least 40% of adults with NF1 have internal neurofibromas as determined by whole-body MRI, whereas most of these tumours are asymptomatic [38, 64, 65].
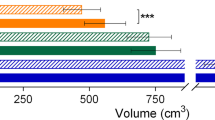
In order to assess the internal tumour load in children and adolescents with NF1 microdeletions, we performed whole-body MRI and volumetric analysis of all internal tumours identified (Supp. Table S2). This was possible for 20 NF1 microdeletion patients investigated in this study, and we compared these results with the internal tumour load of 28 age-matched NF1 patients without large deletions analysed by us in a previous study [39]. Internal tumours were observed in all 20 NF1 microdeletion patients analysed. By contrast, only 17 (61%) of the 28 non-deletion patients had internal tumours (Table 4). Thus, children and adolescents with NF1 microdeletions had more frequently internal neurofibromas than NF1 patients without microdeletions (p = 0.0011; Table 4).
We also determined the total internal tumour load in millilitres (ml) by volumetric analysis in 20 children and adolescents with NF1 microdeletions. The median total internal tumour volume was significantly higher in NF1 microdeletion patients than in age-matched NF1 patients without deletions (p = 0.0002; Table 4). If only non-deletion patients with internal tumours were included in the comparison, the difference remained statistically significant (p = 0.0423; Table 4).
Also the mean total tumour volume was significantly higher in patients with NF1 microdeletions than in NF1 patients without microdeletions (p = 0.0057; Table 4), if non-deletion patients without internal tumours were included in the analysis. Considering only patients with internal tumours, the mean total tumour volume remained to be significantly different comparing patients with and without NF1 microdeletions (p = 0.0216; Table 4). Considerable differences in total internal tumour volume per patient were observed in both groups of patients (Supp. Table S9).
The frequency of a very high internal tumour burden, as arbitrarily defined by total tumour volume of ≥ 800 ml, was not significantly different between children and adolescents with and without NF1 microdeletions (Table 4). Previously, we observed that an extremely high total internal tumour volume of > 3000 ml was significantly more frequent in patients with NF1 microdeletions than in NF1 patients without large deletions [39]. However, such an extremely high volume of internal tumours is likely to be age-related, and our previous study included many adult patients. Of the 38 NF1 deletion patients analysed in this previous study, 23 (61%) were adults. The mean age of these 38 patients was 26.1 years (SD 14.5, 95% CI 21.5–30.6; range: 3–69 years) [39]. The children and adolescents with large NF1 deletions analysed in the present study had a mean age of 8.7 years (SD 5.3; 95% CI 6.24–11.1; range: 1–19 years). We conclude that an extremely high total internal tumour volume of > 3000 ml is significantly more frequent in adults with NF1 microdeletions than in adults without large NF1 deletions. Even although an extremely high internal tumour volume of > 3000 ml was not observed in the children and adolescents analysed here, it is noteworthy that two of the children with type-1 NF1 microdeletions investigated by us exhibited a high internal tumour volume at a young age: female patient 450 had a total internal tumour volume of 1850 ml at the age of 9 years, whereas another girl, patient 1333, exhibited a total internal tumour volume of 2400 ml at the age of 12 years (Supp. Table S2). Equally high total internal tumour volumes were not observed in the group of patients without NF1 microdeletions (Supp. Table S9).
Previous studies indicated that a high burden of internal neurofibromas is a risk factor for malignant peripheral nerve sheath tumour (MPNST) in NF1 [38, 66, 67]. Most MPNSTs in NF1 patients develop from pre-existing externally visible or deep-seated, internal plexiform neurofibromas [68, 69]. Thus, the frequent occurrence of internal tumours in children and adolescents with NF1 microdeletions is important pertaining to their increased MPNST risk. Patients with NF1 microdeletions have a lifetime MPNST risk in the range of 16–26% which is at least twice as high as the risk of patients with intragenic NF1 mutations [18, 24]. Furthermore, MPNSTs seem to occur significantly earlier in patients with NF1 microdeletions as compared with patients with intragenic NF1 mutations [24]. De Raedt et al. [24] observed a substantial difference in the median age at diagnosis between patients with NF1 microdeletion (22 years) and non-deletion cases (30 years). In the study of De Raedt et al. [24], nine patients with large NF1 deletions and MPNSTs were investigated, and the youngest of these patients were 15, 17 and 18 years old.
In the group of patients with NF1 microdeletion investigated by us, 20 were clinically analysed since early childhood and also later in life. These 20 patients were 13–32 years old at the last time of investigation, with a median age of 25 years. Three (15%) of them died because of an MPNST at the age of 13, 26 and 27 years, respectively. All three patients had a high load of internal neurofibromas as concluded from a total internal tumour volume of 1430 ml, 2400 ml and 2500 ml, respectively (Supp. Table S2). An internal tumour load of 2400 ml was observed in a girl with a type-1 NF1 deletion who died at the age of 13 years because of an MPNST.
The increased risk of MPNSTs in patients with NF1 microdeletions is associated with hemizygosity of the SUZ12 gene, located within the NF1 microdeletion region. Both SUZ12 alleles are frequently inactivated in MPNSTs indicating its tumour suppressor function in this tumour type [70,71,72]. Our findings are in accordance with the view that in addition to SUZ12 loss, a high load of internal neurofibromas is causally associated with the increased MPNST risk in patients with NF1 microdeletions who need close monitoring by clinical examination and whole-body MRI performed already at an early age.
Gliomas
The most common central nervous system tumours in NF1 are gliomas [reviewed by 73]. Gliomas usually affect children with NF1; the mean age at diagnosis is 4.5 years. The most common glioma associated with NF1 is pilocytic astrocytoma, a WHO grade I tumour, with the optic pathway (OP) glioma being the hallmark lesion [reviewed by 73]. An estimated 15% of all children with NF1 investigated by MRI have OP gliomas [74]. In our study, three (10 %) of the 29 children and adolescents with NF1 microdeletions investigated by head MRI had non-symptomatic OP gliomas (Supp. Table S2). In the other 26 patients of our study group, neither symptomatic nor non-symptomatic OP gliomas were observed by MRI. Our findings do not indicate an increased OP glioma risk in children with NF1 microdeletions as compared with the general NF1 population (Supp. Table S10).
Non-OP gliomas, located anatomically distinct from the optic pathway, occur in 33–64% of children with NF1 [75,76,77]. In only one of the 29 NF1 microdeletion patients investigated by means of brain MRI, a non-OP glioma located in the brainstem was diagnosed at the age of 18 years (Supp. Table S2). Our findings suggest that also non-OP gliomas are not more frequent in children and adolescents with NF1 microdeletions than in the general NF1 population.
Skeletal anomalies
Scoliosis was observed in 16 (53%) of the 30 children with NF1 microdeletions investigated by us (Supp. Table S11). In previously performed studies including mainly adults with NF1 microdeletions, 43% exhibited scoliosis [17, 18]. The frequency of scoliosis in the general NF1 population is approximately 25% [78]. Our findings confirm that scoliosis is significantly more frequent in patients with NF1 microdeletions than in the general NF1 population (p = 0.0026, two-tailed Fisher’s exact test).
Pes cavus (hollow foot) was observed in 6 (21%) of 29 children with NF1 microdeletions analysed pertaining to this feature (Supp. Table S11). The frequency of this deformity in all patients with NF1 has not assessed as yet. However, pes cavus is not rare in the general population without NF1. In children of the general population at the age of 4–10 years, the frequency of pes cavus is 7%, and in the age group of 10–20 years, the frequency is 14% [79].
Pectus excavatum (PE) presents as a depression in the anterior chest wall resulting of a deviation of the dorsal sternum and the third to seventh rib or costal cartilage. The prevalence of PE in the normal population is 1:300–1000 [80, 81]. The frequency of pectus excavatum in the general NF1 population has been estimated to be up to 30% [82]. By contrast, Miraglia et al. [83] observed PE in only 12 (1%) of 1157 NF1 patients not selected pertaining to NF1 mutation type. We observed PE in 6 (21%) of the 29 children with NF1 microdeletions analysed here (Supp. Table S11). Further studies are necessary to investigate whether PE occurs more frequently in patients with NF1 microdeletions than in patients with intragenic NF1 mutations.
Heart defects
Echocardiography was performed in 18 of the children with NF1 microdeletions investigated here, and heart defects were observed in 9 (50%) of them. The heart defects included ventricular septal defect, pulmonic stenosis, atrial septal defect, aortic insufficiency and hypertrophic cardiomyopathy (Supp. Table S11). In a previous study, we observed cardiac defects in 6 (38%) of 16 NF1 microdeletion patients but none in 16 age-matched NF1 patients with intragenic NF1 mutations [20]. Hence, our present study confirms that congenital heart defects are frequent among patients with NF1 microdeletion and may cause severe clinical complications.
Facial dysmorphic features
Dysmorphic facial features including hypertelorism, broad fleshy nose, coarse facial appearance, flat forehead, deep front hair line, broad neck and low-set ears were observed in many of the NF1 microdeletion patients investigated (Supp. Figure S1). However, in particular in small children, some of these dysmorphic facial features were mild or even missing. In these cases, the facial appearance would not have immediately raised the suspicion of the presence of a large NF1 deletion.
Developmental delay
Infants with NF1 frequently have developmental difficulties across different cognitive and motor domains, and these delays are often apparent already at the end of the first year of life [84, reviewed by 85, 86,87,88]. It has been estimated that 68% of all children with NF1 exhibit delays in at least one of various areas of motor and/or language skills [86]. Longitudinal analysis of developmental delays in children with NF1 which were not preselected with regard to NF1 mutation type indicated that in particular motor delays do not improve over time [89]. Previous studies suggested that global developmental delay even more severe than the delay observed in many children from the general NF1 population is frequent in children with NF1 microdeletions [reviewed by 14]. However, this has not been systematically assessed as yet in a larger number of patients. In the present study, we observed global developmental delay in motor skills and speech in 28 (93%) of the 30 children with NF1 microdeletions investigated by us (Supp. Table S12). These delays were already apparent in young children (1–3 years of age), and the severe delays persisted during later childhood. Our study implies that severe global developmental delay is very frequent in children with NF1 microdeletions. Further detailed studies including children of different age groups, analysed by well-suited and standardized evaluation methods, are necessary to characterize and quantify all areas affected by the developmental delay observed in children with NF1 microdeletions in order to optimize the therapy for this specific group of NF1 patients who needs early intensive and long-term supportive intervention.
Five of the 30 children with NF1 microdeletions investigated here were in the preschool age at the time of investigation. For 25 patients with NF1 microdeletions, longitudinal assessments were available pertaining to their school performance and education. Nine (36%) of these 25 NF1 microdeletion patients attended a school for children with learning disabilities or for children with physical and cognitive deficits, whereas another nine patients (36%) were students with special needs in inclusion education. Seven (28%) patients attended secondary school as regular pupils (Supp. Table S12). We conclude that despite of the global developmental delays observed in most patients with NF1 microdeletions, there is variability pertaining to their performance at school as well as occupation later in life (Supp. Table S12).
FSIQ
Children with NF1 often experience cognitive and behavioural problems [90, 91, reviewed by 85 and 92]. Many studies indicated that the mean full-scale IQ (FSIQ) of children with NF1 is around 90 with a standard deviation of 11–15 (23, 93, reviewed by 85]. Thus, the mean FSIQ in children with NF1 is lower than that of children from the general population. However, variance and heritability of IQ in individuals with NF1 are similar to that of the general population and mostly driven by genetic background differences [23]. Patients with NF1 microdeletions exhibit significantly lower mean FSIQ scores than individuals carrying intragenic pathogenic NF1 mutations [18, 22, 23]. In the study presented here, we determined FSIQ scores in 24 children and adolescents with NF1 microdeletions investigated at the age of 6–18 years. The mean FSIQ in our patient group was 77.7 (SD 12.8) which is comparable with the findings of the previously performed studies as summarized in Table 5. Descheemaeker et al. [22] investigated 11 patients with NF1 microdeletions who exhibited a mean FSIQ of 76 (SD 6.9). By contrast, the mean FSIQ was 88.5 in 106 children without NF1 microdeletion investigated by these authors [22]. Taken together, previous studies and the results presented here indicate that the mean FSIQ in patients with NF1 microdeletions is significantly lower than in patients with intragenic NF1 mutations. However, as emphasized by Descheemaeker et al. [22], there is a substantial overlap between the FSIQ measures in NF1 patients with and without microdeletions. Furthermore, considerable heterogeneity of FSIQ scores is observed comparing individual patients with NF1 microdeletions. In our present study, five (21%) of the 24 patients with NF1 microdeletions analysed had an FSIQ below 70 indicative of mild intellectual disability. Only one of these five patients had an atypical 4.7-Mb spanning deletion, and the others exhibited the frequent type-1 NF1 microdeletions of 1.4 Mb. Previously performed studies indicated an FSIQ < 70 in 18%, 35% and 41% of NF1 microdeletions patients [18, 22, 23] (Table 5). Thus, intellectual disability is observed only in a subgroup of patients with NF1 microdeletions.
In our study, 12 (50%) of the 24 patients with NF1 microdeletions had an FSIQ ≥ 70–< 85. Remarkably, six of our patients with type-1 NF1 deletions had an FSIQ higher than 85 (Table 5, Supp. Table S12). The heterogeneity pertaining to the cognitive capabilities in the group of patients with NF1 microdeletions suggests that cognition in these patients is influenced by additional genetic and also environmental factors. Early therapeutic interventions to promote better achievements of functional potential later in life are very important in this group of patients.
Remarkably, one of our patients (patient 1261) had an FSIQ score of 110 and did not exhibit developmental delay. The patient has a type-1 NF1 deletion of 1.4-Mb as determined by MLPA of blood leucocytes which did not indicate mosaicism with normal cells. However, MLPA is not sensitive enough to detect normal cells without the deletion present at proportions of less than 20%. Hence, we cannot exclude somatic mosaicism with normal cells not harbouring the NF1 microdeletion in patient 1261 which would explain the high FSIQ and the absence of developmental delay in this patient. However, somatic mosaicism with normal cells not harbouring the NF1 microdeletion is very rare in patients with type-1 NF1 deletions [12, 13].
ADHD
It has been estimated that up to 50% of all NF1 patients present with attention deficit hyperactivity disorder (ADHD) [reviewed by 94]. NF1 patients with ADHD score lower in intelligence tests than healthy (sibling) controls and exhibit lower IQ scores than NF1 patients without ADHD [90, 94,95,96,97,98]. In patients with NF1, attention deficits are a specific additional risk factor for suboptimal intellectual performance [99]. Irrespective of cognitive deficits, elevated ADHD symptoms in children with NF1 have been shown to impair considerably daily life functioning [98]. In the study presented here, ADHD was diagnosed in 15 (88%) of 17 children and adolescents with NF1 microdeletion investigated (Supp. Table S12). We conclude that the high frequency of ADHD in patients with NF1 microdeletions contributes considerably to the academic underachievement observed in this group of patients. Early interventions to minimize ADHD symptoms may help to reduce the functional impairment of children with NF1 microdeletions.
ASD
Autism spectrum disorder (ASD) has been estimated to affect 0.8% of the general population [100]. Previous studies demonstrated a high incidence of ASD and a substantially elevated autistic trait burden in individuals with NF1 [93, 101, 102, reviewed by 103]. Social problems in children with NF1 are more prevalent in those with low intellectual functioning [104].
ASD symptoms can be assessed by different means; one of the acknowledged assessment tools is the social responsiveness scale (SRS) questionnaire [44]. The result of this questionnaire is converted into a T-score which indicates the presence or absence and the severity of autistic symptomatology. Clinical ASD, as measured by a T-score ≥ 76, affects 8.7% or 13.2% of NF1 patients according to the study of Morris et al. [105] and Eijk et al. [93], respectively (Table 6). Subclinical autistic symptoms as measured by T-scores ≥ 60–75 have been observed in 16.5% of NF1 patients as reported by Eijk et al. [93] and in 26% of the NF1 patients investigated by Morris et al. [105]. The patients of both cohorts were not selected pertaining to NF1 mutation type and hence are considered to represent the general NF1 population. Here, we determined T-scores in 24 patients with NF1 microdeletions at the age of 4–18 years. T-scores ≥ 76 were observed in two (8.3%) of the patients with NF1 microdeletions analysed in our study. Both patients were male, one with an atypical 4.7-Mb NF1 deletion and the other with a type-1 NF1 deletion of 1.4 Mb (Supp. Table S12). Consequently, severe, clinical ASD does not seem to be more frequent in patients with NF1 microdeletions as compared with the general NF1 population. However, T-scores ≥ 60–75 were observed in 17 (70.8%) of the 24 NF1 microdeletion patients analysed. Hence, T-scores ≥ 60–75, indicative of mild to moderate autistic symptoms, are significantly more frequent in patients with NF1 microdeletions than in the general NF1 population (Table 6, Supp. Tables S12 and S13). Also the mean total T-score was significantly higher in patients with NF1 microdeletions than in the general NF1 population (Table 6).
Among the 24 patients with NF1 microdeletions investigated by us were 15 males and nine females. Previous analyses indicated that male NF1 patients had significantly higher mean total T-scores relative to females [93, 101, 105]. A male predominance of ASD is also observed in the general population [106]. However, the mean T-scores in female and male patients with NF1 microdeletions investigated by us were not significantly different (Supp. Table S14). Nevertheless, the low number of patients analysed by us may mask a putative male predominance among patients with NF1 microdeletions and a high autistic trait burden. Taken together, 19 (79%) of the 24 NF1 microdeletion patients investigated by us had T-scores ≥ 60 and thus exhibited autistic symptomatology. The high frequency of neuropsychiatric phenotypes such as autism and hyperactivity-inattention as well as cognitive deficits observed in patients with NF1 microdeletions emphasizes the need for comprehensive social and therapeutic intervention to improve their executive function and academic performance.
Conclusion
Our study indicates a prevalence of global developmental delay, cognitive deficits, attention deficit and autistic symptomatology in children with NF1 microdeletions which is significantly higher than that observed in the general NF1 population. Our findings advocate an early screening and intensive support for children with NF1 microdeletions who represent an at-risk paediatric subgroup of severely affected NF1 patients. Furthermore, our results indicate a high frequency of cutaneous and subcutaneous neurofibromas in children and adolescents with NF1 microdeletions. Importantly, children and adolescents with NF1 microdeletions exhibit more often externally visible and internal plexiform neurofibromas than age-matched patients with intragenic NF1 mutations. The high prevalence of these tumours and their potential to transform towards malignancy as well as the loss of the SUZ12 gene included in the NF1 microdeletion region represent predisposing factors which increase the MPNST risk of patients with large NF1 deletions. Children and adolescents with NF1 microdeletions are a high-risk group for malignancy; whole-body MRI and long-term follow-up surveillance at a specialist NF1 network clinic are recommended.
References
Crowe FW, Schull WJ, Neel JV (1956) A clinical, pathological and genetic study of multiple neurofibromatosis. Charles C Thomas, Springfield, Illinois
Huson SM, Compston DA, Clark P, Harper PS (1989) A genetic study of von Recklinghausen neurofibromatosis in South East Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet 26:704–711
Lammert M, Friedman JM, Kluwe L, Mautner VF (2005) Prevalence of neurofibromatosis 1 in German children at elementary school enrolment. Arch Dermatol 141:71–74
Uusitalo E, Leppävirta J, Koffert A, Suominen S, Vahtera J, Vahlberg T, Pöyhönen M, Peltonen J, Peltonen S (2015) Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol 135:904–906
Cnossen MH, van der Est MN, Breuning MH, van Asperen CJ, Breslau-Siderius EJ, van der Ploeg AT, de Goede-Bolder A, van den Ouweland AM, Halley DJ, Niermeijer MF (1997) Deletions spanning the neurofibromatosis type 1 gene: implications for genotype-phenotype correlations in neurofibromatosis type 1? Hum Mutat 9:458–464
Rasmussen SA, Colman SD, Ho VT, Abernathy CR, Arn PH, Weiss L, Schwartz C, Saul RA, Wallace MR (1998) Constitutional and mosaic large NF1 gene deletions in neurofibromatosis type 1. J Med Genet 35:468–471
Kluwe L, Siebert R, Gesk S, Friedrich RE, Tinschert S, Kehrer-Sawatzki H, Mautner VF (2004) Screening 500 unselected neurofibromatosis 1 patients for deletions of the NF1 gene. Hum Mutat 23:111–116
Zhang J, Tong H, Fu X, Zhang Y, Liu J, Cheng R, Liang J, Peng J, Sun Z, Liu H, Zhang F, Lu W, Li M, Yao Z (2015) Molecular characterization of NF1 and neurofibromatosis type 1 genotype-phenotype correlations in a Chinese population. Sci Rep 5:11291
Dorschner MO, Sybert VP, Weaver M, Pletcher BA, Stephens K (2000) NF1 microdeletion breakpoints are clustered at flanking repetitive sequences. Hum Mol Genet 9:35–46
Jenne DE, Tinschert S, Reimann H, Lasinger W, Thiel G, Hameister H, Kehrer-Sawatzki H (2001) Molecular characterization and gene content of breakpoint boundaries in patients with neurofibromatosis type 1 with 17q11.2 microdeletions. Am J Hum Genet 69:516–527
López-Correa C, Dorschner M, Brems H, Lázaro C, Clementi M, Upadhyaya M, Dooijes D, Moog U, Kehrer-Sawatzki H, Rutkowski JL, Fryns JP, Marynen P, Stephens K, Legius E (2001) Recombination hotspot in NF1 microdeletion patients. Hum Mol Genet 10:1387–1392
Messiaen L, Vogt J, Bengesser K, Fu C, Mikhail F, Serra E, Garcia-Linares C, Cooper DN, Lazaro C, Kehrer-Sawatzki H (2011) Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental neurofibromatosis type-1 (NF1). Hum Mutat 32:213–219
Summerer A, Schäfer E, Mautner VF, Messiaen L, Cooper DN, Kehrer-Sawatzki H (2019) Ultra-deep amplicon sequencing indicates absence of low-grade mosaicism with normal cells in patients with type-1 NF1 deletions. Hum Genet 138:73–81
Kehrer-Sawatzki H, Mautner VF, Cooper DN (2017) Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum Genet 136:349–376
Venturin M, Guarnieri P, Natacci F, Stabile M, Tenconi R, Clementi M, Hernandez C, Thompson P, Upadhyaya M, Larizza L, Riva P (2004) Mental retardation and cardiovascular malformations in NF1 microdeleted patients point to candidate genes in 17q11.2. J Med Genet 41:35–41
Spiegel M, Oexle K, Horn D, Windt E, Buske A, Albrecht B, Prott EC, Seemanová E, Seidel J, Rosenbaum T, Jenne D, Kehrer-Sawatzki H, Tinschert S (2005) Childhood overgrowth in patients with common NF1 microdeletions. Eur J Hum Genet 13:883–888
Mensink KA, Ketterling RP, Flynn HC, Knudson RA, Lindor NM, Heese BA, Spinner RJ, Babovic-Vuksanovic D (2006) Connective tissue dysplasia in five new patients with NF1 microdeletions: further expansion of phenotype and review of the literature. J Med Genet 43:e8
Mautner VF, Kluwe L, Friedrich RE, Roehl AC, Bammert S, Högel J, Spöri H, Cooper DN, Kehrer-Sawatzki H (2010) Clinical characterisation of 29 neurofibromatosis type-1 patients with molecularly ascertained 1.4 Mb type-1 NF1 deletions. J Med Genet 47:623–630
Pasmant E, Sabbagh A, Spurlock G, Laurendeau I, Grillo E, Hamel MJ, Martin L, Barbarot S, Leheup B, Rodriguez D, Lacombe D, Dollfus H, Pasquier L, Isidor B, Ferkal S, Soulier J, Sanson M, Dieux-Coeslier A, Bièche I, Parfait B, Vidaud M, Wolkenstein P, Upadhyaya M, Vidaud D, members of the NF France Network (2010) NF1 microdeletions in neurofibromatosis type 1: from genotype to phenotype. Hum Mutat 31:E1506–E1518
Nguyen R, Mir TS, Kluwe L, Jett K, Kentsch M, Mueller G, Kehrer-Sawatzki H, Friedman JM, Mautner VF (2013) Cardiac characterization of 16 patients with large NF1 gene deletions. Clin Genet 84:344–349
Ning X, Farschtschi S, Jones A, Kehrer-Sawatzki H, Mautner VF, Friedman JM (2016) Growth in neurofibromatosis 1 microdeletion patients. Clin Genet 89:351–354
Descheemaeker MJ, Roelandts K, De Raedt T, Brems H, Fryns JP, Legius E (2004) Intelligence in individuals with a neurofibromatosis type 1 microdeletion. Am J Med Genet A 131:325–326
Ottenhoff MJ, Rietman AB, Mous SE, Plasschaert E, Gawehns D, Brems H, Oostenbrink R, ENCORE-NF1 Team, van Minkelen R, Nellist M, Schorry E, Legius E, Moll HA, Elgersma Y (2020) Examination of the genetic factors underlying the cognitive variability associated with neurofibromatosis type 1. Genet Med 22:889–897
De Raedt T, Brems H, Wolkenstein P, Vidaud D, Pilotti S, Perrone F, Mautner V, Frahm S, Sciot R, Legius E (2003) Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet 72:1288–1292
Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A (2002) Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 39:311–314
Evans DG, Huson SM, Birch JM (2012) Malignant peripheral nerve sheath tumours in inherited disease. Clin Sarcoma Res 2:17
Uusitalo E, Rantanen M, Kallionpää RA, Pöyhönen M, Leppävirta J, Ylä-Outinen H, Riccardi VM, Pukkala E, Pitkäniemi J, Peltonen S, Peltonen J (2016) Distinctive cancer associations in patients with neurofibromatosis type 1. J Clin Oncol 34:1978–1986
Kayes LM, Riccardi VM, Burke W, Bennett RL, Stephens K (1992) Large de novo DNA deletion in a patient with sporadic neurofibromatosis 1, mental retardation, and dysmorphism. J Med Genet 29:686–690
Kayes LM, Burke W, Riccardi VM, Bennett R, Ehrlich P, Rubenstein A, Stephens K (1994) Deletions spanning the neurofibromatosis 1 gene: identification and phenotype of five patients. Am J Hum Genet 54:424–436
Leppig KA, Kaplan P, Viskochil D, Weaver M, Ortenberg J, Stephens K (1997) Familial neurofibromatosis 1 microdeletions: cosegregation with distinct facial phenotype and early onset of cutaneous neurofibromata. Am J Med Genet 73:197–204
Kehrer-Sawatzki H, Tinschert S, Jenne DE (2003) Heterogeneity of breakpoints in non-LCR-mediated large constitutional deletions of the 17q11.2 NF1 tumour suppressor region. J Med Genet 40:e116
Kehrer-Sawatzki H, Kluwe L, Fünsterer C, Mautner VF (2005) Extensively high load of internal tumors determined by whole body MRI scanning in a patient with neurofibromatosis type 1 and a non-LCR-mediated 2-Mb deletion in 17q11.2. Hum Genet 116:466-475.
Bengesser K, Vogt J, Mussotter T, Mautner VF, Messiaen L, Cooper DN, Kehrer-Sawatzki H (2014) Analysis of crossover breakpoints yields new insights into the nature of the gene conversion events associated with large NF1 deletions mediated by nonallelic homologous recombination. Hum Mutat 35:215–226
Vogt J, Bengesser K, Claes KB, Wimmer K, Mautner VF, van Minkelen R, Legius E, Brems H, Upadhyaya M, Högel J, Lazaro C, Rosenbaum T, Bammert S, Messiaen L, Cooper DN, Kehrer-Sawatzki H (2014) SVA retrotransposon insertion-associated deletion represents a novel mutational mechanism underlying large genomic copy number changes with non-recurrent breakpoints. Genome Biol 15:R80
Hillmer M, Wagner D, Summerer A, Daiber M, Mautner VF, Messiaen L, Cooper DN, Kehrer-Sawatzki H (2016) Fine mapping of meiotic NAHR-associated crossovers causing large NF1 deletions. Hum Mol Genet 25:484–496
Hillmer M, Summerer A, Mautner VF, Högel J, Cooper DN, Kehrer-Sawatzki H (2017) Consideration of the haplotype diversity at nonallelic homologous recombination hotspots improves the precision of rearrangement breakpoint identification. Hum Mutat 38:1711–1722
Summerer A, Mautner VF, Upadhyaya M, Claes KBM, Högel J, Cooper DN, Messiaen L, Kehrer-Sawatzki H (2018) Extreme clustering of type-1 NF1 deletion breakpoints co-locating with G-quadruplex forming sequences. Hum Genet 137:511–520
Mautner VF, Asuagbor FA, Dombi E, Fünsterer C, Kluwe L, Wenzel R, Widemann BC, Friedman JM (2008) Assessment of benign tumour burden by whole-body MRI in patients with neurofibromatosis 1. Neuro-Oncology 10:593–598
Kluwe L, Nguyen R, Vogt J, Bengesser K, Mussotter T, Friedrich RE, Jett K, Kehrer-Sawatzki H, Mautner VF (2012) Internal tumour burden in neurofibromatosis Type I patients with large NF1 deletions. Genes Chromosom Cancer 51:447–451
Hellbrügge T (1994) Münchener Funktionelle Entwicklungsdiagnostik, zweites und drittes Lebensjahr. Durchführungs-, Beurteilungs- und Interpretationshinweise. 4. korrigierte und erweiterte Auflage. Deutsche Akademie für Entwicklungsrehabilitation, München 1994.
Köhler G, Egelkraut H (1984) Münchener Funktionelle Entwicklungsdiagnostik für das zweite und dritte Lebensjahr. Handanweisung. Universität München, Institut für Soziale Pädiatrie und Jugendmedizin, München.
Tewes U, Rossmann P, Schallberger U (1999) Hamburg-Wechsler-Intelligenztest für Kinder, third edn. Huber, Bern
Petermann F, Petermann U (2010) HAWIK-IV. Huber, Bern
Constantino JN, Davis SA, Todd RD, Schindler MK, Gross MM, Brophy SL, Metzger LM, Shoushtari CS, Splinter R, Reich W (2003) Validation of a brief quantitative measure of autistic traits: comparison of the social responsiveness scale with the autism diagnostic interview-revised. J Autism Dev Disord 33:427–433
Constantino J, Gruber C (2005) Social responsiveness scale. CA: Western Psychological Services, Los Angeles
DeBella K, Szudek J, Friedman JM (2000) Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 105:608–614
Roth TM, Petty EM, Barald KF (2008) The role of steroid hormones in the NF1 phenotype: focus on pregnancy. Am J Med Genet A 146A:1624–1633
Well L, Jaeger A, Kehrer-Sawatzki H, Farschtschi S, Avanesov M, Sauer M, de Sousa MT, Bannas P, Derlin T, Adam G, Mautner VF, Salamon JM (2020) The effect of pregnancy on growth-dynamics of neurofibromas in Neurofibromatosis type 1. PLoS One 15:e0232031
Duong TA, Bastuji-Garin S, Valeyrie-Allanore L, Sbidian E, Ferkal S, Wolkenstein P (2011) Evolving pattern with age of cutaneous signs in neurofibromatosis type 1: a cross-sectional study of 728 patients. Dermatology 222:269–273
Huson SM, Harper PS, Compston DA (1988) Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain 111:1355–1381
Tonsgard JH, Yelavarthi KK, Cushner S, Short MP, Lindgren V (1997) Do NF1 gene deletions result in a characteristic phenotype? Am J Med Genet 73:80–86
Riva P, Corrado L, Natacci F, Castorina P, Wu BL, Schneider GH, Clementi M, Tenconi R, Korf BR, Larizza L (2000) NF1 microdeletion syndrome: refined FISH characterization of sporadic and familial deletions with locus-specific probes. Am J Hum Genet 66:100–109
Gao XN, Lin J, Li YH, Gao L, Wang XR, Wang W, Kang HY, Yan GT, Wang LL, Yu L (2011) MicroRNA-193a represses c-kit expression and functions as a methylation-silenced tumor suppressor in acute myeloid leukemia. Oncogene 30:3416–3428
Xing CY, Hu XQ, Xie FY, Yu ZJ, Li HY, Bin-Zhou WJB, Tang LY, Gao SM (2015) Long non-coding RNA HOTAIR modulates c-KIT expression through sponging miR-193a in acute myeloid leukemia. FEBS Lett 589:1981–1987
Liao CP, Booker RC, Brosseau JP, Chen Z, Mo J, Tchegnon E, Wang Y, Clapp DW, Le LQ (2018) Contributions of inflammation and tumor microenvironment to neurofibroma tumorigenesis. J Clin Invest 128:2848–2861
Xu J, Zhang H, Li C, Du H, Shu M, Jia J (2019) Activation of PLCγ/AKT/IκBα/p65 signaling increases inflammation in mast cells to promote growth of cutaneous neurofibroma. Life Sci 239:117079
Jia J, Zhang H, Zhang H, Liu W, Shu M (2020) Infiltrating macrophages induced stem-cell-like features through PI3K/AKT/GSK3β signaling to promote neurofibroma growth. Arch Med Res 51:124–134
Yang FC, Ingram DA, Chen S, Zhu Y, Yuan J, Li X, Yang X, Knowles S, Horn W, Li Y, Zhang S, Yang Y, Vakili ST, Yu M, Burns D, Robertson K, Hutchins G, Parada LF, Clapp DW (2008) Nf1-dependent tumors require a microenvironment containing Nf1- and c-kit-dependent bone marrow. Cell 135:437–448
McGaughran JM, Harris DI, Donnai D, Teare D, MacLeod R, Westerbeek R, Kingston H, Super M, Harris R, Evans DG (1999) A clinical study of type 1 neurofibromatosis in north west England. J Med Genet 36:197–203
Waggoner DJ, Towbin J, Gottesman G, Gutmann DH (2000) Clinic-based study of plexiform neurofibromas in neurofibromatosis 1. Am J Med Genet 92:132–135
Dombi E, Solomon J, Gillespie AJ, Fox E, Balis FM, Patronas N, Korf BR, Babovic-Vuksanovic D, Packer RJ, Belasco J, Goldman S, Jakacki R, Kieran M, Steinberg SM, Widemann BC (2007) NF1 plexiform neurofibroma growth rate by volumetric MRI: relationship to age and body weight. Neurology 68:643–647
Nguyen R, Dombi E, Widemann BC, Solomon J, Fuensterer C, Kluwe L, Friedman JM, Mautner VF (2012) Growth dynamics of plexiform neurofibromas: a retrospective cohort study of 201 patients with neurofibromatosis 1. Orphanet J Rare Dis 7:75
Akshintala S, Baldwin A, Liewehr DJ, Goodwin A, Blakeley JO, Gross AM, Steinberg SM, Dombi E, Widemann BC (2020) Longitudinal evaluation of peripheral nerve sheath tumors in neurofibromatosis type 1: growth analysis of plexiform neurofibromas and distinct nodular lesions. Neuro-oncology, noaa053. Advance online publication. https://doi.org/10.1093/neuonc/noaa053
Tonsgard J, Kwak S, Short M, Dachman A (1998) CT imaging in adults with neurofibromatosis-1: frequent asymptomatic plexiform lesions. Neurology 50:1755–1760
Thakkar SD, Feigen U, Mautner VF (1999) Spinal tumours in neurofibromatosis type 1: an MRI study of frequency, multiplicity and variety. Neuroradiology 41:625–629
Tucker T, Wolkenstein P, Revuz J, Zeller J, Friedman JM (2005) Association between benign and malignant peripheral nerve sheath tumors in NF1. Neurology 65:205–211
Nguyen R, Jett K, Harris GJ, Cai W, Friedman JM (2014) Mautner VF (2014) Benign whole body tumor volume is a risk factor for malignant peripheral nerve sheath tumors in neurofibromatosis type 1. J Neuro-Oncol 116:307–313
Woodruff JM (1999) Pathology of tumors of the peripheral nerve sheath in type 1 neurofibromatosis. Am J Med Genet 89:23–30
Korf BR (2000) Malignancy in neurofibromatosis type 1. Oncologist 5:477–485
De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, Helin K, Hornick JL, Mautner V, Kehrer-Sawatzki H, Clapp W, Bradner J, Vidaud M, Upadhyaya M, Legius E, Cichowski K (2014) PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 514:247–251
Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, Tap WD, Fletcher JA, Huberman KH, Qin LX, Viale A, Singer S, Zheng D, Berger MF, Chen Y, Antonescu CR, Chi P (2014) PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet 46:1227–1232
Zhang M, Wang Y, Jones S, Sausen M, McMahon K, Sharma R, Wang Q, Belzberg AJ, Chaichana K, Gallia GL, Gokaslan ZL, Riggins GJ, Wolinksy JP, Wood LD, Montgomery EA, Hruban RH, Kinzler KW, Papadopoulos N, Vogelstein B, Bettegowda C (2014) Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet 46:1170–1172
Lobbous M, Bernstock JD, Coffee E, Friedman GK, Metrock LK, Chagoya G, Elsayed G, Nakano I, Hackney JR, Korf BR, Nabors LB (2020) An update on neurofibromatosis type 1-associated gliomas. Cancers (Basel) 12:E114
Listernick R, Charrow J, Greenwald MJ, Esterly NB (1989) Optic gliomas in children with neurofibromatosis type 1. J Pediatr 114:788–792
Guillamo JS, Créange A, Kalifa C, Grill J, Rodriguez D, Doz F, Barbarot S, Zerah M, Sanson M, Bastuji-Garin S, Wolkenstein P, Réseau NF, France (2003) Prognostic factors of CNS tumours in neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain 126:152–160
Griffith JL, Morris SM, Mahdi J, Goyal MS, Hershey T, Gutmann DH (2018) Increased prevalence of brain tumors classified as T2 hyperintensities in neurofibromatosis 1. Neurol Clin Pract 8:283–291
Mahdi J, Goyal MS, Griffith J, Morris SM, Gutmann DH (2020) Nonoptic pathway tumors in children with neurofibromatosis type 1. Neurology Apr 16. pii: 10.1212/WNL.0000000000009458. https://doi.org/10.1212/WNL.0000000000009458
Ueda K, Honda O, Satoh Y, Kawai M, Gyobu T, Kanazawa T, Hidaka S, Yanagawa M, Sumikawa H, Tomiyama N (2015) Computed tomography (CT) findings in 88 neurofibromatosis 1 (NF1) patients: prevalence rates and correlations of thoracic findings. Eur J Radiol 84:1191–1195
Troiano G, Nante N, Citarelli GL (2017) Pes planus and pes cavus in Southern Italy: a 5 years study. Ann Ist Super Sanita 53:142–145
Kelly RE Jr, Lawson ML, Paidas CN, Hruban RH (2005) Pectus excavatum in a 112-year autopsy series: anatomic findings and the effect on survival. J Pediatr Surg 40:1275–1278
Westphal FL, Lima LC, Lima Neto JC, Chaves AR, Santos Júnior VL, Ferreira BL (2009) Prevalence of pectus carinatum and pectus excavatum in students in the city of Manaus, Brazil. J Bras Pneumol 35:221–226
Riccardi VM, Eichner JE (1986) Neurofibromatosis: phenotype, natural history and pathogenesis. Johns Hopkins University Press, Baltimore, MD
Miraglia E, Chello C, Calvieri S, Giustini S (2919) Pectus excavatum in neurofibromatosis type 1: single-center experience and review of the literature. [published online ahead of print, 2019 Sep 26]. G Ital Dermatol Venereol. 2019 https://doi.org/10.23736/S0392-0488.19.06456-3
Lorenzo J, Barton B, Acosta MT, North K (2011) Mental, motor, and language development of toddlers with neurofibromatosis type 1. J Pediatr 158:660–665
Lehtonen A, Howie E, Trump D, Huson SM (2013) Behaviour in children with neurofibromatosis type 1: cognition, executive function, attention, emotion, and social competence. Dev Med Child Neurol 55:111–125
Soucy EA, Gao F, Gutmann DH, Dunn CM (2012) Developmental delays in children with neurofibromatosis type 1. J Child Neurol 27:641–644
Kolesnik AM, EJH J, Garg S, Green J, Charman T, Johnson MH, EDEN-BASIS Team (2017) Early development of infants with neurofibromatosis type 1: a case series. Mol Autism 23(8):62
Arnold SS, Payne JM, Lorenzo J, North KN, Barton B (2018) Preliteracy impairments in children with neurofibromatosis type 1. Dev Med Child Neurol 60:703–710
Wessel LE, Gao F, Gutmann DH, Dunn CM (2013) Longitudinal analysis of developmental delays in children with neurofibromatosis type 1. J Child Neurol 28:1689–1693
Hyman SL, Shores A, North KN (2005) The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology 65:1037–1044
Krab LC, Aarsen FK, de Goede-Bolder A, Catsman-Berrevoets CE, Arts WF, Moll HA, Elgersma Y (2008) Impact of neurofibromatosis type 1 on school performance. J Child Neurol 23:1002–1010
Hachon C, Iannuzzi S, Chaix Y (2011) Behavioural and cognitive phenotypes in children with neurofibromatosis type 1 (NF1): the link with the neurobiological level. Brain and Development 33:52–61
Eijk S, Mous SE, Dieleman GC, Dierckx B, Rietman AB, de Nijs PFA, Ten Hoopen LW, van Minkelen R, Elgersma Y, Catsman-Berrevoets CE, Oostenbrink R, Legerstee JS (2018) Autism spectrum disorder in an unselected cohort of children with neurofibromatosis type 1 (NF1). J Autism Dev Disord 48:2278–2285
Heimgärtner M, Granström S, Haas-Lude K, Leark RA, Mautner VF, Lidzba K (2019) Attention deficit predicts intellectual functioning in children with neurofibromatosis type 1. Int J Pediatr 10:9493837
Mautner VF, Kluwe L, Thakker SD, Leark RA (2002) Treatment of ADHD in neurofibromatosis type 1. Dev Med Child Neurol 44:164–170
Pride NA, Payne JM, North KN (2012) The impact of ADHD on the cognitive and academic functioning of children with NF1. Dev Neuropsychol 37:590–600
Garg S, Lehtonen A, Huson SM, Emsley R, Trump D, Evans DG, Green J (2013) Autism and other psychiatric comorbidity in neurofibromatosis type 1: evidence from a population-based study. Dev Med Child Neurol 55:139–145
Payne JM, Haebich KM, MacKenzie R, Walsh KS, Hearps SJC, Coghill D, Barton B, Pride NA, Ullrich NJ, Tonsgard JH, Viskochil D, Schorry EK, Klesse L, Fisher MJ, Gutmann DH, Rosser T, Packer RJ, Korf B, Acosta MT, Bellgrove MA, North KN (2019) Cognition, ADHD symptoms, and functional impairment in children and adolescents with neurofibromatosis type 1. J Atten Disord 2019(14):1087054719894384
Isenberg JC, Templer A, Gao F, Titus JB, Gutmann DH (2013) Attention skills in children with neurofibromatosis type 1. J Child Neurol 28:45–49
Baxter AJ, Brugha TS, Erskine HE, Scheurer RW, Vos T, Scott JG (2015) The epidemiology and global burden of autism spectrum disorders. Psychol Med 45:601–613
Garg S, Heuvelman H, Huson S, Tobin H, Green J, Northern UK NF1 Research Network (2016) Sex bias in autism spectrum disorder in neurofibromatosis type 1. J Neurodev Disord 8:26
Morris SM, Gutmann DH (2018) A genotype-phenotype correlation for quantitative autistic trait burden in neurofibromatosis 1. Neurology 90:377–379
Haebich KM, Pride NA, Walsh KS, Chisholm A, Rouel M, Maier A, Anderson V, Barton B, Silk T, Korgaonkar M, Seal M, Lami F, Lorenzo J, Williams K, Dabscheck G, Rae CD, Kean M, North KN, Payne JM (2019) Understanding autism spectrum disorder and social functioning in children with neurofibromatosis type 1: protocol for a cross-sectional multimodal study. BMJ Open 9:e030601
Huijbregts SC, de Sonneville LM (2011) Does cognitive impairment explain behavioral and social problems of children with neurofibromatosis type 1? Behav Genet 41:430–436
Morris SM, Acosta MT, Garg S, Green J, Huson S, Legius E, North KN, Payne JM, Plasschaert E, Frazier TW, Weiss LA, Zhang Y, Gutmann DH, Constantino JN (2016) Disease burden and symptom structure of autism in neurofibromatosis type 1: a study of the international NF1-ASD consortium team (INFACT). JAMA Psychiatry 73:1276–1284
Halladay AK, Bishop S, Constantino JN, Daniels AM, Koenig K, Palmer K, Messinger D, Pelphrey K, Sanders SJ, Singer AT, Taylor JL, Szatmari P (2015) Sex and gender differences in autism spectrum disorder: summarizing evidence gaps and identifying emerging areas of priority. Mol Autism 6:36
Availability of data and material
All detailed results and relevant data are available as supplementary material.
Code availability
Not applicable.
Funding
Open Access funding provided by Projekt DEAL. This study has been supported by the “Bundesverband Neurofibromatose e.V.” (https://bv-nf.de/).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare not to have any conflict of interest and no disclosures to make.
Ethics approval
Ethics approval for this study was obtained from the authorities at the University Medical Centre Hamburg-Eppendorf, and the patient’s parents gave written informed consent.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kehrer-Sawatzki, H., Kluwe, L., Salamon, J. et al. Clinical characterization of children and adolescents with NF1 microdeletions. Childs Nerv Syst 36, 2297–2310 (2020). https://doi.org/10.1007/s00381-020-04717-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-020-04717-0